
Exploring Regorafenib Responsiveness and Uncovering Molecular Mechanisms in Recurrent Glioblastoma Tumors through Longitudinal In Vitro Sampling
 ,
,  ,
,  , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. GB Tissue Collection
2.2. GB Explant (GB-EXP) Cultures
2.3. GB Cell Lines
2.4. Spheroid Cultures
2.5. Drug Treatments
2.6. Cell Viability
2.7. Nucleic Acids Isolation
2.8. KI67 Expression Analysis
2.9. Confocal Imaging
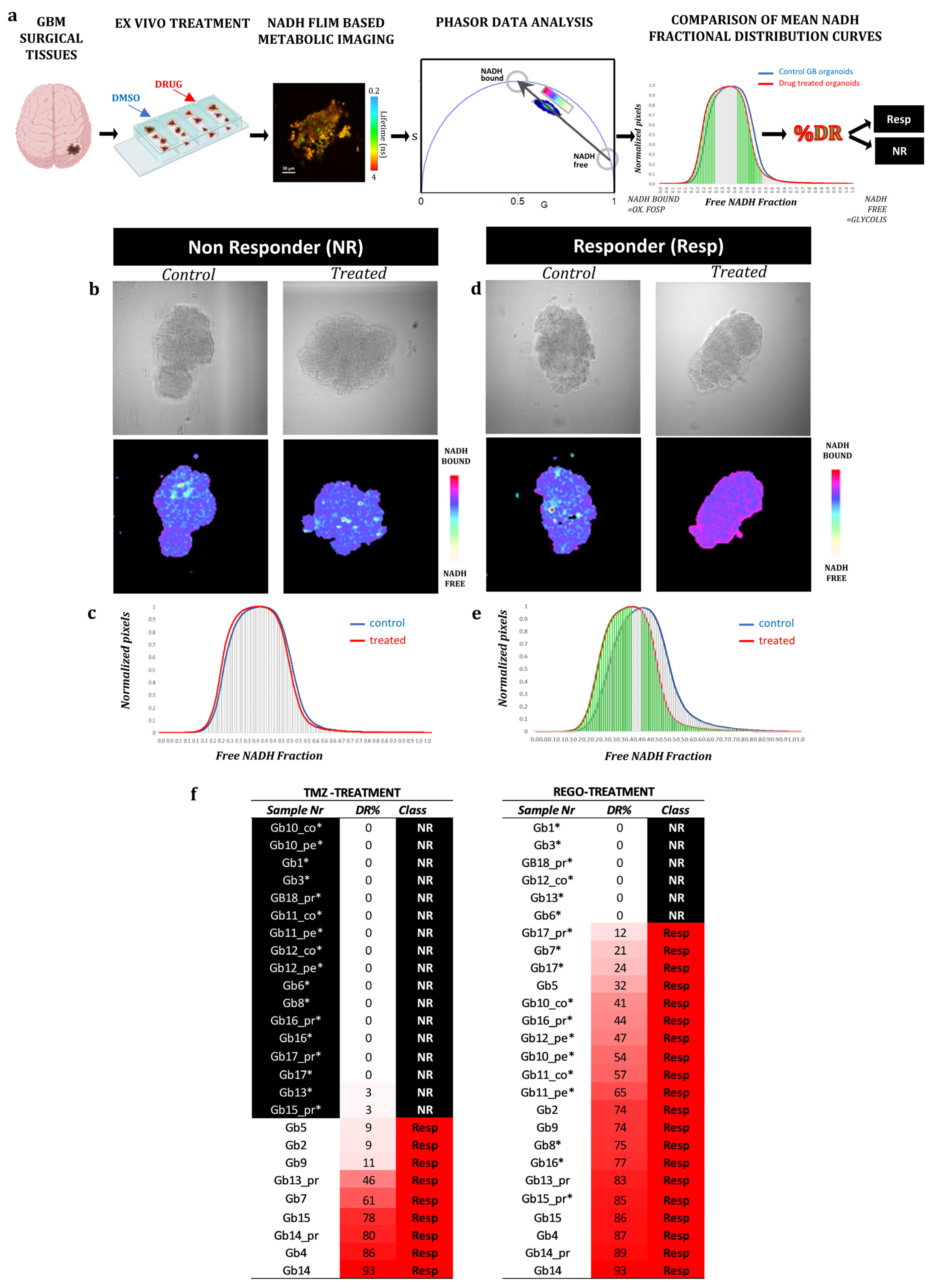
2.10. Lifetime Imaging
2.11. Histology and Staining
2.12. Whole-Transcriptome RNA Analysis (WTA) Libraries
2.13. Whole-Exome Analysis (WEA) Libraries
2.14. Data Analysis
2.14.1. FLIM Data Analysis
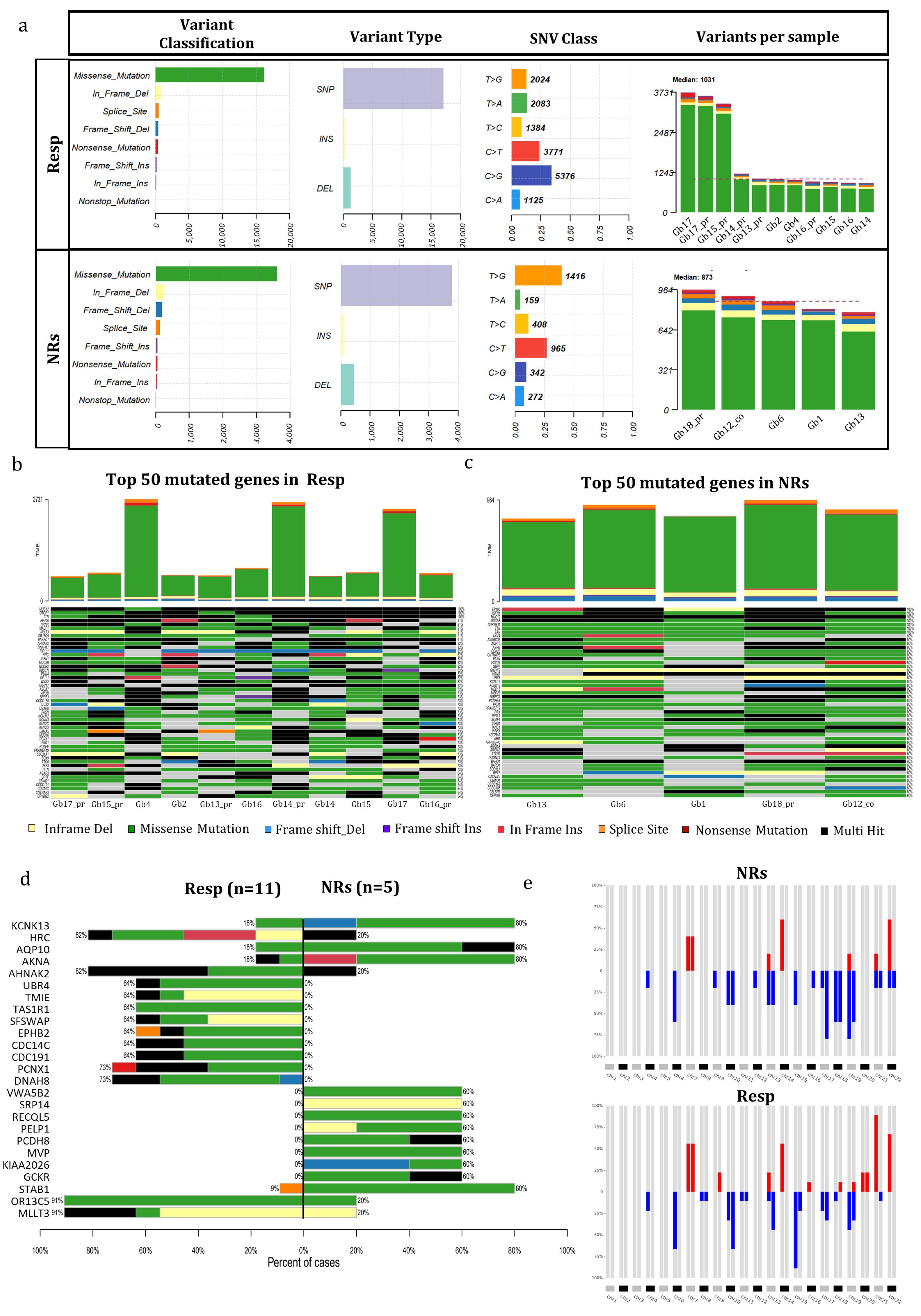
2.14.2. Next-Generation Sequencing Data Analysis
2.14.3. Statistical Analysis
3. Results
3.1. Tumor Samples’ Characteristics
3.2. FLIM Imaging of Patient-Derived Organoids
3.3. Mutational Genetic Background in Regorafenib Resp and NR GB Tumors
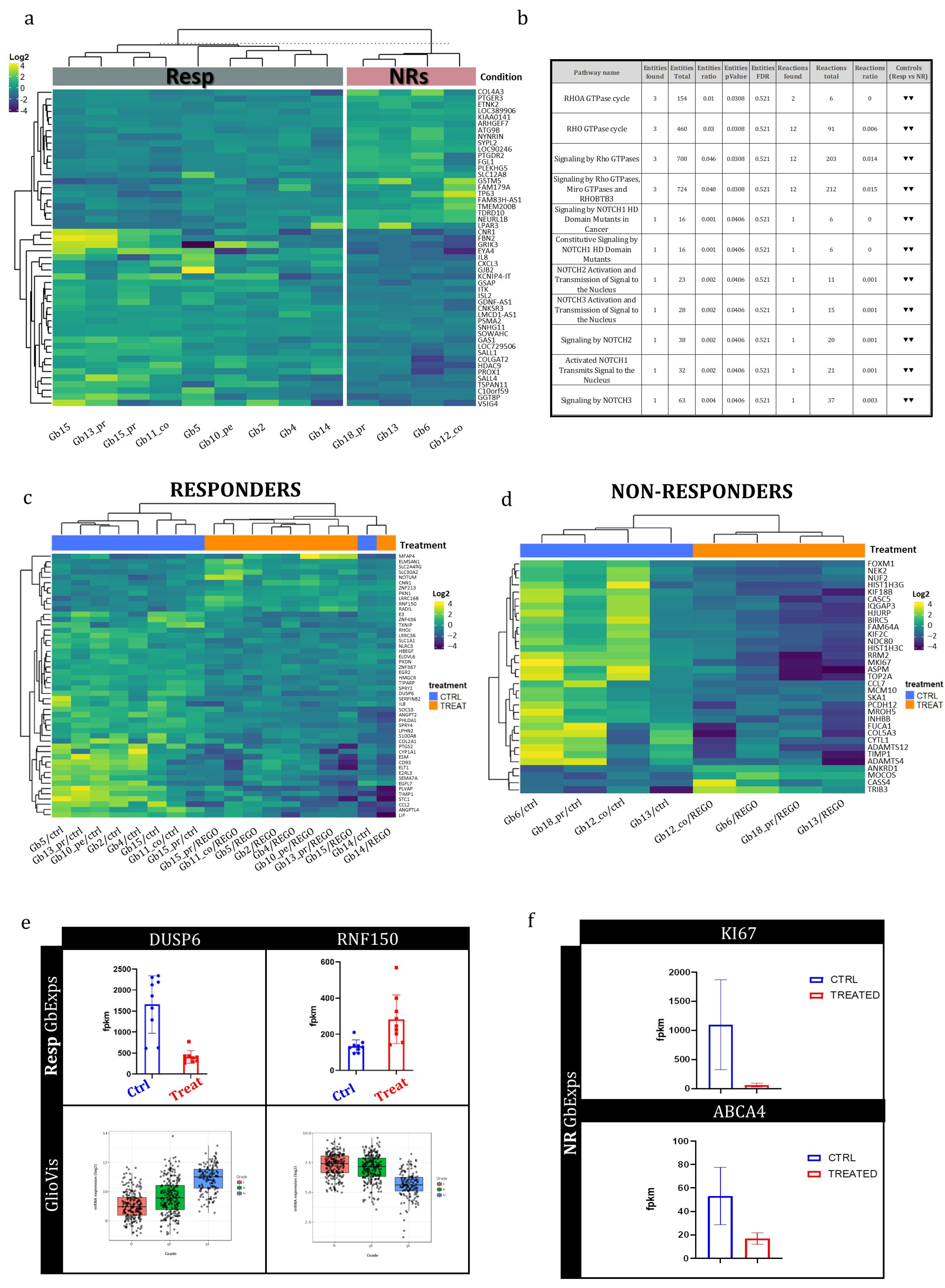
3.4. Transcriptional Genetic Background in Regorafenib Resp and NR GB Tumors
3.5. Early Whole-Transcriptome Changes Induced by Regorafenib Treatment in GB-EXPs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ostrom, Q.T.; Patil, N.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2013–2017. Neuro Oncol. 2020, 22 (Suppl. S1), iv1–iv96. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Rønning, P.A.; Helseth, E.; Meling, T.R.; Johannesen, T.B. A population-based study on the effect of temozolomide in the treatment of glioblastoma multiforme. Neuro Oncol. 2012, 14, 1178–1184. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.B.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Batchelor, T.T.; Gerstner, E.R.; Ye, X.; Desideri, S.; Duda, D.G.; Peereboom, D.; Lesser, G.J.; Chowdhary, S.; Wen, P.Y.; Grossman, S.; et al. Feasibility, phase I, and phase II studies of tandutinib, an oral platelet-derived growth factor receptor-β tyrosine kinase inhibitor, in patients with recurrent glioblastoma. Neuro Oncol. 2017, 19, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Kalpathy-Cramer, J.; Chandra, V.; Da, X.; Ou, Y.; Emblem, K.W.; Muzikansky, A.; Cai, X.; Douw, L.; Evans, J.G.; Dietrich, J.; et al. Phase II study of tivozanib, an oral VEGFR inhibitor, in patients with recurrent glioblastoma. J. Neurooncol. 2017, 131, 603–610. [Google Scholar] [CrossRef]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.-H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Grothey, A.; Van Cutsem, E.; Sobrero, A.; Siena, S.; Falcone, A.; Ychou, M.; Humblet, Y.; Bouché, O.; Mineur, L.; Barone, C.; et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 303–312. [Google Scholar] [CrossRef]
- Demetri, G.D.; Reichardt, P.; Kang, Y.-K.; Blay, J.-Y.; Rutkowski, P.; Gelderblom, H.; Hohenberger, P.; Leahy, M.; Von Mehren, M.; Joensuu, H.; et al. Effi cacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 295–302. [Google Scholar] [CrossRef]
- Lombardi, G.; De Salvo, G.L.; Brandes, A.A.; Eoli, M.; Rudà, R.; Faedi, M.; Lolli, I.; Pace, A.; Daniele, B.; Pasqualetti, F.; et al. Regorafenib compared with lomustine in patients with relapsed glioblastoma (REGOMA): A multicentre, open-label, randomised, controlled, phase 2 trial. Lancet Oncol. 2019, 20, 110–119. [Google Scholar] [CrossRef]
- Mongiardi, M.P.; Buccarelli, M.; Formato, A.; Orecchini, E.; Salbini, M.; Ricci, V.; Orsini, T.; Putti, S.; Chiesa, S.; Ricci-Vitiani, L.; et al. Characterization of Glioblastoma Characterization of Glioblastoma Cells Response to Regorafenib. Cancers 2022, 14, 6193. [Google Scholar] [CrossRef]
- Lombardi, G.; Caccese, M.; Padovan, M.; Cerretti, G.; Pintacuda, G.; Manara, R.; Di Sarra, F.; Zagonel, V. Regorafenib in Recurrent Glioblastoma Patients: A Large and Monocentric Real-Life Study. Cancers 2021, 13, 4731. [Google Scholar] [CrossRef]
- Singh, K.; Hotchkiss, K.M.; Parney, I.F.; De Groot, J.; Sahebjam, S.; Sanai, N.; Platten, M.; Galanis, E.; Lim, M.; Wen, P.Y.; et al. Correcting the drug development paradigm for glioblastoma requires serial tissue sampling. Nat. Med. 2023, 29, 2402–2405. [Google Scholar] [CrossRef]
- Morelli, M.; Lessi, F.; Barachini, S.; Liotti, R.; Montemurro, N.; Perrini, P.; Santonocito, O.S.; Gambacciani, C.; Snuderl, M.; Pieri, F.; et al. Metabolic-imaging of human glioblastoma live tumors: A new precision-medicine approach to predict tumor treatment response early. Front. Oncol. 2022, 12, 969812. [Google Scholar] [CrossRef]
- Foty, R. A simple hanging drop cell culture protocol for generation of 3D spheroids. J. Vis. Exp. 2011, 51, e2720. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Ranjit, S.; Malacrida, L.; Jameson, D.M.; Gratton, E. Fit-free analysis of fluorescence lifetime imaging data using the phasor approach. Nat. Protoc. 2018, 13, 1979–2004. [Google Scholar] [CrossRef]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Ding, X.; Shen, Y.; Lyon, G.J.; Wang, K. SeqMule: Automated pipeline for analysis of human exome/genome sequencing data. Sci. Rep. 2015, 5, srep14283. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, D.; Sato, T.; Cibulskis, K.; Getz, G.; Stewart, C.; Lichtenstein, L. Calling Somatic SNVs and Indels with Mutect2. bioRxiv 2019, 861054. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Mayakonda, A.; Lin, D.C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef] [PubMed]
- Talevich, E.; Shain, A.H.; Botton, T.; Bastian, B.C. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput. Biol. 2016, 12, e1004873. [Google Scholar] [CrossRef]
- Franch-Expósito, S.; Bassaganyas, L.; Vila-Casadesús, M.; Hernández-Illán, E.; Esteban-Fabró, R.; Díaz-Gay, M.; Lozano, J.J.; Castells, A.; Llovet, J.M.; Castellví-Bel, S.; et al. CNApp, a tool for the quantification of copy number alterations and integrative analysis revealing clinical implications. eLife 2020, 9, e50267. [Google Scholar] [CrossRef]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef]
- Liaw, A.; Wiener, M. Classification and Regression by randomForest. R News 2002, 2, 18–22. Available online: http://www.stat.berkeley.edu/ (accessed on 28 December 2023).
- Trinh, A.L.; Chen, H.; Chen, Y.; Hu, Y.; Li, Z.; Siegel, E.R.; Linskey, M.E.; Wang, P.H.; Digman, M.A.; Zhou, Y.-H. Tracking functional tumor cell subpopulations of malignant glioma by phasor fluorescence lifetime imaging microscopy of NADH. Cancers 2017, 9, 168. [Google Scholar] [CrossRef]
- Lukina, M.M.; Dudenkova, V.V.; Ignatova, N.I.; Druzhkova, I.N.; Shimolina, L.E.; Zagaynova, E.V.; Shirmanova, M.V. Metabolic cofactors NAD(P)H and FAD as potential indicators of cancer cell response to chemotherapy with paclitaxel. Biochim. Biophys. Acta—Gen. Subj. 2018, 1862, 1693–1700. [Google Scholar] [CrossRef]
- Xiao, B.; Kuang, Z.; Zhang, W.; Hang, J.; Chen, L.; Lei, T.; He, Y.; Deng, C.; Li, W.; Lu, J.; et al. Glutamate Ionotropic Receptor Kainate Type Subunit 3 (GRIK3) promotes epithelial-mesenchymal transition in breast cancer cells by regulating SPDEF/CDH1 signaling. Mol. Carcinog. 2019, 58, 1314–1323. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.; Shah, S.; Bhattacharya, D.; Toukam, D.K.; Cáceres, R.; Krummel, D.A.P.; Sengupta, S. Ligand-Gated Ion Channels as Targets for Treatment and Management of Cancers. Front. Physiol. 2022, 13, 839437. [Google Scholar] [CrossRef] [PubMed]
- Jou, Y.C.; Wang, S.C.; Dia, Y.C.; Wang, S.T.; Yu, M.H.; Yang, H.Y.; Chen, L.C.; Shen, C.H.; Liu, Y.W. Anti-Cancer Effects and Tumor Marker Role of Glutathione S-Transferase Mu 5 in Human Bladder Cancer. Int. J. Mol. Sci. 2021, 22, 3056. [Google Scholar] [CrossRef]
- Sun, B.; Xu, L.; Bi, W.; Ou, W.B. SALL4 Oncogenic Function in Cancers: Mechanisms and Therapeutic Relevance. Int. J. Mol. Sci. 2022, 23, 2053. [Google Scholar] [CrossRef]
- Yang, C.; Zhang, J.; Ding, M.; Xu, K.; Li, L.; Mao, L.; Zheng, J. Ki67 targeted strategies for cancer therapy. Clin. Transl. Oncol. 2017, 20, 570–575. [Google Scholar] [CrossRef]
- Duan, C.; Yu, M.; Xu, J.; Li, B.-Y.; Zhao, Y.; Kankala, R.K. Overcoming Cancer Multi-drug Resistance (MDR): Reasons, mechanisms, nanotherapeutic solutions, and challenges. Biomed. Pharmacother. 2023, 162, 114643. [Google Scholar] [CrossRef] [PubMed]
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee, S.U. Glioblastoma Multiforme: A Review of its Epidemiology and Pathogenesis through Clinical Presentation and Treatment. Asian Pac. J. Cancer Prev. 2017, 18, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, M.; Tanaka, A.; Namba, K.; Shia, J.; Wang, J.Y.; Roehrl, M.H. Early-Stage Loss of GALNT6 Predicts Poor Clinical Outcome in Colorectal Cancer. Front. Oncol. 2022, 12, 802548. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Y.; Liu, Y.; Wang, Z.M.; Liu, Z.; Wu, M. FLIM as a Promising Tool for Cancer Diagnosis and Treatment Monitoring. Nano-Micro Lett. 2021, 13, 133. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Zhang, L.; Chen, H.; Lei, Y.; Zhang, T.; Wang, Y.; Jin, P.; Lan, J.; Zhou, L.; Huang, Z.; et al. Regorafenib induces lethal autophagy arrest by stabilizing PSAT1 in glioblastoma. Autophagy 2019, 16, 106–122. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Qu, C.; Han, C.; Chen, M.M.; An, L.J.; Zou, W. Potassium channels and their role in glioma: A mini review. Mol. Membr. Biol. 2019, 35, 76–85. [Google Scholar] [CrossRef]
- Felipe, A.; Bielanska, J.; Comes, N.; Vallejo, A.; Roig, S.; Cajal, S.R.Y.; Condom, E.; Hernandez-Losa, J.; Ferreres, J.C. Targeting the voltage-dependent K(+) channels Kv1.3 and Kv1.5 as tumor biomarkers for cancer detection and prevention. Curr. Med. Chem. 2012, 19, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Li, B.; Li, W.; Guo, H.; Zou, F. ATP-sensitive potassium channels control glioma cells proliferation by regulating ERK activity. Carcinogenesis 2009, 30, 737–744. [Google Scholar] [CrossRef]
- Wang, J.; Feng, L.; Zhu, Z.; Zheng, M.; Wang, D.; Chen, Z.; Sun, H. Aquaporins as diagnostic and therapeutic targets in cancer: How far we are? J. Transl. Med. 2015, 13, 96. [Google Scholar] [CrossRef]
- Ramírez-González, A.; Manzo-Merino, J.; Contreras-Ochoa, C.O.; Bahena-Román, M.; Aguilar-Villaseñor, J.M.; Lagunas-Martínez, A.; Rosenstein, Y.; Marina, V.M.; Torres-Poveda, K. Functional role of akna: A scoping review. Biomolecules 2021, 11, 1709. [Google Scholar] [CrossRef]
- David, C.; Nance, J.P.; Hubbard, J.; Hsu, M.; Binder, D.; Wilson, E.H. Stabilin-1 Expression in Tumor Associated Macrophages. Brain Res. 2012, 1481, 71–78. [Google Scholar] [CrossRef]
- Daniel, P.; Meehan, B.; Sabri, S.; Jamali, F.; Sarkaria, J.N.; Choi, D.-S.; Garnier, D.; Kitange, G.; I Glennon, K.; Paccard, A.; et al. Detection of temozolomide-induced hypermutation and response to PD-1 checkpoint inhibitor in recurrent glioblastoma. Neuro-Oncol. Adv. 2022, 4, vdac076. [Google Scholar] [CrossRef]
- Wang, Y.; Ledet, R.J.; Imberg-Kazdan, K.; Logan, S.K.; Garabedian, M.J. Dynein axonemal heavy chain 8 promotes androgen receptor activity and associates with prostate cancer progression. Oncotarget 2016, 7, 49268–49280. Available online: www.impactjournals.com/oncotarget (accessed on 22 December 2023). [CrossRef]
- Zhang, C. COL22A1 and DNAH8 mutations are associated with tumor mutation burden and prognosis of lung adenocarcinoma patients. Res. Sq. 2022, 1–17, preprint. [Google Scholar]
- Cho, H.J.; Koo, J.H. Odorant G protein-coupled receptors as potential therapeutic targets for adult diffuse gliomas: A systematic analysis and review. BMB Rep. 2021, 54, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yekula, A.; Muralidharan, K.; Small, J.L.; Rosh, Z.S.; Kang, K.M.; Carter, B.S.; Balaj, L. Novel Gene Fusions in Glioblastoma Tumor Tissue and Matched Patient Plasma. Cancers 2020, 12, 1219. [Google Scholar] [CrossRef]
- Sun, J.; Ren, P.; Ye, L.; Li, N.; Wang, D. MLLT3 promotes proliferation of osteosarcoma cells by regulating JNK signaling. Int. J. Clin. Exp. Pathol. 2017, 10, 9444–9451. [Google Scholar] [PubMed]
- Salahuddin, M.M.; Omran, G.A.; Helmy, M.W.; Houssen, M.E. Effect of Regorafenib on P2X7 Receptor Expression and Different Oncogenic Signaling Pathways in a Human Breast Cancer Cell Line: A Potential of New Insight of the Antitumor Effects of Regorafenib. Curr. Issues Mol. Biol. 2021, 43, 2199–2209. [Google Scholar] [CrossRef]
- Cheng, S.Y.; Chen, W.-F.; Wen, Z.-H. Glutathione S-transferase M subfamily in TMZ-resistant glioblastoma cells. Ann. Oncol. 2018, 29, viii679. [Google Scholar] [CrossRef]
- Clayton, N.S.; Ridley, A.J. Targeting Rho GTPase Signaling Networks in Cancer. Front. Cell Dev. Biol. 2020, 8, 222. [Google Scholar] [CrossRef]
- Anusewicz, D.; Orzechowska, M.; Bednarek, A.K. Notch Signaling Pathway in Cancer—Review with Bioinformatic Analysis. Cancers 2021, 13, 768. [Google Scholar] [CrossRef]
- Shamel, M.; Mansy, M.; Soliman, M.; Mubarak, R. The role of exogenous epidermal growth factor on Ki-67 proliferation marker expression in the submandibular salivary gland of albino rats receiving doxorubicin. F1000Research 2020, 9, 1393. [Google Scholar] [CrossRef]
- Klein, C.A. Cancer progression and the invisible phase of metastatic colonization. Nat. Rev. Cancer 2020, 20, 681–694. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Rauch, J.; Kolch, W. Targeting MAPK Signaling in Cancer: Mechanisms of Drug Resistance and Sensitivity. Int. J. Mol. Sci. 2020, 21, 1102. [Google Scholar] [CrossRef] [PubMed]
- Guan, H.; Chen, X.; Liu, J.; Sun, J.; Guo, H.; Jiang, Y.; Zhang, H.; Zhang, B.; Lin, J.; Yuan, Q. Molecular characteristics and therapeutic implications of Toll-like receptor signaling pathway in melanoma. Sci. Rep. 2023, 13, 13788. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Mo, R.; Cai, S.; Feng, Y.; Tang, Z.; Ye, J.; Liu, R.; Cai, Z.; Zhu, X.; Deng, Y.; et al. Differential Expression of E2F Transcription Factors and Their Functional and Prognostic Roles in Human Prostate Cancer. Front. Cell Dev. Biol. 2022, 10, 831329. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Patient Nr | Sex | Age at Intervention | Tumor Nr | Tumor Sample Portion Available | Localization | |||
|---|---|---|---|---|---|---|---|---|
| Pr | Rec | Co | Pe | |||||
| Pt.01 | m | 57 | Gb1 | Y | right frontal | |||
| Pt.02 | m | 73 | Gb2 | Y | left temporal | |||
| Pt.03 | m | 52 | Gb3 | Y | left temporal | |||
| Pt.04 | m | 73 | Gb4 | Y | left temporal–parietal | |||
| Pt.05 | m | 73 | Gb5 | Y | left occipital | |||
| Pt.06 | f | 57 | Gb6 | Y | right temporal | |||
| Pt.07 | f | 62 | Gb7 | Y | right parietal | |||
| Pt.08 | m | 61 | Gb8 | Y | left frontoparietal–temporal | |||
| Pt.09 | m | 56 | Gb9 | Y | ||||
| Pt.10 | m | 70 | Gb10_co | Y | Y | left temporal–parietal | ||
| Gb10_pe | Y | Y | ||||||
| Pt.11 | m | 70 | Gb11_co | Y | Y | left fusiform and parahippocampal gyrus | ||
| Gb11_pe | Y | Y | ||||||
| Pt.12 | f | 69 | Gb12_co | Y | Y | left frontoparietal | ||
| Gb12_pe | Y | Y | ||||||
| Pt.13 | m | 55 | Gb13_pr | Y | right frontal | |||
| 56 | Gb13 | Y | right frontal | |||||
| Pt.14 | m | 72 | Gb14_pr | Y | left frontal and insular | |||
| 73 | Gb14 | Y | left temporal | |||||
| Pt.15 | f | 74 | Gb15_pr | Y | left temporal | |||
| 75 | Gb15 | Y | left frontotemporal | |||||
| Pt.16 | m | 78 | Gb16_pr | Y | left temporal | |||
| 79 | Gb16 | Y | left temporal | |||||
| Pt.17 | f | 56 | Gb17_pr | Y | left hemisphere multifocal | |||
| 58 | Gb17 | Y | left temporal | |||||
| Pt.18 | m | 76 | Gb18_pr | Y | left temporal | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morelli, M.; Lessi, F.; Franceschi, S.; Ferri, G.; Giacomarra, M.; Menicagli, M.; Gambacciani, C.; Pieri, F.; Pasqualetti, F.; Montemurro, N.; et al. Exploring Regorafenib Responsiveness and Uncovering Molecular Mechanisms in Recurrent Glioblastoma Tumors through Longitudinal In Vitro Sampling. Cells 2024, 13, 487. https://doi.org/10.3390/cells13060487
Morelli M, Lessi F, Franceschi S, Ferri G, Giacomarra M, Menicagli M, Gambacciani C, Pieri F, Pasqualetti F, Montemurro N, et al. Exploring Regorafenib Responsiveness and Uncovering Molecular Mechanisms in Recurrent Glioblastoma Tumors through Longitudinal In Vitro Sampling. Cells. 2024; 13(6):487. https://doi.org/10.3390/cells13060487
Chicago/Turabian StyleMorelli, Mariangela, Francesca Lessi, Sara Franceschi, Gianmarco Ferri, Manuel Giacomarra, Michele Menicagli, Carlo Gambacciani, Francesco Pieri, Francesco Pasqualetti, Nicola Montemurro, and et al. 2024. "Exploring Regorafenib Responsiveness and Uncovering Molecular Mechanisms in Recurrent Glioblastoma Tumors through Longitudinal In Vitro Sampling" Cells 13, no. 6: 487. https://doi.org/10.3390/cells13060487