
Atypical Neurogenesis, Astrogliosis, and Excessive Hilar Interneuron Loss Are Associated with the Development of Post-Traumatic Epilepsy
 , , , , , , , ,
, , , , , , , ,
Abstract
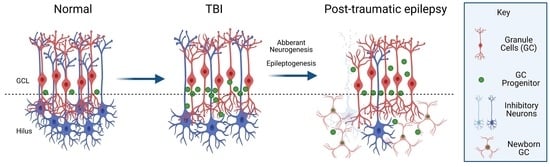
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Controlled Cortical Impact (CCI)
2.3. EEG Implantation
2.4. EEG and Video Analysis
2.5. Brain Tissue Preparation, Serial Sectioning, and Staining
2.6. Brain Tissue Preparation for Astrocyte Isolation and RNA Extraction
2.7. RNA Extraction and Sequencing Analysis
2.8. Immunohistochemistry, Stereo Investigator Analysis, and Lesion Volume
2.9. Astrocyte Morphology Analysis
2.10. Sholl Analysis of Astrocytes in Imaris
2.11. Statistical Analysis
3. Results
3.1. Murine CCI-Induced PTE Is Associated with Injury Severity and Hippocampal Damage
3.2. PTE+ Mice Show Aberrant Migration of Prox1-Neuroblasts in the Dentate Gyrus and Hilus
3.3. PTE+ Mice Display Increased Hilar Expression of c-Fos Alongside an Excessive Loss of Inhibitory Neurons
3.4. Post-Traumatic Epilepsy Alters Hilar Astrogliosis and Astrocyte Morphometric Properties
3.5. Transcriptomic Signature of Forebrain PTE+ Astrocytes following CCI Injury
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Golub, V.M.; Reddy, D.S. Post-Traumatic Epilepsy and Comorbidities: Advanced Models, Molecular Mechanisms, Biomarkers, and Novel Therapeutic Interventions. Pharmacol. Rev. 2022, 74, 387–438. [Google Scholar] [CrossRef] [PubMed]
- Timofeev, I.; Santarius, T.; Kolias, A.G.; Hutchinson, P.J.A. Decompressive craniectomy—Operative technique and perioperative care. In Advances and Technical Standards in Neurosurgery; Springer: Berlin/Heidelberg, Germany, 2012; pp. 115–136. [Google Scholar]
- Taylor, C.A.; Bell, J.M.; Breiding, M.J.; Xu, L. Traumatic Brain Injury-Related Emergency Department Visits, Hospitalizations, and Deaths—United States, 2007 and 2013. MMWR Surveill. Summ. 2017, 66, 1–16. [Google Scholar] [CrossRef] [PubMed]
- DeGrauw, X.; Thurman, D.; Xu, L.; Kancherla, V.; DeGrauw, T. Epidemiology of traumatic brain injury-associated epilepsy and early use of anti-epilepsy drugs: An analysis of insurance claims data, 2004–2014. Epilepsy Res. 2018, 146, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Asikainen, I.; Kaste, M.; Sarna, S. Early and Late Posttraumatic Seizures in Traumatic Brain Injury Rehabilitation Patients: Brain Injury Factors Causing Late Seizures and Influence of Seizures on Long-Term Outcome. Epilepsia 1999, 40, 584–589. [Google Scholar] [CrossRef] [PubMed]
- Frey, L.C. Epidemiology of Posttraumatic Epilepsy: A Critical Review. Epilepsia 2003, 44, 11–17. [Google Scholar] [CrossRef]
- Hauser, W.A.; Annegers, J.F.; Kurland, L.T. Prevalence of Epilepsy in Rochester, Minnesota: 1940–1980. Epilepsia 1991, 32, 429–445. [Google Scholar] [CrossRef]
- Chen, J.W.Y.; Ruff, R.L.; Eavey, R.; Wasterlain, C.G. Posttraumatic epilepsy and treatment. J. Rehabil. Res. Dev. 2009, 46, 685. [Google Scholar] [CrossRef]
- Small, C.; Dagra, A.; Martinez, M.; Williams, E.; Lucke-Wold, B. Examining the role of astrogliosis and JNK signaling in post-traumatic epilepsy. Egypt. J. Neurosurg. 2022, 37, 1. [Google Scholar] [CrossRef]
- Mukherjee, S.; Arisi, G.M.; Mims, K.; Hollingsworth, G.; O’Neil, K.; Shapiro, L.A. Neuroinflammatory mechanisms of post-traumatic epilepsy. J. Neuroinflamm. 2020, 17, 193. [Google Scholar] [CrossRef]
- Ping, X.; Jin, X. Transition from Initial Hypoactivity to Hyperactivity in Cortical Layer V Pyramidal Neurons after Traumatic Brain Injury In Vivo. J. Neurotrauma 2016, 33, 354–361. [Google Scholar] [CrossRef]
- Pitkänen, A.; Ndode-Ekane, X.E.; Lapinlampi, N.; Puhakka, N. Epilepsy biomarkers—Toward etiology and pathology specificity. Neurobiol. Dis. 2019, 123, 42–58. [Google Scholar] [CrossRef] [PubMed]
- Greer, K.; Basso, E.K.G.; Kelly, C.; Cash, A.; Kowalski, E.; Cerna, S.; Ocampo, C.T.; Wang, X.; Theus, M.H. Abrogation of atypical neurogenesis and vascular-derived EphA4 prevents repeated mild TBI-induced learning and memory impairments. Sci. Rep. 2020, 10, 15374. [Google Scholar] [CrossRef] [PubMed]
- Hunt, R.F.I.; Boychuk, J.A.; Smith, B.N. Neural circuit mechanisms of post–traumatic epilepsy. Front. Cell. Neurosci. 2013, 7, 89. [Google Scholar] [CrossRef] [PubMed]
- Dudek, F.E.; Sutula, T.P. Epileptogenesis in the dentate gyrus: A critical perspective. Prog. Brain Res. 2007, 163, 755–773. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.; Meyer, R.M.; Szuflita, N.S.; A Severson, M.; Levine, Z.T. Post-Traumatic, Drug-Resistant Epilepsy and Review of Seizure Control Outcomes from Blinded, Randomized Controlled Trials of Brain Stimulation Treatments for Drug-Resistant Epilepsy. Cureus 2016, 8, e744. [Google Scholar] [CrossRef]
- Parko, K.L.; Rao, V.R. Clinical Approach to Posttraumatic Epilepsy. In Seminars in Neurology; Thieme Medical Publishers: New York, NY, USA, 2015. [Google Scholar]
- Lucke-Wold, B.P.; Nguyen, L.; Turner, R.C.; Logsdon, A.F.; Chen, Y.-W.; Smith, K.E.; Huber, J.D.; Matsumoto, R.; Rosen, C.L.; Tucker, E.S.; et al. Traumatic brain injury and epilepsy: Underlying mechanisms leading to seizure. Seizure 2015, 33, 13–23. [Google Scholar] [CrossRef]
- Agrawal, A.; Timothy, J.; Pandit, L.; Manju, M. Post-traumatic epilepsy: An overview. Clin. Neurol. Neurosurg. 2006, 108, 433–439. [Google Scholar] [CrossRef]
- Englander, J.; Bushnik, T.; Duong, T.T.; Cifu, D.X.; Zafonte, R.; Wright, J.; Hughes, R.; Bergman, W. Analyzing risk factors for late posttraumatic seizures: A prospective, multicenter investigation. Arch. Phys. Med. Rehabil. 2003, 84, 365–373. [Google Scholar] [CrossRef]
- Haltiner, A.M.; Temkin, N.R.; Dikmen, S.S. Risk of seizure recurrence after the first late posttraumatic seizure. Arch. Phys. Med. Rehabil. 1997, 78, 835–840. [Google Scholar] [CrossRef]
- Diaz-Arrastia, R.; Agostini, M.A.; Frol, A.B.; Mickey, B.; Fleckenstein, J.; Bigio, E.; Van Ness, P.C. Neurophysiologic and neuroradiologic features of intractable epilepsy after traumatic brain injury in adults. Arch. Neurol. 2000, 57, 1611–1616. [Google Scholar] [CrossRef]
- Gupta, P.K.; Sayed, N.; Ding, K.; Agostini, M.A.; Van Ness, P.C.; Yablon, S.; Madden, C.; Mickey, B.; D’Ambrosio, R.; Diaz-Arrastia, R. Subtypes of Post-Traumatic Epilepsy: Clinical, Electrophysiological, and Imaging Features. J. Neurotrauma 2014, 31, 1439–1443. [Google Scholar] [CrossRef] [PubMed]
- Temkin, N.R.; Dikmen, S.S.; Wilensky, A.J.; Keihm, J.; Chabal, S.; Winn, H.R. A Randomized, Double-Blind Study of Phenytoin for the Prevention of Post-Traumatic Seizures. N. Engl. J. Med. 1990, 323, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Szaflarski, J.P.; Nazzal, Y.; E Dreer, L. Post-traumatic epilepsy: Current and emerging treatment options. Neuropsychiatr. Dis. Treat. 2014, 10, 1469–1477. [Google Scholar] [CrossRef] [PubMed]
- Zaccara, G.; Lattanzi, S.; Brigo, F. Which treatment strategy in patients with epilepsy with focal seizures uncontrolled by the first anti-seizure medication? Epilepsy Behav. 2021, 121 Pt A, 108031. [Google Scholar] [CrossRef]
- Marion, D.W. Management of traumatic brain injury: Past, present, and future. Clin. Neurosurg. 1999, 45, 184–191. [Google Scholar]
- Brodie, M.J. Road to refractory epilepsy: The Glasgow story. Epilepsia 2013, 54 (Suppl. S2), 5–8. [Google Scholar] [CrossRef]
- Bratton, S.L.; Chestnut, R.M.; Ghajar, J.; Hammond, F.F.M.; Harris, O.A.; Hartl, R.; Manley, G.T.; Nemecek, A.; Newell, D.W.; Rosenthal, G.; et al. XIII. Antiseizure Prophylaxis. J. Neurotrauma 2007, 24 (Suppl. S1), S83–S86. [Google Scholar] [CrossRef]
- Kwan, P.; Schachter, S.C.; Brodie, M.J. Drug-resistant epilepsy. N. Engl. J. Med. 2011, 365, 919–926. [Google Scholar] [CrossRef]
- Semah, F.; Picot, M.C.; Adam, C.; Broglin, D.; Arzimanoglou, A.; Bazin, B.; Cavalcanti, D.; Baulac, M. Is the underlying cause of epilepsy a major prognostic factor for recurrence? Neurology 1998, 51, 1256–1262. [Google Scholar] [CrossRef]
- Irimia, A.; Van Horn, J.D. Epileptogenic focus localization in treatment-resistant post-traumatic epilepsy. J. Clin. Neurosci. 2014, 22, 627–631. [Google Scholar] [CrossRef]
- Pitkänen, A.; Immonen, R.J.; Gröhn, O.H.; Kharatishvili, I. From traumatic brain injury to posttraumatic epilepsy: What animal models tell us about the process and treat-ment options. Epilepsia 2009, 50 (Suppl. S2), 21–29. [Google Scholar] [CrossRef] [PubMed]
- Bolkvadze, T.; Pitkanen, A. Development of post-traumatic epilepsy after controlled cortical impact and lateral flu-id-percussion-induced brain injury in the mouse. J. Neurotrauma 2012, 29, 789–812. [Google Scholar] [CrossRef] [PubMed]
- Butler, C.R.; Boychuk, J.A.; Smith, B.N. Effects of Rapamycin Treatment on Neurogenesis and Synaptic Reorganization in the Dentate Gyrus after Controlled Cortical Impact Injury in Mice. Front. Syst. Neurosci. 2015, 9, 163. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Zeng, L.; Brody, D.L.; Wong, M. Rapamycin Attenuates the Development of Posttraumatic Epilepsy in a Mouse Model of Traumatic Brain Injury. PLoS ONE 2013, 8, e64078. [Google Scholar] [CrossRef] [PubMed]
- Hunt, R.F.; Scheff, S.W.; Smith, B.N. Posttraumatic epilepsy after controlled cortical impact injury in mice. Exp. Neurol. 2009, 215, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Reddy, D.S.; Golub, V.M.; Ramakrishnan, S.; Abeygunaratne, H.; Dowell, S.; Wu, X. A Comprehensive and Advanced Mouse Model of Post-Traumatic Epilepsy with Robust Spontaneous Recurrent Seizures. Curr. Protoc. 2022, 2, e447. [Google Scholar] [CrossRef]
- Kowalski, E.A.; Soliman, E.; Kelly, C.; Basso, E.K.G.; Leonard, J.; Pridham, K.J.; Ju, J.; Cash, A.; Hazy, A.; de Jager, C.; et al. Monocyte proinflammatory phenotypic control by ephrin type A receptor 4 mediates neural tissue damage. JCI Insight 2022, 7, e156319. [Google Scholar] [CrossRef]
- Soliman, E.; Mills, J.; Ju, J.; Kaloss, A.M.; Basso, E.K.G.; Groot, N.; Kelly, C.; Kowalski, E.A.; Elhassanny, M.; Chen, M.; et al. Conditional Deletion of EphA4 on Cx3cr1-Expressing Microglia Fails to Influence Histopathological Outcome and Blood Brain Barrier Disruption Following Brain Injury. Front. Mol. Neurosci. 2021, 14, 747770. [Google Scholar] [CrossRef]
- Kowalski, E.A.; Chen, J.; Hazy, A.; Fritsch, L.E.; Gudenschwager-Basso, E.K.; Chen, M.; Wang, X.; Qian, Y.; Zhou, M.; Byerly, M.; et al. Peripheral loss of EphA4 ameliorates TBI-induced neuroinflammation and tissue damage. J. Neuroinflamm. 2019, 16, 210. [Google Scholar] [CrossRef]
- Theus, M.H.; Brickler, T.; Meza, A.L.; Coutermarsh-Ott, S.; Hazy, A.; Gris, D.; Allen, I.C. Loss of NLRX1 Exacerbates Neural Tissue Damage and NF-kappaB Signaling following Brain Injury. J. Immunol. 2017, 199, 3547–3558. [Google Scholar] [CrossRef]
- Brickler, T.R.; Hazy, A.; Correa, F.G.; Dai, R.; Kowalski, E.J.; Dickerson, R.; Chen, J.; Wang, X.; Morton, P.D.; Whittington, A.; et al. Angiopoietin/Tie2 Axis Regulates the Age-at-Injury Cerebrovascular Response to Traumatic Brain Injury. J. Neurosci. 2018, 38, 9618–9634. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.C.; Thompson, E.G.; Sontheimer, H. Brain-Derived Neurotrophic Factor Inhibits the Function of Cation-Chloride Cotransporter in a Mouse Model of Viral Infection-Induced Epilepsy. Front. Cell Dev. Biol. 2022, 10, 961292. [Google Scholar] [CrossRef] [PubMed]
- Shandra, O.; Winemiller, A.R.; Heithoff, B.P.; Munoz-Ballester, C.; George, K.K.; Benko, M.J.; Zuidhoek, I.A.; Besser, M.N.; Curley, D.E.; Edwards, G.F.; et al. Repetitive Diffuse Mild Traumatic Brain Injury Causes an Atypical Astrocyte Response and Spontaneous Re-current Seizures. J. Neurosci. 2019, 39, 1944–1963. [Google Scholar] [CrossRef] [PubMed]
- Okyere, B.; Mills, W.A.; Wang, X.; Chen, M.; Chen, J.; Hazy, A.; Qian, Y.; Matson, J.B.; Theus, M.H. EphA4/Tie2 crosstalk regulates leptomeningeal collateral remodeling following ischemic stroke. J. Clin. Investig. 2019, 130, 1024–1035. [Google Scholar] [CrossRef]
- Okyere, B.; Creasey, M.; Lebovitz, Y.; Theus, M.H. Temporal remodeling of pial collaterals and functional deficits in a murine model of ischemic stroke. J. Neurosci. Methods 2017, 293, 86–96. [Google Scholar] [CrossRef]
- Holt, L.M.; Olsen, M.L. Novel Applications of Magnetic Cell Sorting to Analyze Cell-Type Specific Gene and Protein Expression in the Central Nervous System. PLoS ONE 2016, 11, e0150290. [Google Scholar] [CrossRef]
- Holt, L.M.; Stoyanof, S.T.; Olsen, M.L. Magnetic Cell Sorting for In Vivo and In Vitro Astrocyte, Neuron, and Microglia Analysis. Curr. Protoc. Neurosci. 2019, 88, e71. [Google Scholar] [CrossRef]
- Holt, L.M.; Hernandez, R.D.; Pacheco, N.L.; Torres Ceja, B.; Hossain, M.; Olsen, M.L. Astrocyte morphogenesis is dependent on BDNF signaling via astrocytic TrkB.T1. Elife 2019, 8, e44667. [Google Scholar] [CrossRef]
- Theus, M.H.; Ricard, J.; Glass, S.J.; Travieso, L.G.; Liebl, D.J. EphrinB3 blocks EphB3 dependence receptor functions to prevent cell death following traumatic brain injury. Cell Death Dis. 2014, 5, e1207. [Google Scholar] [CrossRef]
- Brickler, T.; Gresham, K.; Meza, A.; Coutermarsh-Ott, S.; Williams, T.M.; Rothschild, D.E.; Allen, I.C.; Theus, M.H. Nonessential Role for the NLRP1 Inflammasome Complex in a Murine Model of Traumatic Brain Injury. Mediat. Inflamm. 2016, 2016, 6373506. [Google Scholar] [CrossRef]
- Wang, D.; Fan, L.-S. 2—Particle characterization and behavior relevant to fluidized bed combustion and gasification systems. In Fluidized Bed Technologies for Near-Zero Emission Combustion and Gasification; Scala, F., Ed.; Woodhead Publishing: Sawston, UK, 2013; pp. 42–76. [Google Scholar] [CrossRef]
- Polychronopoulos, N.D.; Gkountas, A.A.; Sarris, I.E.; Spyrou, L.A. A Computational Study on Magnetic Nanoparticles Hyperthermia of Ellipsoidal Tumors. Appl. Sci. 2021, 11, 9526. [Google Scholar] [CrossRef]
- Iwano, T.; Masuda, A.; Kiyonari, H.; Enomoto, H.; Matsuzaki, F. Prox1 postmitotically defines dentate gyrus cells by specifying granule cell identity over CA3 pyramidal cell fate in the hippocampus. Development 2012, 139, 3051–3062. [Google Scholar] [CrossRef] [PubMed]
- Barros, V.N.; Mundim, M.; Galindo, L.T.; Bittencourt, S.; Porcionatto, M.; Mello, L.E. The pattern of c-Fos expression and its refractory period in the brain of rats and monkeys. Front. Cell. Neurosci. 2015, 9, 72. [Google Scholar] [CrossRef] [PubMed]
- Szyndler, J.; Maciejak, P.; Turzyńska, D.; Sobolewska, A.; Taracha, E.; Skórzewska, A.; Lehner, M.; Bidziński, A.; Hamed, A.; Wisłowska-Stanek, A.; et al. Mapping of c-Fos expression in the rat brain during the evolution of pentylenetetrazol-kindled seizures. Epilepsy Behav. 2009, 16, 216–224. [Google Scholar] [CrossRef]
- Dragunow, M.; Robertson, H. Generalized seizures induce c-fos protein(s) in mammalian neurons. Neurosci. Lett. 1987, 82, 157–161. [Google Scholar] [CrossRef]
- Simler, S.; Hirsch, E.; Danober, L.; Motte, J.; Vergnes, M.; Marescaux, C. C-fos expression after single and kindled audiogenic seizures in Wistar rats. Neurosci. Lett. 1994, 175, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Kempermann, G.; Song, H.; Gage, F.H. Neurogenesis in the Adult Hippocampus. Cold Spring Harb. Perspect. Biol. 2015, 7, a018812. [Google Scholar] [CrossRef]
- Lake, B.B.; Ai, R.; Kaeser, G.E.; Salathia, N.S.; Yung, Y.C.; Liu, R.; Wildberg, A.; Gao, D.; Fung, H.-L.; Chen, S.; et al. Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science 2016, 352, 1586–1590. [Google Scholar] [CrossRef]
- Hayatdavoudi, P.; Hosseini, M.; Hajali, V.; Hosseini, A.; Rajabian, A. The role of astrocytes in epileptic disorders. Physiol. Rep. 2022, 10, e15239. [Google Scholar] [CrossRef]
- Aoki, Y.; Hanai, S.; Sukigara, S.; Otsuki, T.; Saito, T.; Nakagawa, E.; Kaido, T.; Kaneko, Y.; Takahashi, A.; Ikegaya, N.; et al. Altered expression of astrocyte-related receptors and channels correlates with epileptogenesis in hippocampal sclerosis. Pediatr. Dev. Pathol. 2019, 22, 532–539. [Google Scholar] [CrossRef]
- Leiter, I.; Bascuñana, P.; Bengel, F.M.; Bankstahl, J.P.; Bankstahl, M. Attenuation of epileptogenesis by 2-deoxy-d-glucose is accompanied by increased cerebral glucose supply, microglial activation and reduced astrocytosis. Neurobiol. Dis. 2019, 130, 104510. [Google Scholar] [CrossRef] [PubMed]
- Torres-Ceja, B.; Olsen, M.L. A closer look at astrocyte morphology: Development, heterogeneity, and plasticity at astrocyte leaflets. Curr. Opin. Neurobiol. 2022, 74, 102550. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Shen, Z.; Zhao, Q.; Yan, L.; Du, L.; Deng, Z. Dynamic Transitions of Epilepsy Waveforms Induced by Astrocyte Dysfunction and Electrical Stimulation. Neural Plast. 2020, 2020, 8867509. [Google Scholar] [CrossRef] [PubMed]
- Steinhauser, C.; Seifert, G.; Bedner, P. Astrocyte dysfunction in temporal lobe epilepsy: K+ channels and gap junction coupling. Glia 2012, 60, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Seifert, G.; Carmignoto, G.; Steinhauser, C. Astrocyte dysfunction in epilepsy. Brain Res. Rev. 2010, 63, 212–221. [Google Scholar] [CrossRef]
- Heinemann, U.; Kaufer, D.; Friedman, A. Blood-brain barrier dysfunction, TGFβ signaling, and astrocyte dysfunction in epilepsy. Glia 2012, 60, 1251–1257. [Google Scholar] [CrossRef]
- Robinson, C.; Apgar, C.; Shapiro, L.A. Astrocyte Hypertrophy Contributes to Aberrant Neurogenesis after Traumatic Brain Injury. Neural Plast. 2016, 2016, 1347987. [Google Scholar] [CrossRef]
- Pirttilä, T.J.; Manninen, A.; Jutila, L.; Nissinen, J.; Kälviäinen, R.; Vapalahti, M.; Immonen, A.; Paljärvi, L.; Karkola, K.; Alafuzoff, I.; et al. Cystatin C expression is associated with granule cell dispersion in epilepsy. Ann. Neurol. 2005, 58, 211–223. [Google Scholar] [CrossRef]
- Liu, X.; Sullivan, K.A.; Madl, J.E.; Legare, M.; Tjalkens, R.B. Manganese-Induced Neurotoxicity: The Role of Astroglial-Derived Nitric Oxide in Striatal Interneuron Degeneration. Toxicol. Sci. 2006, 91, 521–531. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Kharatishvili, I.; Pitkänen, A. Association of the severity of cortical damage with the occurrence of spontaneous seizures and hyperexcitability in an animal model of posttraumatic epilepsy. Epilepsy Res. 2010, 90, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Hunt, R.F.; Scheff, S.W.; Smith, B.N. Regionally Localized Recurrent Excitation in the Dentate Gyrus of a Cortical Contusion Model of Posttraumatic Epilepsy. J. Neurophysiol. 2010, 103, 1490–1500. [Google Scholar] [CrossRef] [PubMed]
- Lothman, E.W.; Bertram, E.H., 3rd; Stringer, J.L. Functional anatomy of hippocampal seizures. Prog. Neurobiol. 1991, 37, 1–82. [Google Scholar] [CrossRef] [PubMed]
- Lowenstein, D.; Thomas, M.; Smith, D.; McIntosh, T. Selective vulnerability of dentate hilar neurons following traumatic brain injury: A potential mechanistic link between head trauma and disorders of the hippocampus. J. Neurosci. 1992, 12, 4846–4853. [Google Scholar] [CrossRef]
- Golarai, G.; Greenwood, A.C.; Feeney, D.M.; Connor, J.A. Physiological and structural evidence for hippocampal involvement in persistent seizure susceptibility after trau-matic brain injury. J. Neurosci. 2001, 21, 8523–8537. [Google Scholar] [CrossRef]
- Shapiro, L.A.; Wang, L.; Ribak, C.E. Rapid astrocyte and microglial activation following pilocarpine-induced seizures in rats. Epilepsia 2008, 49 (Suppl. S2), 33–41. [Google Scholar] [CrossRef]
- Golub, V.M.; Reddy, D.S. Contusion brain damage in mice for modelling of post-traumatic epilepsy with contralateral hippo-campus sclerosis: Comprehensive and longitudinal characterization of spontaneous seizures, neuropathology, and neuropsychiatric comorbidities. Exp. Neurol. 2022, 348, 113946. [Google Scholar] [CrossRef]
- Di Sapia, R.; Moro, F.; Montanarella, M.; Iori, V.; Micotti, E.; Tolomeo, D.; Wang, K.K.W.; Vezzani, A.; Ravizza, T.; Zanier, E.R. In-depth characterization of a mouse model of post-traumatic epilepsy for biomarker and drug discovery. Acta Neuropathol. Commun. 2021, 9, 76. [Google Scholar] [CrossRef]
- Butler, C.R.; Boychuk, J.A.; Smith, B.N. Differential effects of rapamycin treatment on tonic and phasic GABAergic inhibition in dentate granule cells after focal brain injury in mice. Exp. Neurol. 2016, 280, 30–40. [Google Scholar] [CrossRef]
- Bugay, V.; Bozdemir, E.; Vigil, F.A.; Chun, S.H.; Holstein, D.M.; Elliott, W.R.; Sprague, C.J.; Cavazos, J.E.; Zamora, D.O.; Rule, G.; et al. A Mouse Model of Repetitive Blast Traumatic Brain Injury Reveals Post-Trauma Seizures and Increased Neuronal Excitability. J. Neurotrauma 2020, 37, 248–261. [Google Scholar] [CrossRef]
- Partridge, B.; Rossmeisl, J.H.; Kaloss, A.M.; Basso, E.K.G.; Theus, M.H. Novel ablation methods for treatment of gliomas. J. Neurosci. Methods 2020, 336, 108630. [Google Scholar] [CrossRef] [PubMed]
- Frankowski, J.C.; Kim, Y.J.; Hunt, R.F. Selective vulnerability of hippocampal interneurons to graded traumatic brain injury. Neurobiol. Dis. 2018, 129, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Eom, J.; Hunt, R.F. Transplanted interneurons improve memory precision after traumatic brain injury. Nat. Commun. 2019, 10, 5156. [Google Scholar] [CrossRef] [PubMed]
- Kron, M.M.; Zhang, H.; Parent, J.M. The developmental stage of dentate granule cells dictates their contribution to sei-zure-induced plasticity. J. Neurosci. 2010, 30, 2051–2059. [Google Scholar] [CrossRef] [PubMed]
- Parent, J.M.; Timothy, W.Y.; Leibowitz, R.T.; Geschwind, D.H.; Sloviter, R.S.; Lowenstein, D.H. Dentate Granule Cell Neurogenesis Is Increased by Seizures and Contributes to Aberrant Network Reorganization in the Adult Rat Hippocampus. J. Neurosci. 1997, 17, 3727–3738. [Google Scholar] [CrossRef]
- Covolan, L.; Ribeiro, L.T.C.; Longo, B.M.; Mello, L.E.A.M. Cell damage and neurogenesis in the dentate granule cell layer of adult rats after pilocarpine- or kainate-induced status epilepticus. Hippocampus 2000, 10, 169–180. [Google Scholar] [CrossRef]
- Houser, C.R. Granule cell dispersion in the dentate gyrus of humans with temporal lobe epilepsy. Brain Res. 1990, 535, 195–204. [Google Scholar] [CrossRef]
- Jinde, S.; Zsiros, V.; Nakazawa, K. Hilar mossy cell circuitry controlling dentate granule cell excitability. Front. Neural Circuits 2013, 7, 14. [Google Scholar] [CrossRef]
- Cho, K.-O.; Lybrand, Z.R.; Ito, N.; Brulet, R.; Tafacory, F.; Zhang, L.; Good, L.; Ure, K.; Kernie, S.G.; Birnbaum, S.G.; et al. Aberrant hippocampal neurogenesis contributes to epilepsy and associated cognitive decline. Nat. Commun. 2015, 6, 6606. [Google Scholar] [CrossRef]
- Gong, C.; Wang, T.-W.; Huang, H.S.; Parent, J.M. Reelin Regulates Neuronal Progenitor Migration in Intact and Epileptic Hippocampus. J. Neurosci. 2007, 27, 1803–1811. [Google Scholar] [CrossRef]
- Farhy-Tselnicker, I.; Boisvert, M.M.; Liu, H.; Dowling, C.; Erikson, G.A.; Blanco-Suarez, E.; Farhy, C.; Shokhirev, M.N.; Ecker, J.R.; Allen, N.J. Activity-dependent modulation of synapse-regulating genes in astrocytes. Elife 2021, 10, e70514. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, A.; Naruse, M.; Hitoshi, S.; Iwasaki, Y.; Takebayashi, H.; Ikenaka, K. Regulation of glial development by cystatin C. J. Neurochem. 2006, 100, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Pirttilä, T.J.; Lukasiuk, K.; Håkansson, K.; Grubb, A.; Abrahamson, M.; Pitkänen, A. Cystatin C modulates neurodegeneration and neurogenesis following status epilepticus in mouse. Neurobiol. Dis. 2005, 20, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Gavrilovici, C.; Jiang, Y.; Kiroski, I.; Teskey, G.C.; Rho, J.M.; Nguyen, M.D. Postnatal Role of the Cytoskeleton in Adult Epileptogenesis. Cereb. Cortex Commun. 2020, 1, tgaa024. [Google Scholar] [CrossRef] [PubMed]
- Perveen, N.; Ashraf, W.; Alqahtani, F.; Fawad Rasool, M.; Samad, N.; Imran, I. Temporal Lobe Epilepsy: What do we understand about protein alterations? Chem. Biol. Drug Des. 2021, 98, 377–394. [Google Scholar] [CrossRef]
- Do Canto, A.M.; Donatti, A.; Geraldis, J.C.; Godoi, A.B.; Da Rosa, D.C.; Lopes-Cendes, I. Neuroproteomics in Epilepsy: What Do We Know so Far? Front. Mol. Neurosci. 2021, 13, 604158. [Google Scholar] [CrossRef]
- Zhang, S.; Liang, F.; Wang, B.; Le, Y.; Wang, H. Elevated expression of pleiotrophin in pilocarpine-induced seizures of immature rats and in pentylene-tetrazole-induced hippocampal astrocytes in vitro. Acta Histochem. 2014, 116, 415–420. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gudenschwager-Basso, E.K.; Shandra, O.; Volanth, T.; Patel, D.C.; Kelly, C.; Browning, J.L.; Wei, X.; Harris, E.A.; Mahmutovic, D.; Kaloss, A.M.; et al. Atypical Neurogenesis, Astrogliosis, and Excessive Hilar Interneuron Loss Are Associated with the Development of Post-Traumatic Epilepsy. Cells 2023, 12, 1248. https://doi.org/10.3390/cells12091248
Gudenschwager-Basso EK, Shandra O, Volanth T, Patel DC, Kelly C, Browning JL, Wei X, Harris EA, Mahmutovic D, Kaloss AM, et al. Atypical Neurogenesis, Astrogliosis, and Excessive Hilar Interneuron Loss Are Associated with the Development of Post-Traumatic Epilepsy. Cells. 2023; 12(9):1248. https://doi.org/10.3390/cells12091248
Chicago/Turabian StyleGudenschwager-Basso, Erwin Kristobal, Oleksii Shandra, Troy Volanth, Dipan C. Patel, Colin Kelly, Jack L. Browning, Xiaoran Wei, Elizabeth A. Harris, Dzenis Mahmutovic, Alexandra M. Kaloss, and et al. 2023. "Atypical Neurogenesis, Astrogliosis, and Excessive Hilar Interneuron Loss Are Associated with the Development of Post-Traumatic Epilepsy" Cells 12, no. 9: 1248. https://doi.org/10.3390/cells12091248