
Unique Spatial Transcriptomic Profiling of the Murine Femoral Fracture Callus: A Preliminary Report


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Selection and Preparation
2.2. In Vivo Pathological Fracture Model
2.3. Tissue Harvest and Histology Preparation
2.4. Visium Data Processing and Analysis
3. Results
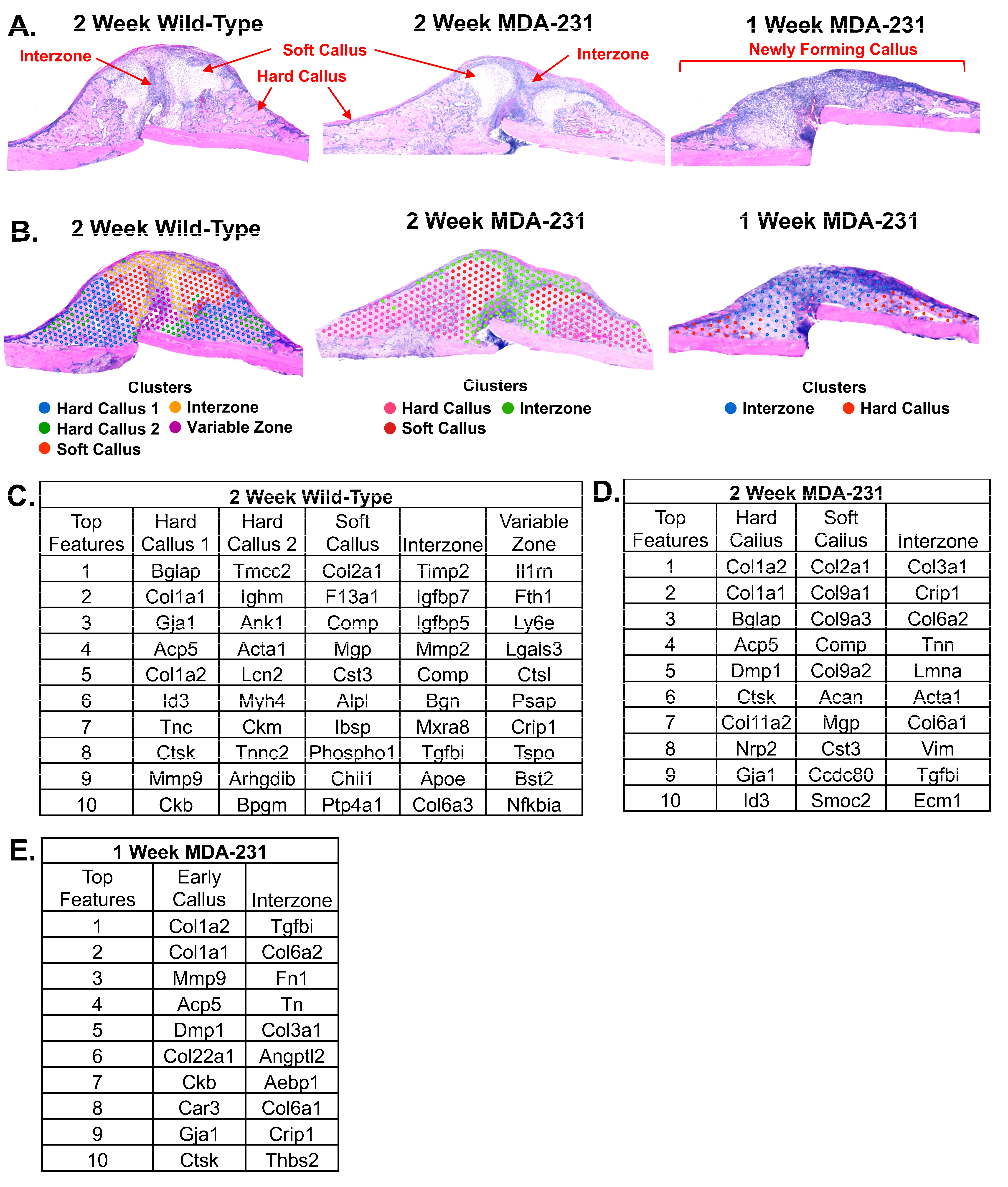
3.1. Validation of Spatial Transcriptomics via Identification of Known Marker Genes
3.2. Unsupervised Global Gene Clustering
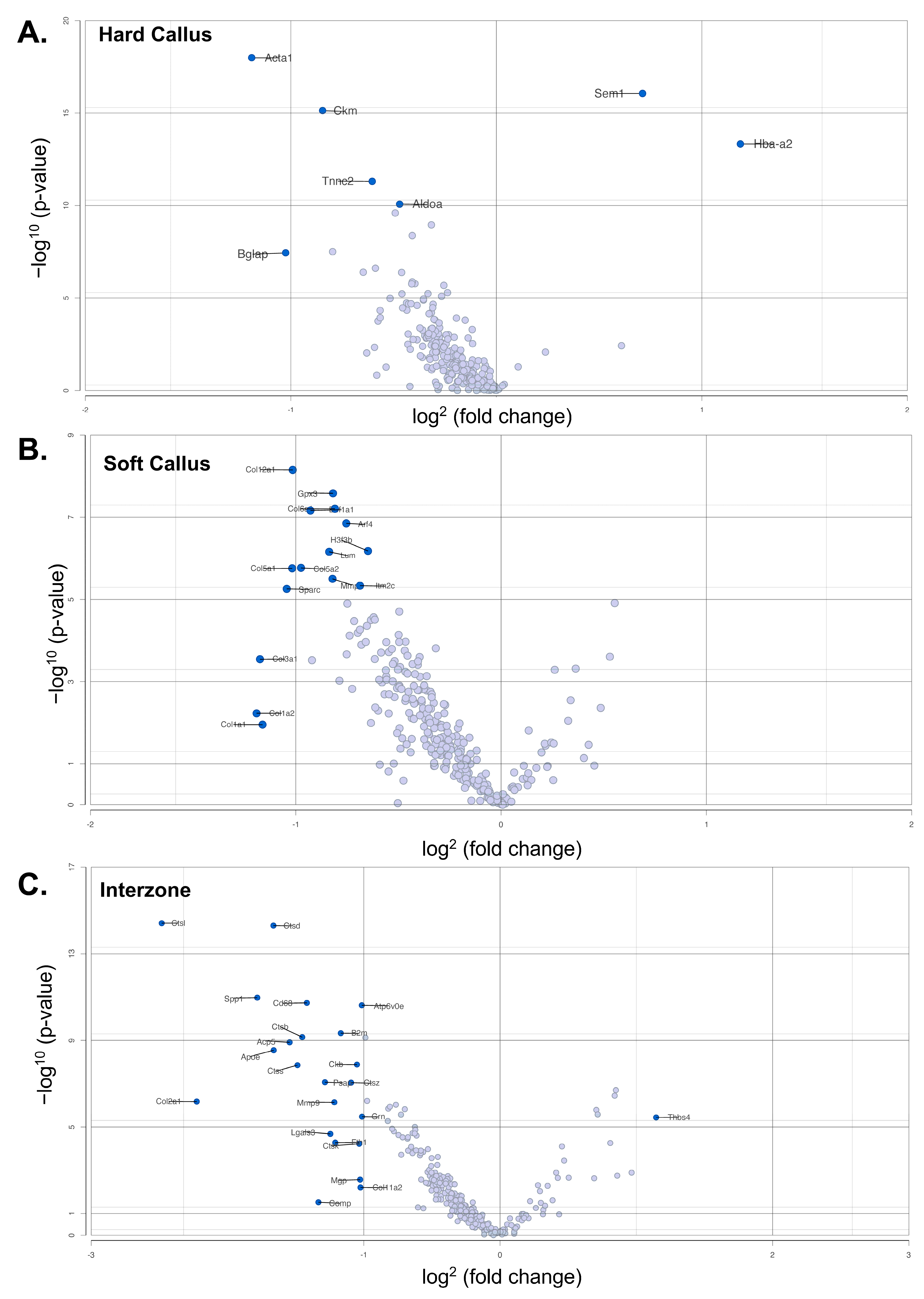
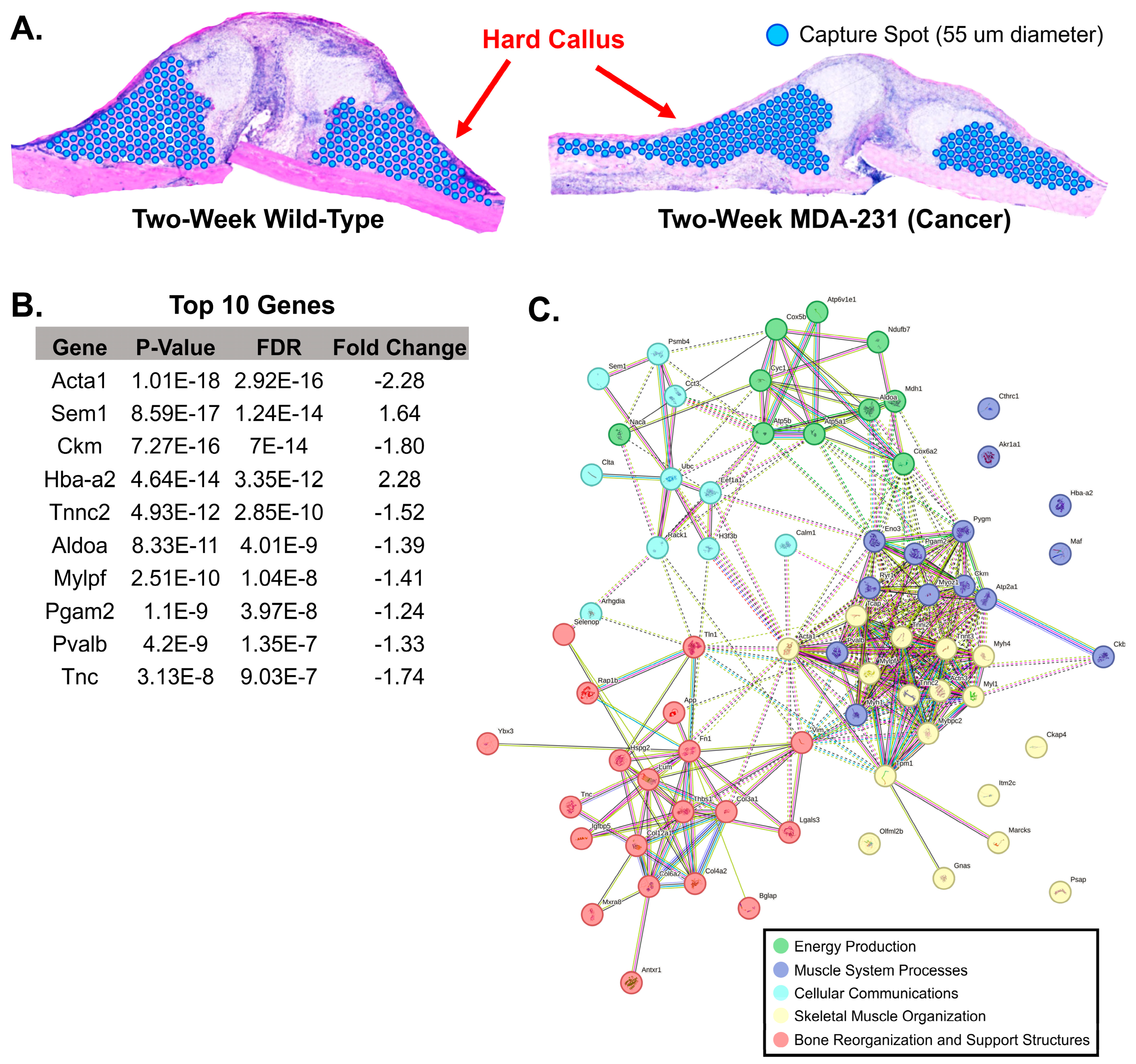
3.3. Comparison of Gene Expression between Two-Week MDA-MB-231 and Wild-Type Fracture Callus
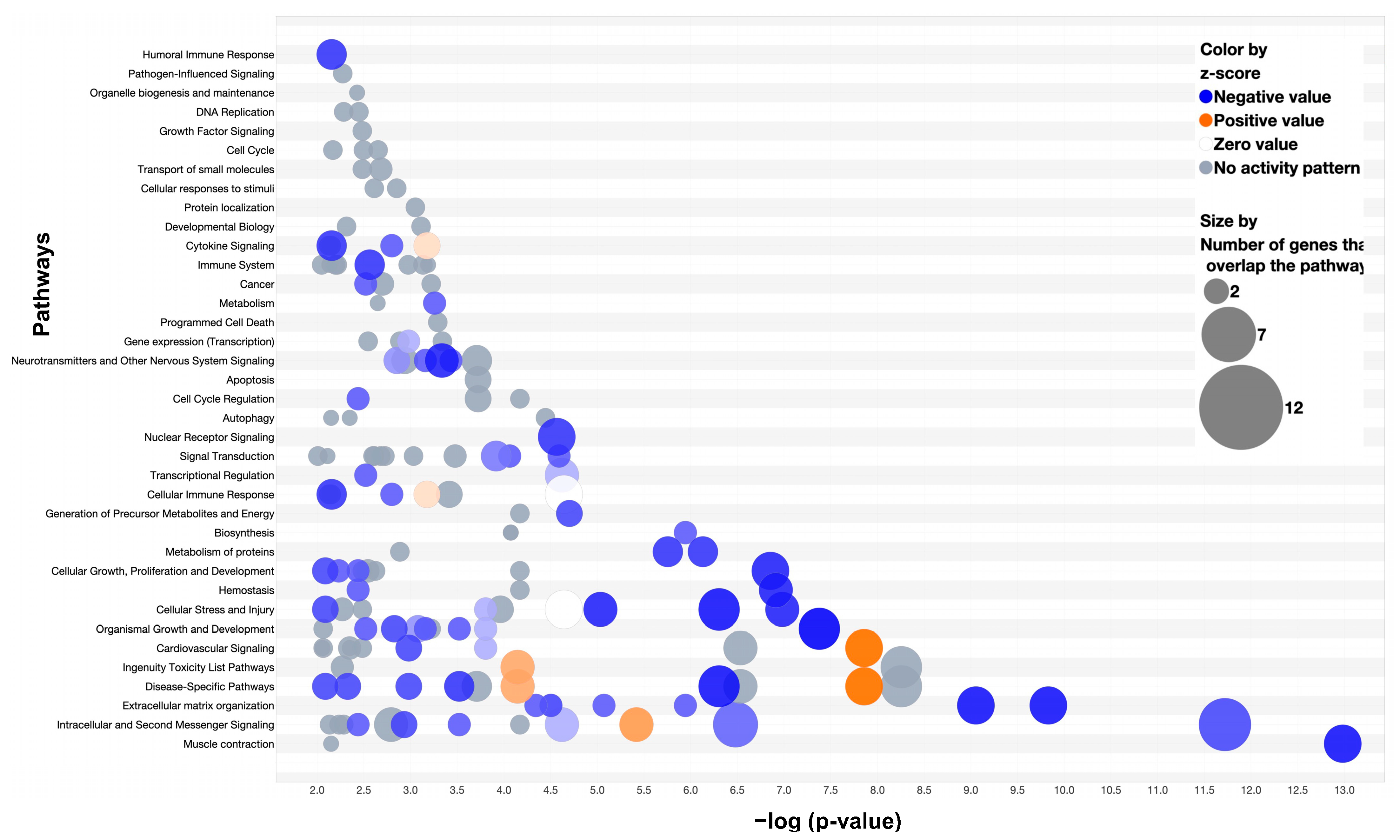
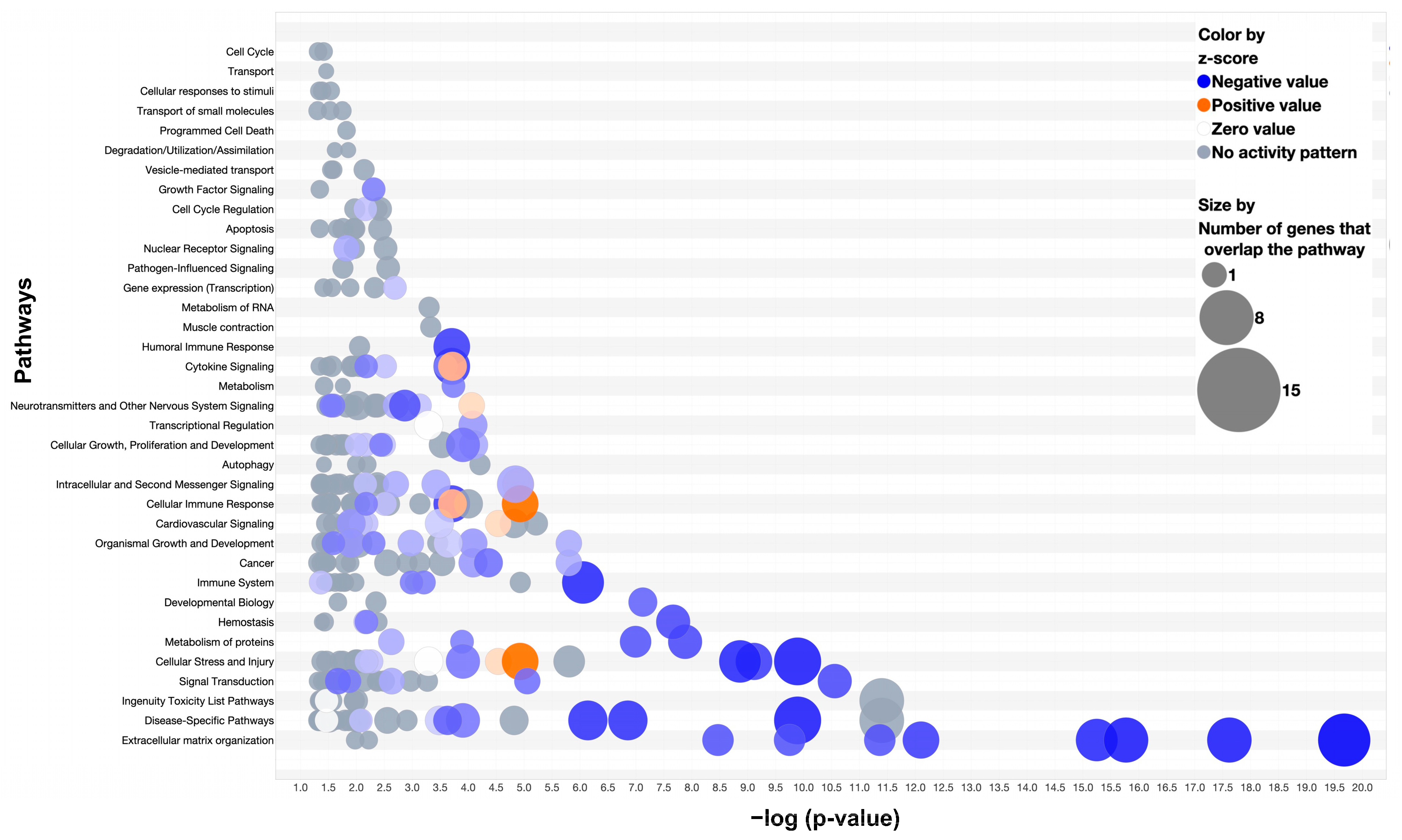
3.4. Pathway Analysis: Comparison of Two-Week MDA-MB-231 and Wild-Type Fracture Callus
3.5. Comparison of One-Week and Two-Week MDA-MB-231 Interzone
4. Discussion
4.1. Pathological Fracture Model: Cell Line Selection
4.2. Unsupervised Clustering Mirrors Fracture Callus Histology: Predictable Genes and Novel Insights into the Interzone
4.3. Using Spatial Transcriptomics for Region of Interest Comparisons
4.4. Downstream Analysis of Biomarker Genes
4.5. Spatial Transcriptomics Shows Temporal Patterning
4.6. Biological Replicates
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xu, Y.; Gong, M.; Wang, Y.; Yang, Y.; Liu, S.; Zeng, Q. Global trends and forecasts of breast cancer incidence and deaths. Sci. Data 2023, 10, 334. [Google Scholar] [CrossRef]
- Lipton, A.; Uzzo, R.; Amato, R.J.; Ellis, G.K.; Hakimian, B.; Roodman, G.D.; Smith, M.R. The science and practice of bone health in oncology: Managing bone loss and metastasis in patients with solid tumors. J. Natl. Compr. Cancer Netw. 2009, 7 (Suppl. 7), S1–S29. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.J.; Selander, K.; Chirgwin, J.M.; Dallas, M.; Grubbs, B.G.; Wieser, R.; Massagué, J.; Mundy, G.R.; Guise, T.A. TGF-beta signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J Clin. Investig. 1999, 103, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Hiraga, T.; Kizaka-Kondoh, S.; Hirota, K.; Hiraoka, M.; Yoneda, T. Hypoxia and hypoxia-inducible factor-1 expression enhance osteolytic bone metastases of breast cancer. Cancer Res. 2007, 67, 4157–4163. [Google Scholar] [CrossRef] [PubMed]
- Manore, S.G.; Doheny, D.L.; Wong, G.L.; Lo, H.W. IL-6/JAK/STAT3 Signaling in Breast Cancer Metastasis: Biology and Treatment. Front. Oncol. 2022, 12, 866014. [Google Scholar] [CrossRef] [PubMed]
- Ghiasi, M.S.; Chen, J.; Vaziri, A.; Rodriguez, E.K.; Nazarian, A. Bone fracture healing in mechanobiological modeling: A review of principles and methods. Bone Rep. 2017, 6, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Back, J.; Nguyen, M.N.; Li, L.; Lee, S.; Lee, I.; Chen, F.; Gillinov, L.; Chung, Y.-H.; Alder, K.D.; Kwon, H.-K.; et al. Inflammatory conversion of quiescent osteoblasts by metastatic breast cancer cells through pERK1/2 aggravates cancer-induced bone destruction. Bone Res. 2021, 9, 43. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.N.; Solaris, J.; Ramsey, K.E.; Yang, X.; Bostrom, M.P.; Stephan, D.; Daluiski, A. Identification of novel gene expression in healing fracture callus tissue by DNA microarray. HSS J. 2008, 4, 149–160. [Google Scholar] [CrossRef]
- Butezloff, M.M.; Volpon, J.B.; Ximenez, J.P.B.; Astolpho, K.; Correlo, V.M.; Reis, R.L.; Silva, R.B.; Zamarioli, A. Gene expression changes are associated with severe bone loss and deficient fracture callus formation in rats with complete spinal cord injury. Spinal Cord 2020, 58, 365–376. [Google Scholar] [CrossRef]
- Bostrom, M.P.; Lane, J.M.; Berberian, W.S.; Missri, A.A.; Tomin, E.; Weiland, A.; Doty, S.B.; Glaser, D.; Rosen, V.M. Immunolocalization and expression of bone morphogenetic proteins 2 and 4 in fracture healing. J. Orthop. Res. 1995, 13, 357–367. [Google Scholar] [CrossRef]
- Zimmermann, G.; Schmeckenbecher, K.H.K.; Boeuf, S.; Weiss, S.; Bock, R.; Moghaddam, A.; Richter, W. Differential gene expression analysis in fracture callus of patients with regular and failed bone healing. Injury 2012, 43, 347–356. [Google Scholar] [CrossRef]
- Li, X.; Wang, C.-Y. From bulk, single-cell to spatial RNA sequencing. Int. J. Oral Sci. 2021, 13, 36. [Google Scholar] [CrossRef] [PubMed]
- Thrane, K.; Eriksson, H.; Maaskola, J.; Hansson, J.; Lundeberg, J. Spatially Resolved Transcriptomics Enables Dissection of Genetic Heterogeneity in Stage III Cutaneous Malignant Melanoma. Cancer Res. 2018, 78, 5970–5979. [Google Scholar] [CrossRef]
- Hafemeister, C.; Satija, R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019, 20, 296. [Google Scholar] [CrossRef] [PubMed]
- Zaira, Y.G.-C.; David, G.-C.; Carmen Parra, C.; Rocío, A.-G.; Cristina, V.; Clemente, I.; Javier, S.C.C. Cartilage Tissue Engineering: The Role of Extracellular Matrix (ECM) and Novel Strategies. In Regenerative Medicine and Tissue Engineering; Jose, A.A., Ed.; IntechOpen: Rijeka, Yugoslavia, 2013; p. Ch. 15. [Google Scholar]
- Nakajima, F.; Ogasawara, A.; Goto, K.; Moriya, H.; Ninomiya, Y.; Einhorn, T.A.; Yamazaki, M. Spatial and temporal gene expression in chondrogenesis during fracture healing and the effects of basic fibroblast growth factor. J. Orthop. Res. 2001, 19, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Toyosawa, S.; Kanatani, N.; Shintani, S.; Kobata, M.; Yuki, M.; Kishino, M.; Ijuhin, N.; Komori, T. Expression of dentin matrix protein 1 (DMP1) during fracture healing. Bone 2004, 35, 553–561. [Google Scholar] [CrossRef]
- Qin, C.; D’Souza, R.; Feng, J.Q. Dentin Matrix Protein 1 (DMP1): New and Important Roles for Biomineralization and Phosphate Homeostasis. J. Dent. Res. 2007, 86, 1134–1141. [Google Scholar] [CrossRef] [PubMed]
- Suen, P.K.; Qin, L. Sclerostin, an emerging therapeutic target for treating osteoporosis and osteoporotic fracture: A general review. J. Orthop. Transl. 2016, 4, 1–13. [Google Scholar] [CrossRef]
- Diegel, C.R.; Hann, S.; Ayturk, U.M.; Hu, J.C.W.; Lim, K.E.; Droscha, C.J.; Madaj, Z.B.; Foxa, G.E.; Izaguirre, I.; Transgenics Core, V.V.A.; et al. An osteocalcin-deficient mouse strain without endocrine abnormalities. PLoS Genet. 2020, 16, e1008361. [Google Scholar] [CrossRef]
- Mendoza-Londono, R.; Fahiminiya, S.; Majewski, J.; Tétreault, M.; Nadaf, J.; Kannu, P.; Sochett, E.; Howard, A.; Stimec, J.; Dupuis, L.; et al. Recessive osteogenesis imperfecta caused by missense mutations in SPARC. Am. J. Hum. Genet. 2015, 96, 979–985. [Google Scholar] [CrossRef]
- Bragdon, B.C.; Bahney, C.S. Origin of Reparative Stem Cells in Fracture Healing. Curr. Osteoporos. Rep. 2018, 16, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Aronson, J.; Good, B.; Stewart, C.; Harrison, B.; Harp, J. Preliminary studies of mineralization during distraction osteogenesis. Clin. Orthop. Relat. Res. 1990, 43–49. [Google Scholar] [CrossRef]
- Cailleau, R.; Olivé, M.; Cruciger, Q.V. Long-term human breast carcinoma cell lines of metastatic origin: Preliminary characterization. Vitro 1978, 14, 911–915. [Google Scholar] [CrossRef]
- Kang, Y.; Siegel, P.M.; Shu, W.; Drobnjak, M.; Kakonen, S.M.; Cordón-Cardo, C.; Guise, T.A.; Massagué, J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003, 3, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Grano, M.; Mori, G.; Minielli, V.; Cantatore, F.P.; Colucci, S.; Zallone, A.Z. Breast Cancer Cell Line MDA-231 Stimulates Osteoclastogenesis and Bone Resorption in Human Osteoclasts. Biochem. Biophys. Res. Commun. 2000, 270, 1097–1100. [Google Scholar] [CrossRef]
- Sandberg, M.; Aro, H.; Multimäki, P.; Aho, H.; Vuorio, E. In situ localization of collagen production by chondrocytes and osteoblasts in fracture callus. J. Bone Joint Surg. Am. 1989, 71, 69–77. [Google Scholar] [CrossRef]
- Luo, Y.Y.; Karsdal, M.A. Chapter 11-Type XI Collagen. In Biochemistry of Collagens, Laminins and Elastin; Karsdal, M.A., Ed.; Academic Press: Cambridge, MA, USA, 2016; pp. 77–80. [Google Scholar]
- Colnot, C.; Thompson, Z.; Miclau, T.; Werb, Z.; Helms, J.A. Altered fracture repair in the absence of MMP9. Development 2003, 130, 4123–4133. [Google Scholar] [CrossRef]
- Murphy, G.; Lee, M.H. What are the roles of metalloproteinases in cartilage and bone damage? Ann. Rheum. Dis. 2005, 64, iv44. [Google Scholar] [CrossRef]
- Lee, J.M.; Lee, E.H.; Kim, I.S.; Kim, J.E. Tgfbi deficiency leads to a reduction in skeletal size and degradation of the bone matrix. Calcif. Tissue Int. 2015, 96, 56–64. [Google Scholar] [CrossRef]
- Alves De Souza, C.; Queiroz Alves De Souza, A.; Queiroz Alves De Souza, M.D.S.; Dias Leite, J.A.; Silva De Morais, M.; Barem Rabenhorst, S.H. A link between osteomyelitis and IL1RN and IL1B polymorphisms-a study in patients from Northeast Brazil. Acta Orthop. 2017, 88, 556–561. [Google Scholar] [CrossRef]
- Gainor, B.J.; Buchert, P. Fracture healing in metastatic bone disease. Clin. Orthop. Relat. Res. 1983, 178, 297–302. [Google Scholar] [CrossRef]
- Amend, S.R.; Uluckan, O.; Hurchla, M.; Leib, D.; Novack, D.V.; Silva, M.; Frazier, W.; Weilbaecher, K.N. Thrombospondin-1 regulates bone homeostasis through effects on bone matrix integrity and nitric oxide signaling in osteoclasts. J. Bone Miner. Res. 2015, 30, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Kwan, K.M.; Zhang, Z.; Deng, J.M.; Darnay, B.G.; Behringer, R.R.; Nakamura, T.; de Crombrugghe, B.; Akiyama, H. Cthrc1 is a positive regulator of osteoblastic bone formation. PLoS ONE 2008, 3, e3174. [Google Scholar] [CrossRef]
- Pugh, J.; Sherry, H.S.; Futterman, B.; Frankel, V.H. Biomechanics of pathologic fractures. Clin. Orthop. Relat. Res. 1982, 169, 109–114. [Google Scholar] [CrossRef]
- Chiquet, M.; Birk, D.E.; Bönnemann, C.G.; Koch, M. Collagen XII: Protecting bone and muscle integrity by organizing collagen fibrils. Int. J. Biochem. Cell. Biol. 2014, 53, 51–54. [Google Scholar] [CrossRef]
- Rosset, E.M.; Bradshaw, A.D. SPARC/osteonectin in mineralized tissue. Matrix Biol. 2016, 52-54, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Potts, W.; Bowyer, J.; Jones, H.; Tucker, D.; Freemont, A.J.; Millest, A.; Martin, C.; Vernon, W.; Neerunjun, D.; Slynn, G.; et al. Cathepsin L-deficient mice exhibit abnormal skin and bone development and show increased resistance to osteoporosis following ovariectomy. Int. J. Exp. Pathol. 2004, 85, 85–96. [Google Scholar] [CrossRef]
- Ashley, J.W.; Shi, Z.; Zhao, H.; Li, X.; Kesterson, R.A.; Feng, X. Genetic ablation of CD68 results in mice with increased bone and dysfunctional osteoclasts. PLoS ONE 2011, 6, e25838. [Google Scholar] [CrossRef]
- Denhardt, D.T.; Noda, M. Osteopontin expression and function: Role in bone remodeling. J. Cell. Biochem. Suppl. 1998, 30-31, 92–102. [Google Scholar] [CrossRef]
- Chen, J.; Sun, Y.; Mao, X.; Liu, Q.; Wu, H.; Chen, Y. RANKL up-regulates brain-type creatine kinase via poly(ADP-ribose) polymerase-1 during osteoclastogenesis. J. Biol. Chem. 2010, 285, 36315–36321. [Google Scholar] [CrossRef]
- Rauner, M.; Föger-Samwald, U.; Kurz, M.F.; Brünner-Kubath, C.; Schamall, D.; Kapfenberger, A.; Varga, P.; Kudlacek, S.; Wutzl, A.; Höger, H.; et al. Cathepsin S controls adipocytic and osteoblastic differentiation, bone turnover, and bone microarchitecture. Bone 2014, 64, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Jones, J.C.; Shankavaram, U.; Sproull, M.; Camphausen, K.; Krauze, A.V. Analytical Considerations of Large-Scale Aptamer-Based Datasets for Translational Applications. Cancers 2022, 14, 2227. [Google Scholar] [CrossRef] [PubMed]
- Bhattacherjee, A.; Zhang, C.; Watson, B.R.; Djekidel, M.N.; Moffitt, J.R.; Zhang, Y. Spatial transcriptomics reveals the distinct organization of mouse prefrontal cortex and neuronal subtypes regulating chronic pain. Nat. Neurosci. 2023, 26, 1880–1893. [Google Scholar] [CrossRef]
- Powell, E.; Piwnica-Worms, D.; Piwnica-Worms, H. Contribution of p53 to metastasis. Cancer Discov. 2014, 4, 405–414. [Google Scholar] [CrossRef]
- Corona, A.; Blobe, G.C. The role of the extracellular matrix protein TGFBI in cancer. Cell. Signal. 2021, 84, 110028. [Google Scholar] [CrossRef]
- Klavert, J.; van der Eerden, B.C.J. Fibronectin in Fracture Healing: Biological Mechanisms and Regenerative Avenues. Front. Bioeng. Biotechnol. 2021, 9, 663357. [Google Scholar] [CrossRef]
- Williams, C.G.; Lee, H.J.; Asatsuma, T.; Vento-Tormo, R.; Haque, A. An introduction to spatial transcriptomics for biomedical research. Genome Med. 2022, 14, 68. [Google Scholar] [CrossRef]
- Matsubara, T.; Soh, J.; Morita, M.; Uwabo, T.; Tomida, S.; Fujiwara, T.; Kanazawa, S.; Toyooka, S.; Hirasawa, A. DV200 Index for Assessing RNA Integrity in Next-Generation Sequencing. BioMed Res. Int. 2020, 2020, 9349132. [Google Scholar] [CrossRef]
- Patil, A.T.; Bennett, D.D.; Xu, J.; Weisman, P.; Matson, D.R. Spatial transcriptomics of a giant pilomatricoma. J. Cutan. Pathol. 2023, 50, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Shek, D.; Gloss, B.; Lai, J.; Ma, L.; Zhang, H.E.; Carlino, M.S.; Mahajan, H.; Nagrial, A.; Gao, B.; Read, S.A.; et al. Identification and Characterisation of Infiltrating Immune Cells in Malignant Pleural Mesothelioma Using Spatial Transcriptomics. Methods Protoc. 2023, 6, 35. [Google Scholar] [CrossRef] [PubMed]
- Parigi, S.M.; Larsson, L.; Das, S.; Ramirez Flores, R.O.; Frede, A.; Tripathi, K.P.; Diaz, O.E.; Selin, K.; Morales, R.A.; Luo, X.; et al. The spatial transcriptomic landscape of the healing mouse intestine following damage. Nat. Commun. 2022, 13, 828. [Google Scholar] [CrossRef] [PubMed]
- Navarro, J.F.; Croteau, D.L.; Jurek, A.; Andrusivova, Z.; Yang, B.; Wang, Y.; Ogedegbe, B.; Riaz, T.; Støen, M.; Desler, C.; et al. Spatial Transcriptomics Reveals Genes Associated with Dysregulated Mitochondrial Functions and Stress Signaling in Alzheimer Disease. iScience 2020, 23, 101556. [Google Scholar] [CrossRef]
- Chen, W.T.; Lu, A.; Craessaerts, K.; Pavie, B.; Sala Frigerio, C.; Corthout, N.; Qian, X.; Laláková, J.; Kühnemund, M.; Voytyuk, I.; et al. Spatial Transcriptomics and In Situ Sequencing to Study Alzheimer’s Disease. Cell 2020, 182, 976–991.e919. [Google Scholar] [CrossRef]
- Baker, E.A.G.; Schapiro, D.; Dumitrascu, B.; Vickovic, S.; Regev, A. In silico tissue generation and power analysis for spatial omics. Nat. Methods 2023, 20, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Rapp, A.E.; Bindl, R.; Recknagel, S.; Erbacher, A.; Müller, I.; Schrezenmeier, H.; Ehrnthaller, C.; Gebhard, F.; Ignatius, A. Fracture Healing Is Delayed in Immunodeficient NOD/scid-IL2Rγcnull Mice. PLoS ONE 2016, 11, e0147465. [Google Scholar] [CrossRef] [PubMed]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, W.; Caruana, D.L.; Back, J.; Lee, F.Y. Unique Spatial Transcriptomic Profiling of the Murine Femoral Fracture Callus: A Preliminary Report. Cells 2024, 13, 522. https://doi.org/10.3390/cells13060522
Jiang W, Caruana DL, Back J, Lee FY. Unique Spatial Transcriptomic Profiling of the Murine Femoral Fracture Callus: A Preliminary Report. Cells. 2024; 13(6):522. https://doi.org/10.3390/cells13060522
Chicago/Turabian StyleJiang, Will, Dennis L. Caruana, Jungho Back, and Francis Y. Lee. 2024. "Unique Spatial Transcriptomic Profiling of the Murine Femoral Fracture Callus: A Preliminary Report" Cells 13, no. 6: 522. https://doi.org/10.3390/cells13060522