
Peroxisome Proliferator-Activated Receptor-γ as a Target and Regulator of Epigenetic Mechanisms in Nonalcoholic Fatty Liver Disease

Abstract
:1. Introduction
2. The Role of Epigenetic Mechanisms in Regulating PPARγ Regulatory Function in NAFLD
2.1. DNA Methylation
2.2. Histone Modifications
2.2.1. Histone Methylation/Demethylation
2.2.2. Histone Acetylation/Deacetylation
2.3. Noncoding RNAs
2.3.1. miRNAs-PPARγ Axis
2.3.2. lncRNAs-PPARγ Axis
2.3.3. circRNAs-PPARγ Axis
3. Limitations and Future Perspectives
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Galoosian, A.; Kaswala, D.; Li, A.A.; Gadiparthi, C.; Cholankeril, G.; Kim, D.; Ahmed, A. Nonalcoholic Fatty Liver Disease: Epidemiology, Liver Transplantation Trends and Outcomes, and Risk of Recurrent Disease in the Graft. J. Clin. Transl. Hepatol. 2018, 6, 420–424. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Mingolla, L.; Rigolon, R.; Pichiri, I.; Cavalieri, V.; Zoppini, G.; Lippi, G.; Bonora, E.; Targher, G. Nonalcoholic fatty liver disease is independently associated with an increased incidence of cardiovascular disease in adult patients with type 1 diabetes. Int. J. Cardiol. 2016, 225, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R.; Roden, M. NAFLD and diabetes mellitus. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 32–42. [Google Scholar] [CrossRef]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef]
- Ratziu, V.; Rinella, M.; Beuers, U.; Loomba, R.; Anstee, Q.M.; Harrison, S.; Francque, S.; Sanyal, A.; Newsome, P.N.; Younossi, Z. The times they are a-changin’ (for NAFLD as well). J. Hepatol. 2020, 73, 1307–1309. [Google Scholar] [CrossRef]
- Kim, D.; Ahmed, A. Reply to: “NAFLD vs. MAFLD-It is not the name but the disease that decides the outcome in fatty liver”. J. Hepatol. 2022, 76, 477–478. [Google Scholar] [CrossRef]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Machado, M.V.; Diehl, A.M. Pathogenesis of Nonalcoholic Steatohepatitis. Gastroenterology 2016, 150, 1769–1777. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef]
- Loomba, R.; Friedman, S.L.; Shulman, G.I. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 2021, 184, 2537–2564. [Google Scholar] [CrossRef]
- Ratziu, V. Starting the battle to control non-alcoholic steatohepatitis. Lancet 2015, 385, 922–924. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef]
- Braissant, O.; Foufelle, F.; Scotto, C.; Dauça, M.; Wahli, W. Differential expression of peroxisome proliferator-activated receptors (PPARs): Tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology 1996, 137, 354–366. [Google Scholar] [CrossRef]
- Tontonoz, P.; Hu, E.; Graves, R.A.; Budavari, A.I.; Spiegelman, B.M. mPPAR gamma 2: Tissue-specific regulator of an adipocyte enhancer. Genes Dev. 1994, 10, 1224–1234. [Google Scholar] [CrossRef]
- Greene, M.E.; Blumberg, B.; McBride, O.W.; Yi, H.F.; Kronquist, K.; Kwan, K.; Hsieh, L.; Greene, G.; Nimer, S.D. Isolation of the human peroxisome proliferator activated receptor gamma cDNA: Expression in hematopoietic cells and chromosomal mapping. Gene Expr. 1995, 4, 281–299. [Google Scholar]
- Fajas, L.; Auboeuf, D.; Raspé, E.; Schoonjans, K.; Lefebvre, A.M.; Saladin, R.; Najib, J.; Laville, M.; Fruchart, J.C.; Deeb, S.; et al. The organization, promoter analysis, and expression of the human PPARgamma gene. J. Biol. Chem. 1997, 272, 18779–18789. [Google Scholar] [CrossRef]
- Hernandez-Quiles, M.; Broekema, M.F.; Kalkhoven, E. PPARgamma in Metabolism, Immunity, and Cancer: Unified and Diverse Mechanisms of Action. Front. Endocrinol. 2021, 12, 624112. [Google Scholar] [CrossRef] [PubMed]
- Tontonoz, P.; Spiegelman, B.M. Fat and beyond: The diverse biology of PPARgamma. Annu. Rev. Biochem. 2008, 77, 289–312. [Google Scholar] [CrossRef] [PubMed]
- Lehrke, M.; Lazar, M.A. The many faces of PPARgamma. Cell 2005, 123, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Chawla, A.; Schwarz, E.J.; Dimaculangan, D.D.; Lazar, M.A. Peroxisome proliferator-activated receptor (PPAR) gamma: Adipose-predominant expression and induction early in adipocyte differentiation. Endocrinology 1994, 135, 798–800. [Google Scholar] [CrossRef]
- Vidal-Puig, A.; Jimenez-Liñan, M.; Lowell, B.B.; Hamann, A.; Hu, E.; Spiegelman, B.; Flier, J.S.; Moller, D.E. Regulation of PPAR gamma gene expression by nutrition and obesity in rodents. J. Clin. Investig. 1996, 97, 2553–2561. [Google Scholar] [CrossRef]
- Chen, Y.; Jimenez, A.R.; Medh, J.D. Identification and regulation of novel PPAR-gamma splice variants in human THP-1 macrophages. Biochim. Biophys. Acta 2006, 1759, 32–43. [Google Scholar] [CrossRef]
- Wu, C.W.; Chu, E.S.; Lam, C.N.; Cheng, A.S.; Lee, C.W.; Wong, V.W.; Sung, J.J.; Yu, J. PPARgamma is essential for protection against nonalcoholic steatohepatitis. Gene Ther. 2010, 17, 790–798. [Google Scholar] [CrossRef]
- Pettinelli, P.; Videla, L.A. Up-Regulation of PPAR-γ mRNA Expression in the Liver of Obese Patients: An Additional Reinforcing Lipogenic Mechanism to SREBP-1c Induction. J. Clin. Endocrinol. Metab. 2011, 96, 1424–1430. [Google Scholar] [CrossRef]
- Lima-Cabello, E.; García-Mediavilla, M.V.; Miquilena-Colina, M.E.; Vargas-Castrillón, J.; Lozano-Rodríguez, T.; Fernández-Bermejo, M.; Olcoz, J.L.; González-Gallego, J.; García-Monzón, C.; Sánchez-Campos, S. Enhanced expression of pro-inflammatory mediators and liver X-receptor-regulated lipogenic genes in non-alcoholic fatty liver disease and hepatitis C. Clin. Sci. 2011, 120, 239–250. [Google Scholar] [CrossRef]
- Yamazaki, T.; Shiraishi, S.; Kishimoto, K.; Miura, S.; Ezaki, O. An increase in liver PPARγ2 is an initial event to induce fatty liver in response to a diet high in butter: PPARγ2 knockdown improves fatty liver induced by high-saturated fat. J. Nutr. Biochem. 2011, 22, 543–553. [Google Scholar] [CrossRef]
- Lutchman, G.; Promrat, K.; Kleiner, D.E.; Heller, T.; Ghany, M.G.; Yanovski, J.A.; Liang, T.J.; Hoofnagle, J.H. Changes in serum adipokine levels during pioglitazone treatment for nonalcoholic steatohepatitis: Relationship to histological improvement. Clin. Gastroenterol. Hepatol. 2006, 4, 1048–1052. [Google Scholar] [CrossRef]
- Ratziu, V.; Giral, P.; Jacqueminet, S.; Charlotte, F.; Hartemann-Heurtier, A.; Serfaty, L.; Podevin, P.; Lacorte, J.M.; Bernhardt, C.; Bruckert, E.; et al. Rosiglitazone for nonalcoholic steatohepatitis: One-year results of the randomized placebo-controlled Fatty Liver Improvement with Rosiglitazone Therapy (FLIRT) Trial. Gastroenterology 2008, 135, 100–110. [Google Scholar] [CrossRef]
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes Dev. 2009, 23, 781–783. [Google Scholar] [CrossRef]
- Foley, D.L.; Craig, J.M.; Morley, R.; Olsson, C.A.; Dwyer, T.; Smith, K.; Saffery, R. Prospects for epigenetic epidemiology. Am. J. Epidemiol. 2009, 169, 389–400. [Google Scholar] [CrossRef]
- Eslam, M.; Valenti, L.; Romeo, S. Genetics and epigenetics of NAFLD and NASH: Clinical impact. J. Hepatol. 2018, 68, 268–279. [Google Scholar] [CrossRef]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef]
- Hyun, J.; Jung, Y. DNA Methylation in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2020, 21, 8138. [Google Scholar] [CrossRef]
- Zaiou, M.; Amrani, R.; Rihn, B.; Hajri, T. Dietary Patterns Influence Target Gene Expression through Emerging Epigenetic Mechanisms in Nonalcoholic Fatty Liver Disease. Biomedicines 2021, 9, 1256. [Google Scholar] [CrossRef]
- Tian, Y.; Wong, V.W.-S.; Chan, H.L.-Y.; Cheng, A.S.-L. Epigenetic regulation of hepatocellular carcinoma in non-alcoholic fatty liver disease. Semin. Cancer Biol. 2013, 23, 471–482. [Google Scholar] [CrossRef]
- Ahrens, M.; Ammerpohl, O.; von Schönfels, W.; Kolarova, J.; Bens, S.; Itzel, T.; Teufel, A.; Herrmann, A.; Brosch, M.; Hinrichsen, H.; et al. DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease-specific and remodeling signatures after bariatric surgery. Cell Metab. 2013, 18, 296–302. [Google Scholar] [CrossRef] [PubMed]
- De Mello, V.D.; Matte, A.; Perfilyev, A.; Männistö, V.; Rönn, T.; Nilsson, E.; Käkelä, P.; Ling, C.; Pihlajamäki, J. Human liver epigenetic alterations in non-alcoholic steatohepatitis are related to insulin action. Epigenetics 2017, 12, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Sookoian, S.; Rosselli, M.S.; Gemma, C.; Burgueño, A.L.; Fernández Gianotti, T.; Castaño, G.O.; Pirola, C.J. Epigenetic regulation of insulin resistance in nonalcoholic fatty liver disease: Impact of liver methylation of the peroxisome proliferator–activated receptor γ coactivator 1α promoter. Hepatology 2010, 52, 1992–2000. [Google Scholar] [CrossRef] [PubMed]
- Zeybel, M.; Hardy, T.; Wong, Y.K.; Mathers, J.C.; Fox, C.R.; Gackowska, A.; Oakley, F.; Burt, A.D.; Wilson, C.L.; Anstee, Q.M.; et al. Multigenerational epigenetic adaptation of the hepatic wound-healing response. Nat. Med. 2012, 18, 1369–1377. [Google Scholar] [CrossRef]
- Zeybel, M.; Hardy, T.; Robinson, S.M.; Fox, C.; Anstee, Q.M.; Ness, T.; Masson, S.; Mathers, J.C.; French, J.; White, S.; et al. Differential DNA methylation of genes involved in fibrosis progression in non-alcoholic fatty liver disease and alcoholic liver disease. Clin. Epigenet. 2015, 7, 25. [Google Scholar] [CrossRef]
- Yigit, B.; Boyle, M.; Ozler, O.; Erden, N.; Tutucu, F.; Hardy, T.; Bergmann, C.; Distler, J.H.W.; Adali, G.; Dayangac, M.; et al. Plasma cell-free DNA methylation: A liquid biomarker of hepatic fibrosis. Gut 2018, 67, 1907–1908. [Google Scholar] [CrossRef]
- Hajri, T.; Zaiou, M.; Fungwe, T.V.; Ouguerram, K.; Besong, S. Epigenetic Regulation of Peroxisome Proliferator-Activated Receptor Gamma Mediates High-Fat Diet-Induced Non-Alcoholic Fatty Liver Disease. Cells 2021, 10, 1355. [Google Scholar] [CrossRef]
- Hardy, T.; Zeybel, M.; Day, C.P.; Dipper, C.; Masson, S.; McPherson, S.; Henderson, E.; Tiniakos, D.; White, S.; French, J.; et al. Plasma DNA methylation: A potential biomarker for stratification of liver fibrosis in non-alcoholic fatty liver disease. Gut 2017, 66, 1321–1328. [Google Scholar] [CrossRef]
- Laird, A.; Thomson, J.P.; Harrison, D.J.; Meehan, R.R. 5-hydroxymethylcytosine profiling as an indicator of cellular state. Epigenomics 2013, 5, 655–669. [Google Scholar] [CrossRef]
- Pirola, C.J.; Scian, R.; Gianotti, T.F.; Dopazo, H.; Rohr, C.; Martino, J.S.; Castaño, G.O.; Sookoian, S. Epigenetic Modifications in the Biology of Nonalcoholic Fatty Liver Disease: The Role of DNA Hydroxymethylation and TET Proteins. Medicine 2015, 94, e1480. [Google Scholar] [CrossRef]
- Tong, J.; Han, C.J.; Zhang, J.Z.; He, W.Z.; Zhao, G.J.; Cheng, X.; Zhang, L.; Deng, K.Q.; Liu, Y.; Fan, H.F.; et al. Hepatic Interferon Regulatory Factor 6 Alleviates Liver Steatosis and Metabolic Disorder by Transcriptionally Suppressing Peroxisome Proliferator-Activated Receptor γ in Mice. Hepatology 2019, 69, 2471–2488. [Google Scholar] [CrossRef]
- Lee, J.; Song, J.H.; Park, J.H.; Chung, M.Y.; Lee, S.H.; Jeon, S.B.; Park, S.H.; Hwang, J.T.; Choi, H.K. Dnmt1/Tet2-mediated changes in Cmip methylation regulate the development of nonalcoholic fatty liver disease by controlling the Gbp2-Pparγ-CD36 axis. Exp. Mol. Med. 2023, 55, 143–157. [Google Scholar] [CrossRef]
- Jenuwein, T.; Allis, C.D. Translating the Histone Code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef]
- Lee, J.; Kim, Y.; Friso, S.; Choi, S.W. Epigenetics in non-alcoholic fatty liver disease. Mol. Asp. Med. 2017, 54, 78–88. [Google Scholar] [CrossRef]
- Ling, C.; Groop, L. Epigenetics: A molecular link between environmental factors and type 2 diabetes. Diabetes 2009, 58, 2718–2725. [Google Scholar] [CrossRef]
- Kim, D.H.; Kim, J.; Kwon, J.S.; Sandhu, J.; Tontonoz, P.; Lee, S.K.; Lee, S.; Lee, J.W. Critical Roles of the Histone Methyltransferase MLL4/KMT2D in Murine Hepatic Steatosis Directed by ABL1 and PPARγ2. Cell Rep. 2016, 17, 1671–1682. [Google Scholar] [CrossRef]
- Jun, H.J.; Kim, J.; Hoang, M.H.; Lee, S.J. Hepatic lipid accumulation alters global histone h3 lysine 9 and 4 trimethylation in the peroxisome proliferator-activated receptor alpha network. PLoS ONE 2012, 7, e44345. [Google Scholar] [CrossRef]
- Fan, Z.; Li, L.; Li, M.; Zhang, X.; Hao, C.; Yu, L.; Zeng, S.; Xu, H.; Fang, M.; Shen, A.; et al. The histone methyltransferase Suv39h2 contributes to nonalcoholic steatohepatitis in mice. Hepatology 2017, 65, 1904–1919. [Google Scholar] [CrossRef]
- Mosammaparast, N.; Shi, Y. Reversal of histone methylation: Biochemical and molecular mechanisms of histone demethylases. Annu. Rev. Biochem. 2010, 79, 155–179. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, S.; Zhao, Y.; Lin, C.; Zhong, F.; Jin, L.; He, F.; Wang, H. Histone H3K9 demethylase JMJD1A modulates hepatic stellate cells activation and liver fibrosis by epigenetically regulating peroxisome proliferator-activated receptor γ. FASEB J. 2015, 29, 1830–1841. [Google Scholar] [CrossRef]
- Kim, J.H.; Jung, D.Y.; Nagappan, A.; Jung, M.H. Histone H3K9 demethylase JMJD2B induces hepatic steatosis through upregulation of PPARγ2. Sci. Rep. 2018, 8, 13734. [Google Scholar] [CrossRef]
- Colak, Y.; Yesil, A.; Mutlu, H.H.; Caklili, O.T.; Ulasoglu, C.; Senates, E.; Takir, M.; Kostek, O.; Yilmaz, Y.; Yilmaz Enc, F.; et al. A potential treatment of non-alcoholic fatty liver disease with SIRT1 activators. J. Gastrointestin. Liver Dis. 2014, 23, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Bricambert, J.; Miranda, J.; Benhamed, F.; Girard, J.; Postic, C.; Dentin, R. Salt-inducible kinase 2 links transcriptional coactivator p300 phosphorylation to the prevention of ChREBP-dependent hepatic steatosis in mice. Clin. Investig. 2010, 120, 4316–4331. [Google Scholar] [CrossRef] [PubMed]
- Lefterova, M.I.; Zhang, Y.; Steger, D.J.; Schupp, M.; Schug, J.; Cristancho, A.; Feng, D.; Zhuo, D.; Stoeckert, C.J.; Liu, X.S.; et al. PPARγ and C/EBP Factors Orchestrate Adipocyte Biology via Adjacent Binding on a Genome-Wide Scale. Genes Dev. 2008, 22, 2941–2952. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, T.S.; Xu, Z.; Zhang, X.; Wang, L.; Gimble, J.M.; Lander, E.S.; Rosen, E.D. Comparative epigenomic analysis of murine and human adipogenesis. Cell 2010, 143, 156–169. [Google Scholar] [CrossRef]
- Kraakman, M.J.; Liu, Q.; Postigo-Fernandez, J.; Ji, R.; Kon, N.; Larrea, D.; Namwanje, M.; Fan, L.; Chan, M.; Area-Gomez, E.; et al. PPARγ deacetylation dissociates thiazolidinedione’s metabolic benefits from its adverse effects. J. Clin. Investig. 2018, 128, 2600–2612. [Google Scholar] [CrossRef]
- Zahr, T.; Liu, L.; Chan, M.; Zhou, Q.; Cai, B.; He, Y.; Aaron, N.; Accili, D.; Sun, L.; Qiang, L. PPARγ (Peroxisome Proliferator-Activated Receptor γ) Deacetylation Suppresses Aging-Associated Atherosclerosis and Hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 2023, 43, 30–44. [Google Scholar] [CrossRef]
- Liu, L.; Fan, L.; Chan, M.; Kraakman, M.J.; Yang, J.; Fan, Y.; Aaron, N.; Wan, Q.; Carrillo-Sepulveda, M.A.; Tall, A.R.; et al. PPARγ Deacetylation Confers the Antiatherogenic Effect and Improves Endothelial Function in Diabetes Treatment. Diabetes 2020, 69, 1793–1803. [Google Scholar] [CrossRef]
- Rodrigues, B.; McNeill, J.H. Cardiac dysfunction in isolated perfused hearts from spontaneously diabetic BB rats. Can. J. Physiol. Pharmacol. 1990, 68, 514–518. [Google Scholar] [CrossRef]
- Emmett, M.J.; Lazar, M.A. Integrative regulation of physiology by histone deacetylase 3. Nat. Rev. Mol. Cell. Biol. 2019, 20, 102–115. [Google Scholar] [CrossRef]
- Knutson, S.K.; Chyla, B.J.; Amann, J.M.; Bhaskara, S.; Huppert, S.S.; Hiebert, S.W. Liver-specific deletion of histone deacetylase 3 disrupts metabolic transcriptional networks. EMBO J. 2008, 27, 1017–1028. [Google Scholar] [CrossRef]
- Ferrari, A.; Longo, R.; Fiorino, E.; Silva, R.; Mitro, N.; Cermenati, G.; Gilardi, F.; Desvergne, B.; Andolfo, A.; Magagnotti, C.; et al. HDAC3 is a molecular brake of the metabolic switch supporting white adipose tissue browning. Nat. Commun. 2017, 8, 93. [Google Scholar] [CrossRef]
- Whitt, J.; Woo, V.; Lee, P.; Moncivaiz, J.; Haberman, Y.; Denson, L.; Tso, P.; Alenghat, T. Disruption of Epithelial HDAC3 in Intestine Prevents Diet-Induced Obesity in Mice. Gastroenterology 2018, 155, 501–513. [Google Scholar] [CrossRef]
- Li, D.; Wang, X.; Ren, W.; Ren, J.; Lan, X.; Wang, F.; Li, H.; Zhang, F.; Han, Y.; Song, T.; et al. High expression of liver histone deacetylase 3 contributes to high-fat-diet-induced metabolic syndrome by suppressing the PPAR-γ and LXR-α-pathways in E3 rats. Mol. Cell. Endocrinol. 2011, 344, 69–80. [Google Scholar] [CrossRef]
- Jiang, X.; Ye, X.; Guo, W.; Lu, H.; Gao, Z. Inhibition of HDAC3 promotes ligand-independent PPARγ activation by protein acetylation. J. Mol. Endocrinol. 2014, 53, 191–200. [Google Scholar] [CrossRef]
- Ding, R.-B.; Bao, J.; Deng, C.-X. Emerging roles of SIRT1 in fatty liver diseases. Int. J. Biol. Sci. 2017, 13, 852–867. [Google Scholar] [CrossRef]
- Cao, Y.; Xue, Y.; Xue, L.; Jiang, X.; Wang, X.; Zhang, Z.; Yang, J.; Lu, J.; Zhang, C.; Wang, W.; et al. Hepatic menin recruits SIRT1 to control liver steatosis through histone deacetylation. J. Hepatol. 2013, 59, 1299–1306. [Google Scholar] [CrossRef]
- de Gregorio, E.; Colell, A.; Morales, A.; Marí, M. Relevance of SIRT1-NF-κB Axis as Therapeutic Target to Ameliorate Inflammation in Liver Disease. Int. J. Mol. Sci. 2020, 21, 3858. [Google Scholar] [CrossRef]
- Vilà, L.; Elias, I.; Roca, C.; Ribera, A.; Ferré, T.; Casellas, A.; Lage, R.; Franckhauser, S.; Bosch, F. AAV8-mediated Sirt1 gene transfer to the liver prevents high carbohydrate diet-induced nonalcoholic fatty liver disease. Molecular therapy. Mol. Ther. Methods Clin. Dev. 2014, 1, 14039. [Google Scholar] [CrossRef]
- Purushotham, A.; Schug, T.T.; Xu, Q.; Surapureddi, S.; Guo, X.; Li, X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 2009, 9, 327–338. [Google Scholar] [CrossRef]
- Deng, X.Q.; Chen, L.L.; Li, N.X. The expression of SIRT1 in nonalcoholic fatty liver disease induced by high-fat diet in rats. Liver Int. 2007, 27, 708–715. [Google Scholar] [CrossRef] [PubMed]
- Mariani, S.; Fiore, D.; Basciani, S.; Persichetti, A.; Contini, S.; Lubrano, C.; Salvatori, L.; Lenzi, A.; Gnessi, L. Plasma levels of SIRT1 associate with non-alcoholic fatty liver disease in obese patients. Endocrine 2015, 49, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Hu, M.; Liang, X.; Ajmo, J.M.; Li, X.; Bataller, R.; Odena, G.; Stevens, S.M., Jr.; You, M. Deletion of SIRT1 from hepatocytes in mice disrupts lipin-1 signaling and aggravates alcoholic fatty liver. Gastroenterology 2014, 146, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Mayoral, R.; Osborn, O.; McNelis, J.; Johnson, A.M.; Oh, D.Y.; Izquierdo, C.L.; Chung, H.; Li, P.; Traves, P.G.; Bandyopadhyay, G.; et al. Adipocyte SIRT1 knockout promotes PPARγ activity, adipogenesis and insulin sensitivity in chronic-HFD and obesity. Mol. Metab. 2015, 4, 378–391. [Google Scholar] [CrossRef]
- Li, P.; Ruan, X.; Yang, L.; Kiesewetter, K.; Zhao, Y.; Luo, H.; Chen, Y.; Gucek, M.; Zhu, J.; Cao, H. A Liver-Enriched Long Non-Coding RNA, lncLSTR, Regulates Systemic Lipid Metabolism in Mice. Cell Metab. 2015, 21, 455–467. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, C.; Barbier, O.; Smalling, R.; Tsuchiya, H.; Lee, S.; Delker, D.; Zou, A.; Hagedorn, C.H.; Wang, L. Bcl2 is a critical regulator of bile acid homeostasis by dictating Shp and lncRNA H19 function. Sci. Rep. 2016, 6, 20559. [Google Scholar] [CrossRef]
- Zaiou, M. Noncoding RNAs as additional mediators of epigenetic regulation in nonalcoholic fatty liver disease. World J. Gastroenterol. 2022, 28, 5111–5128. [Google Scholar] [CrossRef]
- Liu, C.H.; Ampuero, J.; Gil-Gómez, A.; Montero-Vallejo, R.; Rojas, Á.; Muñoz-Hernández, R.; Gallego-Durán, R.; Romero-Gómez, M. miRNAs in patients with non-alcoholic fatty liver disease: A systematic review and meta-analysis. J. Hepatol. 2018, 69, 1335–1348. [Google Scholar] [CrossRef]
- Nobili, V.; Alisi, A.; Valenti, L.; Miele, L.; Feldstein, A.E.; Alkhouri, N. NAFLD in children: New genes, new diagnostic modalities and new drugs. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 517–530. [Google Scholar] [CrossRef]
- Zhu, M.; Wang, Q.; Zhou, W.; Liu, T.; Yang, L.; Zheng, P.; Zhang, L.; Ji, G. Integrated analysis of hepatic mRNA and miRNA profiles identified molecular networks and potential biomarkers of NAFLD. Sci. Rep. 2018, 8, 7628. [Google Scholar] [CrossRef]
- Lv, X.; Yan, J.; Jiang, J.; Zhou, X.; Lu, Y.; Jiang, H. MicroRNA-27a-3p suppression of peroxisome proliferator-activated receptor-γ contributes to cognitive impairments resulting from sevoflurane treatment. J. Neurochem. 2017, 143, 306–319. [Google Scholar] [CrossRef]
- Baffy, G. MicroRNAs in Nonalcoholic Fatty Liver Disease. J. Clin. Med. 2015, 4, 1977–1988. [Google Scholar] [CrossRef]
- Wu, H.; Ng, R.; Chen, X.; Steer, C.J.; Song, G. MicroRNA-21 is a potential link between non-alcoholic fatty liver disease and hepatocellular carcinoma via modulation of the HBP1-p53-Srebp1c pathway. Gut 2016, 65, 1850–1860. [Google Scholar] [CrossRef]
- Sun, C.; Huang, F.; Liu, X.; Xiao, X.; Yang, M.; Hu, G.; Liu, H.; Liao, L. miR-21 regulates triglyceride and cholesterol metabolism in non-alcoholic fatty liver disease by targeting HMGCR. Int. J. Mol. Med. 2015, 35, 847–853. [Google Scholar] [CrossRef]
- Loyer, X.; Paradis, V.; Hénique, C.; Vion, A.-C.; Colnot, N.; Guerin, C.L.; Devue, C.; On, S.; Scetbun, J.; Romain, M.; et al. Liver microRNA-21 is overexpressed in non-alcoholic steatohepatitis and contributes to the disease in experimental models by inhibiting PPARα expression. Gut 2015, 65, 1882–1894. [Google Scholar] [CrossRef]
- Benhamouche-Trouillet, S.; Postic, C. Emerging role of miR-21 in non-alcoholic fatty liver disease. Gut 2016, 65, 1781–1783. [Google Scholar] [CrossRef]
- Pillai, S.S.; Lakhani, H.V.; Zehra, M.; Wang, J.; Dilip, A.; Puri, N.; O’Hanlon, K.; Sodhi, K. Predicting Nonalcoholic Fatty Liver Disease through a Panel of Plasma Biomarkers and MicroRNAs in Female West Virginia Population. Int. J. Mol. Sci. 2020, 21, 6698. [Google Scholar] [CrossRef]
- Liu, J.; Xiao, Y.; Wu, X.; Jiang, L.; Yang, S.; Ding, Z.; Fang, Z.; Hua, H.; Kirby, M.S.; Shou, J. A circulating microRNA signature as noninvasive diagnostic and prognostic biomarkers for nonalcoholic steatohepatitis. BMC Genom. 2018, 19, 188. [Google Scholar] [CrossRef]
- Rodrigues, P.M.; Afonso, M.B.; Simão, A.L.; Carvalho, C.C.; Trindade, A.; Duarte, A.; Borralho, P.M.; Machado, M.V.; Cortez-Pinto, H.; Rodrigues, C.M.; et al. miR-21 ablation and obeticholic acid ameliorate nonalcoholic steatohepatitis in mice. Cell Death Dis. 2017, 8, e2825. [Google Scholar] [CrossRef]
- Almohawes, Z.N.; El-Kott, A.; Morsy, K.; Shati, A.A.; El-Kenawy, A.E.; Khalifa, H.S.; Elsaid, F.G.; Abd-Lateif, A.M.; Abu-Zaiton, A.; Ebealy, E.R.; et al. Salidroside inhibits insulin resistance and hepatic steatosis by downregulating miR-21 and subsequent activation of AMPK and upregulation of PPARα in the liver and muscles of high fat diet-fed rats. Arch. Physiol. Biochem. 2022, 1–18. [Google Scholar] [CrossRef]
- Zhang, X.; Deng, F.; Zhang, Y.; Zhang, X.; Chen, J.; Jiang, Y. PPARγ attenuates hepatic inflammation and oxidative stress of non-alcoholic steatohepatitis via modulating the miR-21-5p/SFRP5 pathway. Mol. Med. Rep. 2021, 24, 823. [Google Scholar] [CrossRef] [PubMed]
- Shirasaki, T.; Honda, M.; Shimakami, T.; Horii, R.; Yamashita, T.; Sakai, Y.; Sakai, A.; Okada, H.; Watanabe, R.; Murakami, S.; et al. MicroRNA-27a Regulates Lipid Metabolism and Inhibits Hepatitis C Virus Replication in Human Hepatoma Cells. J. Virol. 2013, 87, 5270–5286. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Kim, A.Y.; Lee, H.W.; Son, Y.H.; Lee, G.Y.; Lee, J.W.; Lee, Y.K.; Kim, J.B. miR-27a is a negative regulator of adipocyte differentiation via suppressing PPARgamma expression. Biochem. Biophys. Res. Commun. 2010, 392, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Sun, W.; Zhou, M.; Tang, Y. MicroRNA-27a regulates hepatic lipid metabolism and alleviates NAFLD via repressing FAS and SCD1. Sci. Rep. 2017, 7, 14493. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Ge, G.; Pan, T.; Wen, D.; Gan, J. A pilot study of serum microRNAs panel as potential biomarkers for diagnosis of nonalcoholic fatty liver disease. PLoS ONE 2014, 9, e105192. [Google Scholar] [CrossRef]
- Gerhard, G.S. Micro RNAs in the development of non-alcoholic fatty liver disease. World J. Hepatol. 2015, 7, 226–234. [Google Scholar] [CrossRef]
- Zhang, J.; Powell, C.; Kay, M.; Sonkar, R.; Meruvu, S.; Choudhury, M. Effect of Chronic Western Diets on Non-Alcoholic Fatty Liver of Male Mice Modifying the PPAR-γ Pathway via miR-27b-5p Regulation. Int. J. Mol. Sci. 2021, 22, 1822. [Google Scholar] [CrossRef]
- Zhu, D.; Lyu, L.; Shen, P.; Wang, J.; Chen, J.; Sun, X.; Chen, L.; Zhang, L.; Zhou, Q.; Duan, Y. rSjP40 protein promotes PPARγ expression in LX-2 cells through microRNA-27b. FASEB J. 2018, 32, 4798–4803. [Google Scholar] [CrossRef]
- Jennewein, C.; von Knethen, A.; Schmid, T.; Brüne, B. MicroRNA-27b contributes to lipopolysaccharide-mediated peroxisome proliferator-activated receptor gamma (PPARgamma) mRNA destabilization. J. Biol. Chem. 2010, 285, 11846–11853. [Google Scholar] [CrossRef]
- Hsu, C.C.; Lai, C.Y.; Lin, C.Y.; Yeh, K.Y.; Her, G.M. MicroRNA-27b Depletion Enhances Endotrophic and Intravascular Lipid Accumulation and Induces Adipocyte Hyperplasia in Zebrafish. Int. J. Mol. Sci. 2017, 19, 93. [Google Scholar] [CrossRef]
- Li, S.; Li, J.; Fei, B.Y.; Shao, D.; Pan, Y.; Mo, Z.H.; Sun, B.Z.; Zhang, D.; Zheng, X.; Zhang, M.; et al. MiR-27a promotes hepatocellular carcinoma cell proliferation through suppression of its target gene peroxisome proliferator-activated receptor γ Chin. Med. J. 2015, 128, 941–947. [Google Scholar] [CrossRef]
- Zheng, X.; Zhang, F.; Zhao, Y.; Zhang, J.; Dawulieti, J.; Pan, Y.; Cui, L.; Sun, M.; Shao, D.; Li, M.; et al. Self-assembled dual fluorescence nanoparticles for CD44-targeted delivery of anti-miR-27a in liver cancer theranostics. Theranostics 2018, 8, 3808–3823. [Google Scholar] [CrossRef]
- Zarfeshani, A.; Ngo, S.; Sheppard, A.M. MicroRNA Expression Relating to Dietary-Induced Liver Steatosis and NASH. J. Clin. Med. 2015, 4, 1938–1950. [Google Scholar] [CrossRef]
- Castro, R.E.; Ferreira, D.M.; Afonso, M.B.; Borralho, P.M.; Machado, M.V.; Cortez-Pinto, H.; Rodrigues, C.M. miR-34a/SIRT1/p53 is suppressed by ursodeoxycholic acid in the rat liver and activated by disease severity in human non-alcoholic fatty liver disease. J. Hepatol. 2013, 58, 119–125. [Google Scholar] [CrossRef]
- Meng, F.; Glaser, S.S.; Francis, H.; Yang, F.; Han, Y.; Stokes, A.; Staloch, D.; McCarra, J.; Liu, J.; Venter, J.; et al. Epigenetic regulation of miR-34a expression in alcoholic liver injury. Am. J. Pathol. 2012, 181, 804–817. [Google Scholar] [CrossRef]
- Cermelli, S.; Ruggieri, A.; Marrero, J.A.; Ioannou, G.N.; Beretta, L. Circulating MicroRNAs in Patients with Chronic Hepatitis C and Non-Alcoholic Fatty Liver Disease. PLoS ONE 2011, 6, e23937. [Google Scholar] [CrossRef]
- Salvoza, N.C.; Klinzing, D.C.; Gopez-Cervantes, J.; Baclig, M.O. Association of Circulating Serum miR-34a and miR-122 with Dyslipidemia among Patients with Non-Alcoholic Fatty Liver Disease. PLoS ONE 2016, 11, e0153497. [Google Scholar] [CrossRef]
- Braza-Boïls, A.; Marí-Alexandre, J.; Molina, P.; Arnau, M.A.; Barceló-Molina, M.; Domingo, D.; Girbes, J.; Giner, J.; Martínez-Dolz, L.; Zorio, E. Deregulated hepatic microRNAs underlie the association between non-alcoholic fatty liver disease and coronary artery disease. Liver Int. 2016, 36, 1221–1229. [Google Scholar] [CrossRef]
- Ding, J.; Li, M.; Wan, X.; Jin, X.; Chen, S.; Yu, C.; Li, Y. Effect of miR-34a in regulating steatosis by targeting PPARα expression in nonalcoholic fatty liver disease. Sci. Rep. 2015, 5, 13729. [Google Scholar] [CrossRef]
- Guo, X.-Y.; Sun, F.; Chen, J.-N.; Wang, Y.-Q.; Pan, Q.; Fan, J.-G. circRNA_0046366 inhibits hepatocellular steatosis by normalization of PPAR signaling. World J. Gastroenterol. 2018, 24, 323–337. [Google Scholar] [CrossRef]
- Derdak, Z.; Villegas, K.A.; Harb, R.; Wu, A.M.; Sousa, A.; Wands, J.R. Inhibition of p53 attenuates steatosis and liver injury in a mouse model of non-alcoholic fatty liver disease. J. Hepatol. 2013, 58, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chen, Y.; Wu, S.; He, J.; Lou, L.; Ye, W.; Wang, J. microRNA-34a and microRNA-34c promote the activation of human hepatic stellate cells by targeting peroxisome proliferator-activated receptor γ. Mol. Med. Rep. 2015, 11, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Estep, M.; Armistead, D.; Hossain, N.; Elarainy, H.; Goodman, Z.; Baranova, A.; Chandhoke, V.; Younossi, Z.M. Differential expression of miRNAs in the visceral adipose tissue of patients with non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2010, 32, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Hanin, G.; Yayon, N.; Tzur, Y.; Haviv, R.; Bennett, E.R.; Udi, S.; Krishnamoorthy, Y.R.; Kotsiliti, E.; Zangen, R.; Efron, B.; et al. miRNA-132 induces hepatic steatosis and hyperlipidaemia by synergistic multitarget suppression. Gut 2018, 67, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.; Chu, D.C.; Maxwell, A.; Oakley, F.; Zhu, N.L.; Tsukamoto, H.; Mann, D.A. MeCP2 controls an epigenetic pathway that promotes myofibroblast transdifferentiation and fibrosis. Gastroenterology 2010, 138, 705–714. [Google Scholar] [CrossRef]
- Csak, T.; Bala, S.; Lippai, D.; Kodys, K.; Catalano, D.; Iracheta-Vellve, A.; Szabo, G. MicroRNA-155 Deficiency Attenuates Liver Steatosis and Fibrosis without Reducing Inflammation in a Mouse Model of Steatohepatitis. PLoS ONE 2015, 10, e0129251. [Google Scholar] [CrossRef]
- Miller, A.M.; Gilchrist, D.S.; Nijjar, J.; Araldi, E.; Ramírez, C.M.; Lavery, C.A.; Fernandez-Hernando, C.; McInnes, I.B.; Kurowska-Stolarska, M. MiR-155 Has a Protective Role in the Development of Non-Alcoholic Hepatosteatosis in Mice. PLoS ONE 2013, 8, e72324. [Google Scholar] [CrossRef]
- Bala, S.; Csak, T.; Saha, B.; Zatsiorsky, J.; Kodys, K.; Catalano, D.; Satishchandran, A.; Szabo, G. The pro-inflammatory effects of miR-155 promote liver fibrosis and alcohol-induced steatohepatitis. J. Hepatol. 2016, 64, 1378–1387. [Google Scholar] [CrossRef]
- Wang, Y.; Zheng, Z.J.; Jia, Y.J.; Yang, Y.L.; Xue, Y.M. Role of p53/miR-155-5p/sirt1 loop in renal tubular injury of diabetic kidney disease. J. Transl. Med. 2018, 16, 146. [Google Scholar] [CrossRef]
- Skårn, M.; Namløs, H.M.; Noordhuis, P.; Wang, M.-Y.; Meza-Zepeda, L.A.; Myklebost, O. Adipocyte Differentiation of Human Bone Marrow-Derived Stromal Cells Is Modulated by MicroRNA-155, MicroRNA-221, and MicroRNA-222. Stem Cells Dev. 2012, 21, 873–883. [Google Scholar] [CrossRef]
- Ying, W.; Riopel, M.; Bandyopadhyay, G.; Dong, Y.; Birmingham, A.; Seo, J.B.; Ofrecio, J.M.; Wollam, J.; Hernandez-Carretero, A.; Fu, W.; et al. Adipose Tissue Macrophage-Derived Exosomal miRNAs Can Modulate In Vivo and In Vitro Insulin Sensitivity. Cell 2017, 171, 372–384.e12. [Google Scholar] [CrossRef]
- Tryggestad, J.B.; Teague, A.M.; Sparling, D.P.; Jiang, S.; Chernausek, S.D. Macrophage-Derived MicroRNA-155 Increases in Obesity and Influences Adipocyte Metabolism by Targeting Peroxisome Proliferator-Activated Receptor Gamma. Obesity 2019, 27, 1856–1864. [Google Scholar] [CrossRef]
- Wang, D.R.; Wang, B.; Yang, M.; Liu, Z.L.; Sun, J.; Wang, Y.; Sun, H.; Xie, L.J. Suppression of miR-30a-3p Attenuates Hepatic Steatosis in Non-alcoholic Fatty Liver Disease. Biochem. Genet. 2020, 58, 691–704. [Google Scholar] [CrossRef]
- Mittal, S.; Inamdar, S.; Acharya, J.; Pekhale, K.; Kalamkar, S.; Boppana, R.; Ghaskadbi, S. miR-3666 inhibits development of hepatic steatosis by negatively regulating PPARγ. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158777. [Google Scholar] [CrossRef]
- Mattick, J.S.; Amaral, P.P.; Carninci, P.; Carpenter, S.; Chang, H.Y.; Chen, L.L.; Chen, R.; Dean, C.; Dinger, M.E.; Fitzgerald, K.A.; et al. Long non-coding RNAs: Definitions, functions, challenges and recommendations. Nat. Rev. Mol. Cell Biol. 2023, 1–17. [Google Scholar] [CrossRef]
- Huarte, M. The emerging role of lncRNAs in cancer. Nat. Med. 2015, 21, 1253–1261. [Google Scholar] [CrossRef]
- Sookoian, S.; Rohr, C.; Salatino, A.; Dopazo, H.; Fernandez Gianotti, T.; Castaño, G.O.; Pirola, C.J. Genetic variation in long noncoding RNAs and the risk of nonalcoholic fatty liver disease. Oncotarget 2017, 8, 22917–22926. [Google Scholar] [CrossRef]
- Lan, X.; Wu, L.; Wu, N.; Chen, Q.; Li, Y.; Du, X.; Wei, C.; Feng, L.; Li, Y.; Osoro, E.K.; et al. Long Noncoding RNA lnc-HC Regulates PPARγ-Mediated Hepatic Lipid Metabolism through miR-130b-3p. Mol. Ther. Nucleic Acids 2019, 18, 954–965. [Google Scholar] [CrossRef]
- Xu, B.M.; Xiao, L.; Kang, C.M.; Ding, L.; Guo, F.X.; Li, P.; Lu, Z.F.; Wu, Q.; Xu, Y.J.; Bai, H.L.; et al. LncRNA AC096664.3/PPAR-γ/ABCG1-dependent signal transduction pathway contributes to the regulation of cholesterol homeostasis. J. Cell. Biochem. 2019, 120, 13775–13782. [Google Scholar] [CrossRef]
- Liu, J.; Tang, T.; Wang, G.D.; Liu, B. LncRNA-H19 promotes hepatic lipogenesis by directly regulating miR-130a/PPARγ axis in non-alcoholic fatty liver disease. Biosci. Rep. 2019, 39, BSR20181722. [Google Scholar] [CrossRef]
- Wang, H.; Cao, Y.; Shu, L.; Zhu, Y.; Peng, Q.; Ran, L.; Wu, J.; Luo, Y.; Zuo, G.; Luo, J.; et al. Long non-coding RNA (lncRNA) H19 induces hepatic steatosis through activating MLXIPL and mTORC1 networks in hepatocytes. J. Cell. Mol. Med. 2020, 24, 1399–1412. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Yu, D.; Nian, X.; Liu, J.; Koenig, R.J.; Xu, B.; Sheng, L. LncRNA SRA promotes hepatic steatosis through repressing the expression of adipose triglyceride lipase (ATGL). Sci. Rep. 2016, 6, 35531. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.X.; Li, B.Z.; Mitchell, L.A.; Wu, Y.; Qi, X.; Jin, Z.; Jia, B.; Wang, X.; Zeng, B.X.; Liu, H.M.; et al. “Perfect” designer chromosome V and behavior of a ring derivative. Science 2017, 355, eaaf4704. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Gerin, I.; Miao, H.; Vu-Phan, D.; Johnson, C.N.; Xu, R.; Chen, X.W.; Cawthorn, W.P.; MacDougald, O.A.; Koenig, R.J. Multiple roles for the non-coding RNA SRA in regulation of adipogenesis and insulin sensitivity. PLoS ONE 2010, 5, e14199. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Ding, J.; Chen, A.; Song, Y.; Xu, C.; Tian, F.; Zhao, J. Aerobic exercise improves adipogenesis in diet-induced obese mice via the lncSRA/p38/JNK/PPARγ pathway. Nutr. Res. 2022, 105, 20–32. [Google Scholar] [CrossRef]
- Guy, C.D.; Suzuki, A.; Zdanowicz, M.; Abdelmalek, M.F.; Burchette, J.; Unalp, A.; Diehl, A.M.; NASH CRN. Hedgehog pathway activation parallels histologic severity of injury and fibrosis in human nonalcoholic fatty liver disease. Hepatology 2012, 55, 1711–1721. [Google Scholar] [CrossRef]
- Kwon, H.; Song, K.; Han, C.; Chen, W.; Wang, Y.; Dash, S.; Lim, K.; Wu, T. Inhibition of hedgehog signaling ameliorates hepatic inflammation in mice with nonalcoholic fatty liver disease. Hepatology 2016, 63, 1155–1169. [Google Scholar] [CrossRef]
- Jiang, Y.; Peng, J.; Song, J.; He, J.; Jiang, M.; Wang, J.; Ma, L.; Wang, Y.; Lin, M.; Wu, H.; et al. Loss of Hilnc prevents diet-induced hepatic steatosis through binding of IGF2BP2. Nat. Metab. 2021, 3, 1569–1584. [Google Scholar] [CrossRef]
- Barrett, S.P.; Salzman, J. Circular RNAs: Analysis, expression and potential functions. Development 2016, 143, 1838–1847. [Google Scholar] [CrossRef]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef]
- Zaiou, M. The Emerging Role and Promise of Circular RNAs in Obesity and Related Metabolic Disorders. Cells 2020, 9, 1473. [Google Scholar] [CrossRef]
- Jin, X.; Feng, C.Y.; Xiang, Z.; Chen, Y.P.; Li, Y.M. CircRNA expression pattern and circRNA-miRNA-mRNA network in the pathogenesis of nonalcoholic steatohepatitis. Oncotarget 2016, 7, 66455–66467. [Google Scholar] [CrossRef]
- Guo, X.Y.; Chen, J.N.; Sun, F.; Wang, Y.Q.; Pan, Q.; Fan, J.G. circRNA_0046367 Prevents Hepatoxicity of Lipid Peroxidation: An Inhibitory Role against Hepatic Steatosis. Oxid. Med. Cell. Longev. 2017, 2017, 3960197. [Google Scholar] [CrossRef]
- Cai, H.; Jiang, Z.; Yang, X.; Lin, J.; Cai, Q.; Li, X. Circular RNA HIPK3 contributes to hyperglycemia and insulin homeostasis by sponging miR-192-5p and upregulating transcription factor forkhead box O1. Endocr. J. 2020, 67, 397–408. [Google Scholar] [CrossRef]
- Lu, J.; Pang, L.; Zhang, B.; Gong, Z.; Song, C. Silencing circANKRD36 inhibits streptozotocin-induced insulin resistance and inflammation in diabetic rats by targeting miR-145 via XBP1. Inflamm. Res. 2021, 70, 695–704. [Google Scholar] [CrossRef]
- Guo, X.Y.; He, C.X.; Wang, Y.Q.; Sun, C.; Li, G.M.; Su, Q.; Pan, Q.; Fan, J.G. Circular RNA Profiling and Bioinformatic Modeling Identify Its Regulatory Role in Hepatic Steatosis. BioMed Res. Int. 2017, 2017, 5936171. [Google Scholar] [CrossRef]
- Yuan, X.; Li, Y.; Wen, S.; Xu, C.; Wang, C.; He, Y.; Zhou, L. CircLDLR acts as a sponge for miR-667-5p to regulate SIRT1 expression in non-alcoholic fatty liver disease. Lipids Health Dis. 2022, 21, 127. [Google Scholar] [CrossRef]
- Picard, F.; Kurtev, M.; Chung, N.; Topark-Ngarm, A.; Senawong, T.; Machado De Oliveira, R.; Leid, M.; McBurney, M.W.; Guarente, L. Sirt1 Promotes Fat Mobilization in White Adipocytes by Repressing PPAR-Gamma. Nature 2004, 429, 771–776. [Google Scholar] [CrossRef]
- Lin, X.; Du, Y.; Lu, W.; Gui, W.; Sun, S.; Zhu, Y.; Wang, G.; Eserberg, D.T.; Zheng, F.; Zhou, J.; et al. CircRNF111 Protects Against Insulin Resistance and Lipid Deposition via Regulating miR-143-3p/IGF2R Axis in Metabolic Syndrome. Front. Cell Dev. Biol. 2021, 9, 663148. [Google Scholar] [CrossRef]
- Lee, Y.H.; Bae, S.C.; Song, G.G. Meta-analysis of associations between the peroxisome proliferator-activated receptor-γ Pro12Ala polymorphism and susceptibility to nonalcoholic fatty liver disease, rheumatoid arthritis, and psoriatic arthritis. Genet. Test. Mol. Biomark. 2014, 18, 341–348. [Google Scholar] [CrossRef]
- Xie, R.; Tang, S.; Yang, Y. Associations of peroxisome proliferator-activated receptor-γ Pro12Ala polymorphism with non-alcoholic fatty liver disease: A meta-analysis. J. Diabetes Complicat. 2022, 36, 108261. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Li, Y.-Y.; Nie, Y.-Q.; Sha, W.-H.; Du, Y.-L.; Lai, X.-B.; Zhou, Y.-J. Effect of peroxisome proliferator-activated receptors-gamma and co-activator-1alpha genetic polymorphisms on plasma adiponectin levels and susceptibility of non-alcoholic fatty liver disease in Chinese people. Liver Int. 2008, 28, 385–392. [Google Scholar] [CrossRef]
- Zhou, Y.J.; Li, Y.Y.; Nie, Y.Q.; Yang, H.; Zhan, Q.; Huang, J.; Shi, S.L.; Lai, X.B.; Huang, H.L. Influence of polygenetic polymorphisms on the susceptibility to non-alcoholic fatty liver disease of Chinese people. J. Gastroenterol. Hepatol. 2010, 25, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Flessa, C.M.; Nasiri-Ansari, N.; Kyrou, I.; Leca, B.M.; Lianou, M.; Chatzigeorgiou, A.; Kaltsas, G.; Kassi, E.; Randeva, H.S. Genetic and Diet-Induced Animal Models for Non-Alcoholic Fatty Liver Disease (NAFLD) Research. Int. J. Mol. Sci. 2022, 23, 15791. [Google Scholar] [CrossRef]
- Santhekadur, P.K.; Kumar, D.P.; Sanyal, A.J. Preclinical models of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 230–237. [Google Scholar] [CrossRef]
- Xu, X.; Tan, X.; Tampe, B.; Wilhelmi, T.; Hulshoff, M.S.; Saito, S.; Moser, T.; Kalluri, R.; Hasenfuss, G.; Zeisberg, E.M.; et al. High-fidelity CRISPR/Cas9- based gene-specific hydroxymethylation rescues gene expression and attenuates renal fibrosis. Nat. Commun. 2018, 9, 3509. [Google Scholar] [CrossRef]
- Horii, T.; Morita, S.; Hatada, I. Generation of Epigenetic Disease Model Mice by Targeted Demethylation of the Epigenome. Methods Mol. Biol. 2023, 2577, 255–268. [Google Scholar] [CrossRef]
- Ratziu, V.; Charlotte, F.; Bernhardt, C.; Giral, P.; Halbron, M.; Lenaour, G.; Hartmann-Heurtier, A.; Bruckert, E.; Poynard, T.; LIDO Study Group. Long-term efficacy of rosiglitazone in nonalcoholic steatohepatitis: Results of the fatty liver improvement by rosiglitazone therapy (FLIRT 2) extension trial. Hepatology 2010, 51, 445–453. [Google Scholar] [CrossRef]
- Skat-Rørdam, J.; Højland Ipsen, D.; Lykkesfeldt, J.; Tveden-Nyborg, P. A role of peroxisome proliferator-activated receptor γ in non-alcoholic fatty liver disease. Basic Clin. Pharmacol. Toxicol. 2019, 124, 528–537. [Google Scholar] [CrossRef]
- Yang, X.; Frank, J.; Gonzalez, F.J.; Huang, M.; Bi, H. Nuclear receptors and non-alcoholic fatty liver disease: An update. Liver Res. 2020, 4, 88–93. [Google Scholar] [CrossRef]
- Ahmadian, M.; Myoung Suh, J.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARgamma signaling and metabolism: The good, the bad and the future. Nat. Med. 2013, 19, 557–566. [Google Scholar] [CrossRef]
- Romualdo, G.R.; Valente, L.C.; Sprocatti, A.C.; Bacil, G.P.; de Souza, I.P.; Rodrigues, J.; Rodrigues, M.A.M.; Vinken, M.; Cogliati, B.; Barbisan, L.F. Western diet-induced mouse model of non-alcoholic fatty liver disease associated with metabolic outcomes: Features of gut microbiome-liver-adipose tissue axis. Nutrition 2022, 103–104, 111836. [Google Scholar] [CrossRef]



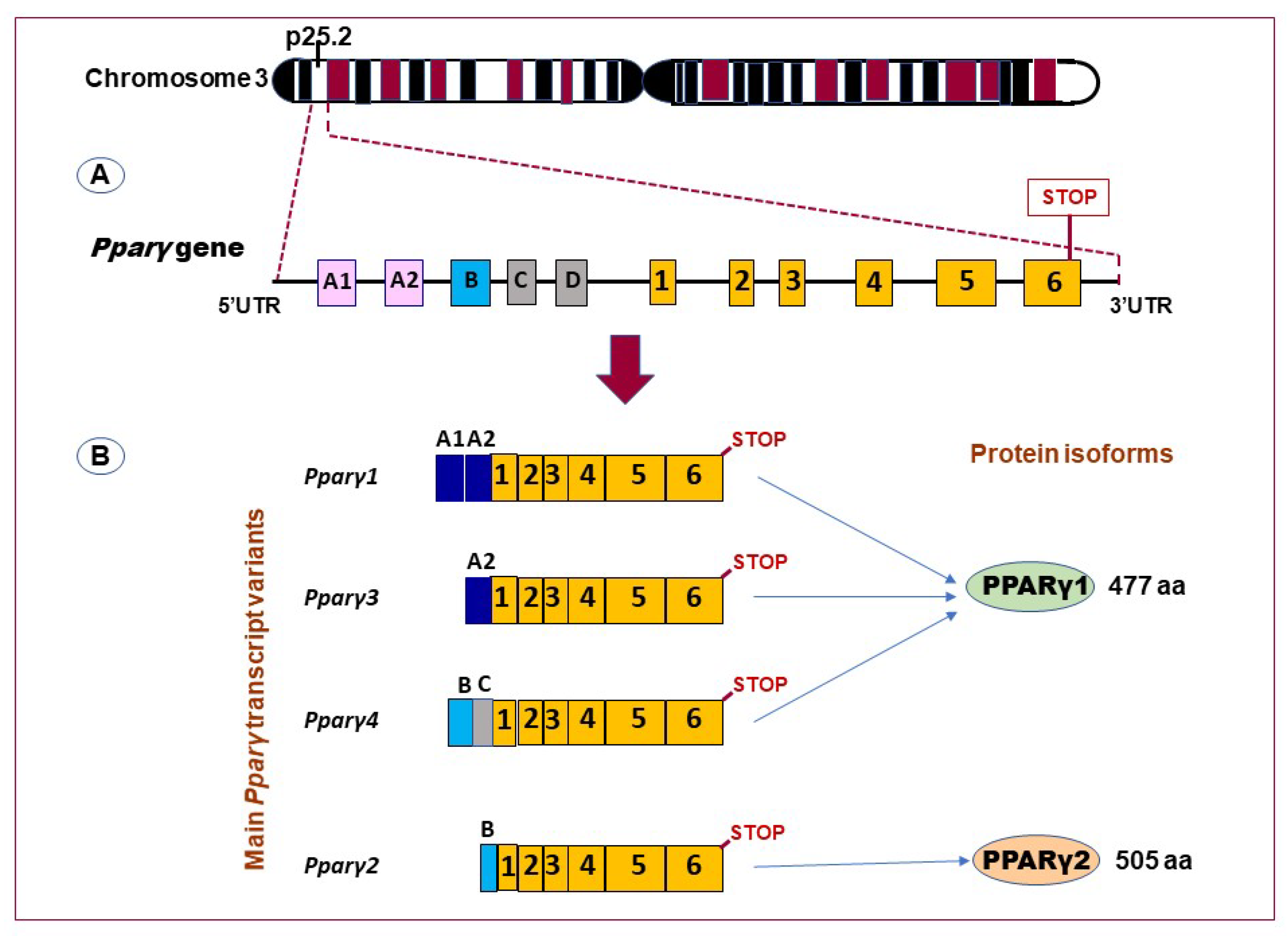{kind=link}
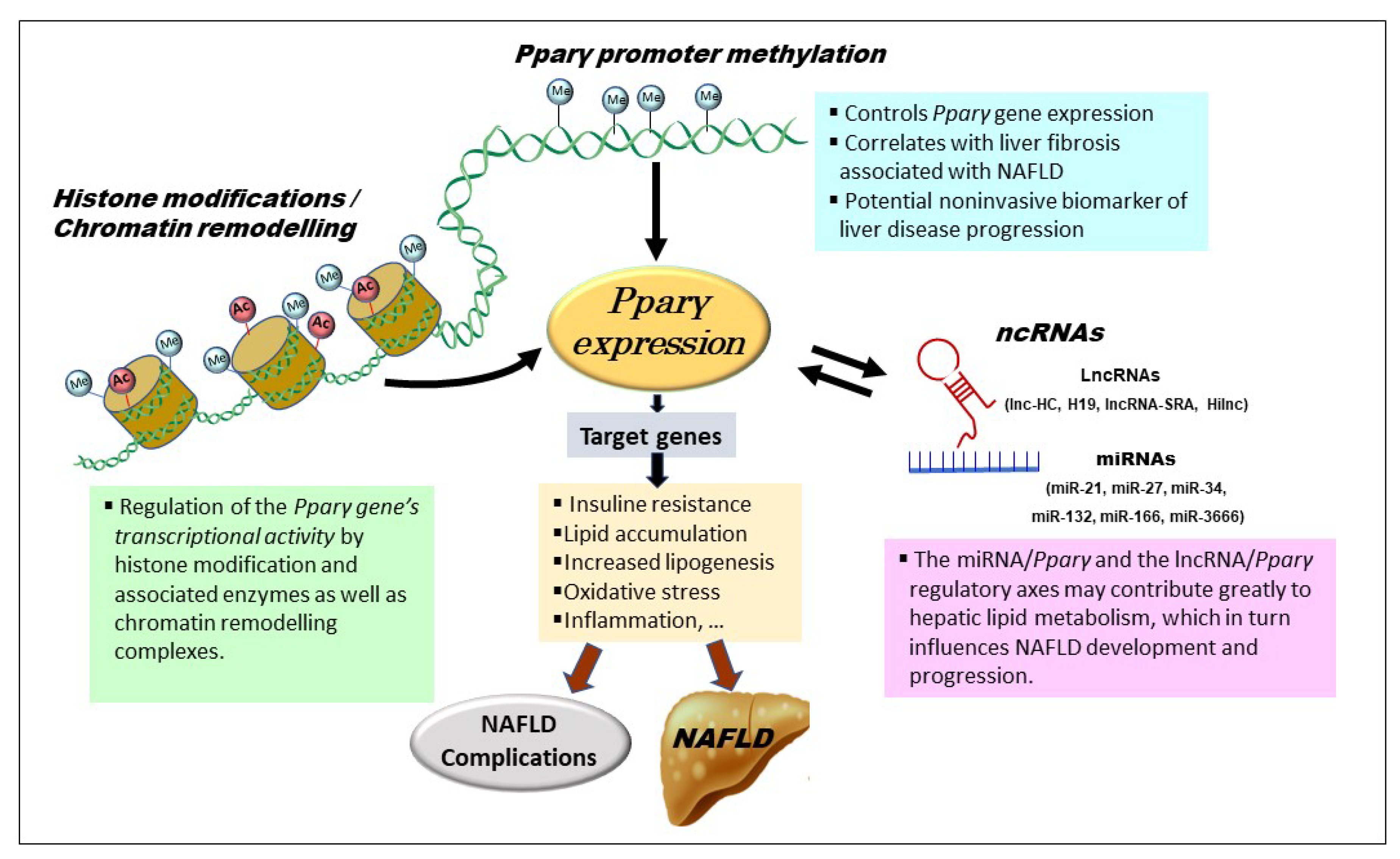{kind=link}
| Epigenetic Change | Biological Effect | Reference |
|---|---|---|
| Pparα/Pparγ/ methylation | DNA methylation of Pparα and Pparγ can distinguish between mild and severe NAFLD-associated fibrosis | [45] |
| Pparγ promoter methylation | HFD and palmitic acid alter global and Pparγ promoter DNA methylation, resulting in Pparγ expression and enhanced lipid retention in the liver, which leads to the development of NAFLD | [47] |
| Pparγ promoter methylation | Pparγ promoter hypermethylation levels in plasma-free DNA could be used as a non-invasive method to differentiate between NAFLD patients with mild and severe fibrosis | [48] |
| Pparγ promoter methylation | Methylation levels of Pparγ correlate with liver fibrosis in rat model as well as in NAFLD patients | [44,48] |
| Epigenetic Effector | Biological Effect | Reference |
|---|---|---|
| MLL4 | Murine steatosis caused by excessive feeding is regulated by the histone H3 lysine 4 methyltransferase MLL4/KMT2D via PPARγ2 | [56] |
| Suv39h2 | SUV39H2 expression in hepatocytes, mice, and human livers is induced by pro-NASH stimuli, and thus contributes to NASH pathogenesis by suppressing Pparγ and Sirt1 expression. | [58] |
| JMJD1A | JMJD1A promotes PPARγ expression by regulating the demethylation of Pparγ gene and thus inhibit HSCs activation and fibrosis | [60] |
| JMJD2B | JMJD2B promotes the development of hepatic steatosis by upregulating PPARγ2 and steatosis target genes. | [61] |
| miRNA | Target or Pathway | Pathophysiological Processes | References |
|---|---|---|---|
| miR-21 | PPARγ/SFR5 | PPARγ prevents inflammation and oxidative stress in mouse models and human tissue samples from NASH patients by modulating the miR21-5p/SFRP5 pathway | [101] |
| miR-27 | PPARγ | Disrupting endogenous miR-27b activity in Zebrafish causes lipid accumulation and increased PPARγ expression in the liver, resulting in the early onset of NAFLD and NASH | [110] |
| miR-34 | PPARγ | By targeting PPARγ, miR-34a/c activation may be associated with liver fibrosis | [122] |
| miR-132 | PPARγ/MeCP/EZH2 | miR132/MeCP2/EZH2 axis is involved in the regulation of liver fibrosis | [125] |
| miR-155 | PPARγ | miR-155 contained in obese ATM-exosomes, contributes to insulin resistance and glucose intolerance via a mechanism that is most likely related to direct suppression of its target gene Pparγ | [131] |
| miR-3666 | PPARγ | miR-3666 inhibits the development of hepatic steatosis by negatively regulating PPARγ | [134] |
| LncRNA | Targeted or Pathway | Pathophysiological Processes | References |
|---|---|---|---|
| Lnc-HC | miR-130-3p/PPARγ | LncRNA regulates hepatic lipid droplets accumulation via miR-130-3p/PPARγ pathway, which may help to prevent NAFLD | [138] |
| H19 | miR-130a/PPARγ | H19 promotes hepatic lipogenesis and the progression of NAFLD via miR-130a/PPARγ axis | [140] |
| LncRNA-SRA | PPARγ | LncRNA-SRA promotes hepatic steatosis by repressing the expression of adipose triglyceride lipase | [141] |
| Hilnc | IGF22BP2/PPARγ | The loss of Hilnc prevents diet-induced hepatic steatosis by inhibiting PPARγ | [148] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaiou, M. Peroxisome Proliferator-Activated Receptor-γ as a Target and Regulator of Epigenetic Mechanisms in Nonalcoholic Fatty Liver Disease. Cells 2023, 12, 1205. https://doi.org/10.3390/cells12081205
Zaiou M. Peroxisome Proliferator-Activated Receptor-γ as a Target and Regulator of Epigenetic Mechanisms in Nonalcoholic Fatty Liver Disease. Cells. 2023; 12(8):1205. https://doi.org/10.3390/cells12081205
Chicago/Turabian StyleZaiou, Mohamed. 2023. "Peroxisome Proliferator-Activated Receptor-γ as a Target and Regulator of Epigenetic Mechanisms in Nonalcoholic Fatty Liver Disease" Cells 12, no. 8: 1205. https://doi.org/10.3390/cells12081205