

Glucocorticoid Hormones as Modulators of the Kynurenine Pathway in Chronic Pain Conditions



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Pathomechanisms of Chronic Pain
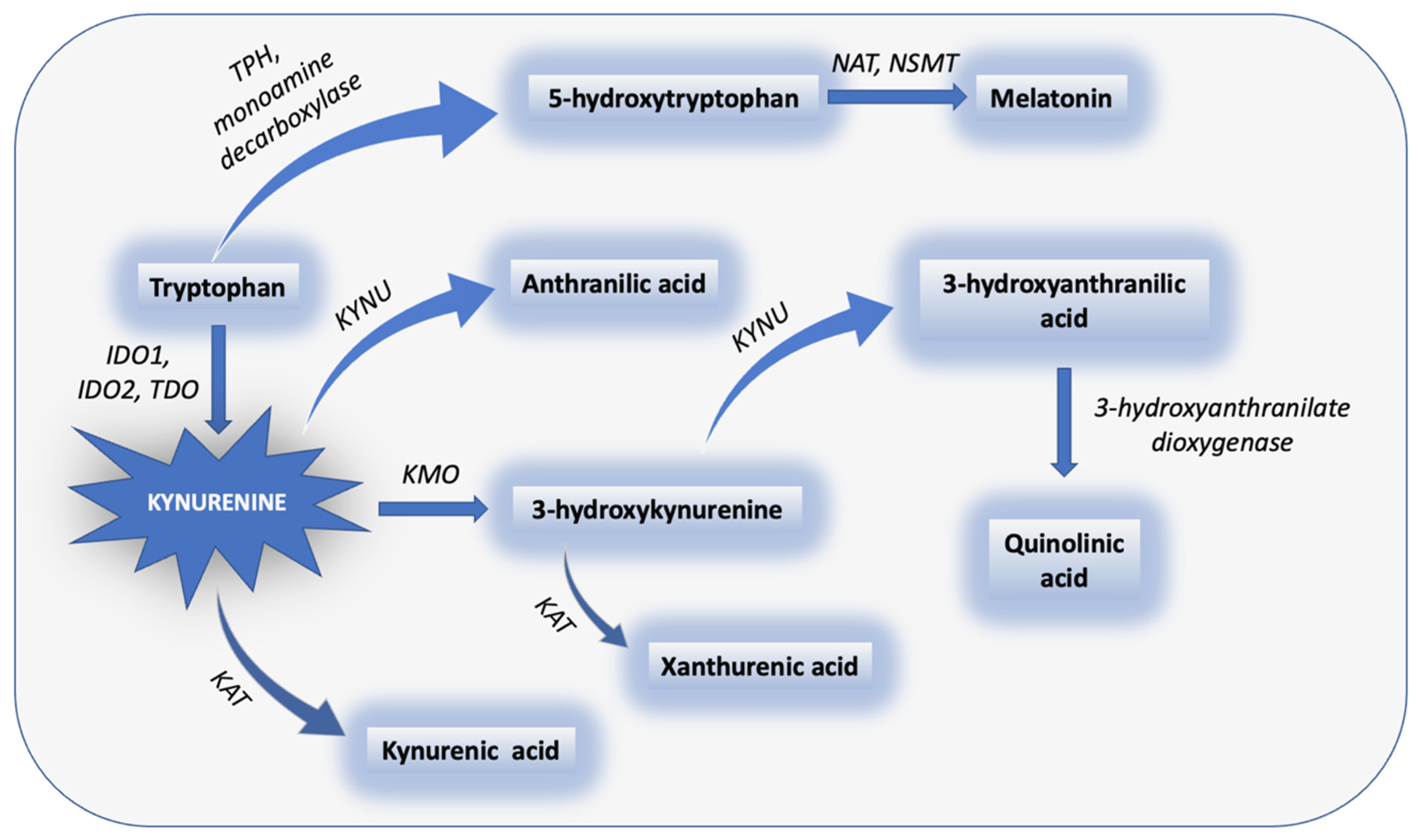
3. The Metabolic Pathways of TRP: KP and Serotonin Pathway
KP in Chronic Pain and Comorbid Depression
4. Pain Quantification Using Biomarkers
4.1. KYN Metabolites as Biomarkers for Chronic Pain
4.2. KYN Metabolites and the Blood–Brain Barrier
4.3. KYNA as a Target Compound in Nociceptive Processing
4.4. The KP in Migraine and Neuropathic Pain
4.5. The KP Compounds as Analgesic Targets in Chronic Pain
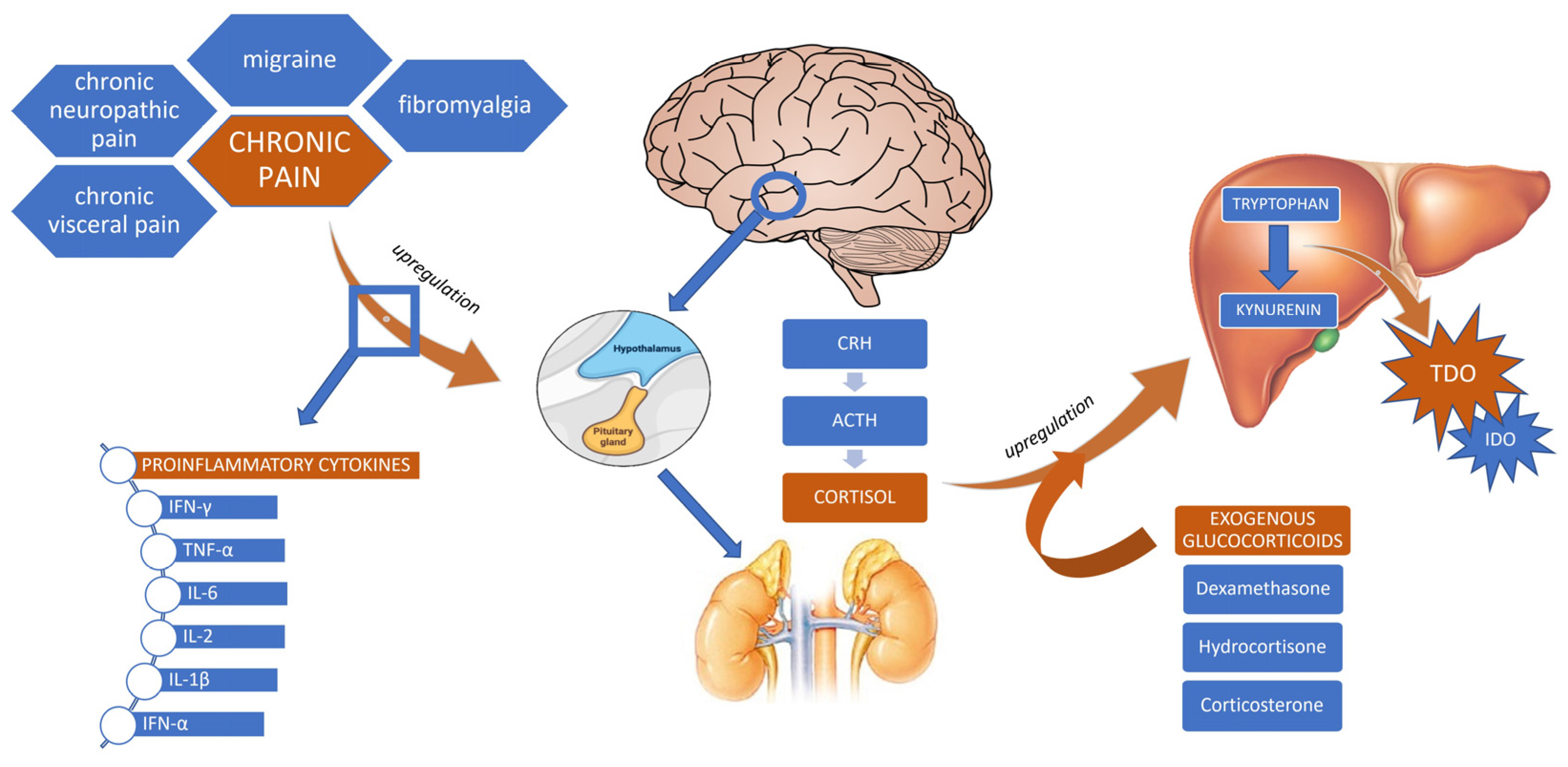
5. The Effects of Chronic Pain on the HPA Axis
5.1. Diurnal Cortisol Slope
5.2. Cortisol and Chronic Pain
5.3. Glucocorticoids and the KP
6. Discussion
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Raja, S.N.; Carr, D.B.; Cohen, M.; Finnerup, N.B.; Flor, H.; Gibson, S.; Keefe, F.J.; Mogil, J.S.; Ringkamp, M.; Sluka, K.A.; et al. The revised International Association for the Study of Pain definition of pain: Concepts, challenges, and compromises. Pain 2020, 161, 1976–1982. [Google Scholar] [CrossRef] [PubMed]
- Morrison, I.; Perini, I.; Dunham, J. Facets and mechanisms of adaptive pain behavior: Predictive regulation and action. Front. Hum. Neurosci. 2013, 7, 755. [Google Scholar] [CrossRef] [PubMed]
- Price, T.J.; Dussor, G. Evolution: The Advantage of ‘Maladaptive’ Pain Plasticity. Curr. Biol. 2014, 24, R384–R386. [Google Scholar] [CrossRef] [PubMed]
- Griep, E.N.; Boersma, J.W.; Lentjes, E.G.; Prins, A.P.; Van Der Korst, J.K.; De Kloet, E.R. Function of the hypothalamic-pituitary-adrenal axis in patients with fibromyalgia and low back pain. J. Rheumatol. 1998, 25, 1374–1381. [Google Scholar]
- Kuner, R.; Flor, H. Structural plasticity and reorganisation in chronic pain. Nat. Rev. Neurosci. 2016, 18, 20–30. [Google Scholar] [CrossRef]
- Chapman, C.R.; Tuckett, R.P.; Song, C.W. Pain and Stress in a Systems Perspective: Reciprocal Neural, Endocrine, and Immune Interactions. J. Pain 2008, 9, 122–145. [Google Scholar] [CrossRef]
- Jovanovic, F.; Candido, K.D.; Knezevic, N.N. The Role of the Kynurenine Signaling Pathway in Different Chronic Pain Conditions and Potential Use of Therapeutic Agents. Int. J. Mol. Sci. 2020, 21, 6045. [Google Scholar] [CrossRef]
- Besedovsky, H.O.; Del Rey, A. The cytokine-HPA axis feed-back circuit. Z. Rheumatol. 2000, 59 (Suppl. 2), II26–II30. [Google Scholar] [CrossRef]
- Rivest, S. How circulating cytokines trigger the neural circuits that control the hypothalamic–pituitary–adrenal axis. Psychoneuroendocrinology 2001, 26, 761–788. [Google Scholar] [CrossRef]
- Andersson, J. The inflammatory reflex—Introduction. J. Intern. Med. 2005, 257, 122–125. [Google Scholar] [CrossRef]
- Silverman, M.N.; Pearce, B.D.; Biron, C.A.; Miller, A.H. Immune modulation of the hypothalamic-pituitary-adrenal (HPA) axis during viral infection. Viral Immunol. 2005, 18, 41–78. [Google Scholar] [CrossRef] [PubMed]
- Zelaya, C.E.; Dahlhamer, J.M.; Lucas, J.W.; Connor, E.M. Chronic Pain and High-impact Chronic Pain Among U.S. Adults, 2019. NCHS Data Brief 2020, 390, 1–8. [Google Scholar]
- Tanaka, M.; Torok, N.; Toth, F.; Szabo, A.; Vecsei, L. Co-Players in Chronic Pain: Neuroinflammation and the Tryptophan-Kynurenine Metabolic Pathway. Biomedicines 2021, 9, 897. [Google Scholar] [CrossRef]
- Pinho-Ribeiro, F.A.; Verri, W.A., Jr.; Chiu, I.M. Nociceptor Sensory Neuron–Immune Interactions in Pain and Inflammation. Trends Immunol. 2017, 38, 5–19. [Google Scholar] [CrossRef]
- Edwards, R.R.; Dworkin, R.H.; Sullivan, M.D.; Turk, D.C.; Wasan, A.D. The Role of Psychosocial Processes in the Development and Maintenance of Chronic Pain. J. Pain 2016, 17 (Suppl. S9), T70–T92. [Google Scholar] [CrossRef] [PubMed]
- Mifflin, K.; Kerr, B.J. The transition from acute to chronic pain: Understanding how different biological systems interact. Can. J. Anesth. 2013, 61, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Lamana, M.S.; Miranda, J.; Tobaldini, G.; Fischer, L.; Tambeli, C.H. Pain chronification and chronic pain impair a defensive behavior, but not the ability of acute pain to facilitate it, through the activation of an endogenous analgesia circuit. Behav. Neurosci. 2018, 132, 614–623. [Google Scholar] [CrossRef]
- Basbaum, A.I.; Bautista, D.M.; Scherrer, G.; Julius, D. Cellular and molecular mechanisms of pain. Cell 2009, 139, 267–284. [Google Scholar] [CrossRef]
- Turnbull, A.V.; Rivier, C.L. Regulation of the hypothalamic-pituitary-adrenal axis by cytokines: Actions and mechanisms of action. Physiol. Rev. 1999, 79, 1–71. [Google Scholar] [CrossRef]
- Liu, C.; Chu, D.; Kalantar-Zadeh, K.; George, J.; Young, H.A.; Liu, G. Cytokines: From Clinical Significance to Quantification. Adv. Sci. 2021, 8, e2004433. [Google Scholar] [CrossRef]
- Koch, A.; Zacharowski, K.; Boehm, O.; Stevens, M.; Lipfert, P.; von Giesen, H.-J.; Wolf, A.; Freynhagen, R. Nitric oxide and pro-inflammatory cytokines correlate with pain intensity in chronic pain patients. Inflamm. Res. 2007, 56, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-K.; Jeon, S.W. Neuroinflammation and the Immune-Kynurenine Pathway in Anxiety Disorders. Curr. Neuropharmacol. 2018, 16, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Tóth, F.; Polyák, H.; Szabó, Á.; Mándi, Y.; Vécsei, L. Immune Influencers in Action: Metabolites and Enzymes of the Tryptophan-Kynurenine Metabolic Pathway. Biomedicines 2021, 9, 734. [Google Scholar] [CrossRef]
- Höglund, E.; Øverli, Ø.; Winberg, S. Tryptophan Metabolic Pathways and Brain Serotonergic Activity: A Comparative Review. Front. Endocrinol. 2019, 10, 158. [Google Scholar] [CrossRef] [PubMed]
- Agus, A.; Planchais, J.; Sokol, H. Gut Microbiota Regulation of Tryptophan Metabolism in Health and Disease. Cell Host Microbe 2018, 23, 716–724. [Google Scholar] [CrossRef]
- Qin, Y.; Wang, N.; Zhang, X.; Han, X.; Zhai, X.; Lu, Y. IDO and TDO as a potential therapeutic target in different types of depression. Metab. Brain Dis. 2018, 33, 1787–1800. [Google Scholar] [CrossRef]
- Phillips, R.S. Structure and mechanism of kynureninase. Arch. Biochem. Biophys. 2014, 544, 69–74. [Google Scholar] [CrossRef]
- Nematollahi, A.; Sun, G.; Jayawickrama, G.S.; Church, W.B. Kynurenine Aminotransferase Isozyme Inhibitors: A Review. Int. J. Mol. Sci. 2016, 17, 946. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, J.; Wang, C.; Liu, Y.; Li, M.; Li, Y.; Li, R.; Han, Z.; Wang, J.; Chen, L.; et al. Kynurenine-3-monooxygenase (KMO) broadly inhibits viral infections via triggering NMDAR/Ca2+ influx and CaMKII/ IRF3-mediated IFN-β production. PLOS Pathog. 2022, 18, e1010366. [Google Scholar] [CrossRef]
- Tanaka, M.; Bohár, Z.; Vécsei, L. Are Kynurenines Accomplices or Principal Villains in Dementia? Maintenance of Kynurenine Metabolism. Molecules 2020, 25, 564. [Google Scholar] [CrossRef]
- Karmakar, S.; Lal, G. Role of serotonin receptor signaling in cancer cells and anti-tumor immunity. Theranostics 2021, 11, 5296–5312. [Google Scholar] [CrossRef]
- Walther, D.J.; Peter, J.U.; Bashammakh, S.; Hörtnagl, H.; Voits, M.; Fink, H.; Bader, M. Synthesis of Serotonin by a Second Tryptophan Hydroxylase Isoform. Science 2003, 299, 76. [Google Scholar] [CrossRef] [PubMed]
- Herr, N.; Bode, C.; Duerschmied, D. The Effects of Serotonin in Immune Cells. Front. Cardiovasc. Med. 2017, 4, 48. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Yu, Y.; Shen, Y.; Liu, Q.; Zhao, Z.; Sharma, R.; Reiter, R.J. Melatonin Synthesis and Function: Evolutionary History in Animals and Plants. Front. Endocrinol. 2019, 10, 249. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, S.; Sawhney, G.; Pandhi, P. The therapeutic potential of melatonin: A review of the science. MedGenMed Medscape Gen. Med. 2004, 6, 46. [Google Scholar]
- Carlin, J.M.; Borden, E.C.; Sondel, P.M.; Byrne, G.I. Biologic-response-modifier-induced indoleamine 2,3-dioxygenase activity in human peripheral blood mononuclear cell cultures. J. Immunol. 1987, 139, 2414–2418. [Google Scholar] [CrossRef]
- Salter, M.; Pogson, C.; A Smith, S.; Cook, J.S.; A Bender, D. The role of tryptophan 2,3-dioxygenase in the hormonal control of tryptophan metabolism in isolated rat liver cells. Effects of glucocorticoids and experimental diabetes. Biochem. J. 1985, 229, 499–504. [Google Scholar] [CrossRef]
- Oxenkrug, G. Serotonin—Kynurenine hypothesis of depression: Historical overview and recent developments. Curr. Drug Targets 2013, 14, 514–521. [Google Scholar] [CrossRef]
- Athnaiel, O.; Ong, C.; Knezevic, N.N. The Role of Kynurenine and Its Metabolites in Comorbid Chronic Pain and Depression. Metabolites 2022, 12, 950. [Google Scholar] [CrossRef]
- Marx, W.; McGuinness, A.J.; Rocks, T.; Ruusunen, A.; Cleminson, J.; Walker, A.J.; Gomes-Da-Costa, S.; Lane, M.; Sanches, M.; Diaz, A.P.; et al. The kynurenine pathway in major depressive disorder, bipolar disorder, and schizophrenia: A meta-analysis of 101 studies. Mol. Psychiatry 2020, 26, 4158–4178. [Google Scholar] [CrossRef]
- Tappe-Theodor, A.; Kuner, R. A common ground for pain and depression. Nat. Neurosci. 2019, 22, 1612–1614. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Jin, Y.; Meng, Q.; Zhu, X.; Bai, T.; Tian, Y.; Mao, Y.; Wang, L.; Xie, W.; Zhong, H.; et al. A neural circuit for comorbid depressive symptoms in chronic pain. Nat. Neurosci. 2019, 22, 1649–1658. [Google Scholar] [CrossRef] [PubMed]
- Zádor, F.; Joca, S.; Nagy-Grócz, G.; Dvorácskó, S.; Szűcs, E.; Tömböly, C.; Benyhe, S.; Vécsei, L. Pro-Inflammatory Cytokines: Potential Links between the Endocannabinoid System and the Kynurenine Pathway in Depression. Int. J. Mol. Sci. 2021, 22, 5903. [Google Scholar] [CrossRef]
- Gunn, J.; Hill, M.; Cotten, B.M.; Deer, T.R. An Analysis of Biomarkers in Patients with Chronic Pain. Pain Physician 2020, 23, E41–E49. [Google Scholar] [CrossRef]
- Kalso, E. Biomarkers for pain. Pain 2004, 107, 199–201. [Google Scholar] [CrossRef] [PubMed]
- Dantzer, R.; O′Connor, J.C.; Freund, G.G.; Johnson, R.W.; Kelley, K.W. From inflammation to sickness and depression: When the immune system subjugates the brain. Nat. Rev. Neurosci. 2008, 9, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Ong, W.-Y.; Stohler, C.S.; Herr, D.R. Role of the Prefrontal Cortex in Pain Processing. Mol. Neurobiol. 2018, 56, 1137–1166. [Google Scholar] [CrossRef]
- Biomarkers Definitions Working Group; Atkinson, A.J., Jr.; Colburn, W.A.; DeGruttola, V.G.; DeMets, D.L.; Downing, G.J.; Hoth, D.F.; Oates, J.A.; Peck, C.C.; Spilker, B.A.; et al. Biomarkers and surrogate endpoints: Preferred definitions and conceptual framework. Clin. Pharmacol. Ther. 2001, 69, 89–95. [Google Scholar] [CrossRef]
- Pope, J.E.; Fishman, M.A.; Gunn, J.A.; Cotten, B.M.; Hill, M.M.; Deer, T.R. Cross-Validation of the Foundation Pain Index with PROMIS-29 in Chronic Pain Patients. J. Pain Res. 2021, 14, 2677–2685. [Google Scholar] [CrossRef]
- Tu, H.; Rady, P.L.; Juelich, T.; Smith, E.M.; Tyring, S.K.; Hughes, T.K. Cytokine regulation of tryptophan metabolism in the hypothalamic-pituitary-adrenal (HPA) axis: Implications for protective and toxic consequences in neuroendocrine regulation. Cell. Mol. Neurobiol. 2005, 25, 673–680. [Google Scholar] [CrossRef]
- Kadry, H.; Noorani, B.; Cucullo, L. A blood–brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS 2020, 17, 69. [Google Scholar] [CrossRef] [PubMed]
- Ciapała, K.; Mika, J.; Rojewska, E. The Kynurenine Pathway as a Potential Target for Neuropathic Pain Therapy Design: From Basic Research to Clinical Perspectives. Int. J. Mol. Sci. 2021, 22, 11055. [Google Scholar] [CrossRef] [PubMed]
- Fukui, S.; Schwarcz, R.; Rapoport, S.I.; Takada, Y.; Smith, Q.R. Blood? Brain barrier transport of kynurenines: Implications for brain synthesis and metabolism. J. Neurochem. 1991, 56, 2007–2017. [Google Scholar] [CrossRef] [PubMed]
- Vécsei, L.; Miller, J.; MacGarvey, U.; Beal, M.F. Kynurenine and probenecid inhibit pentylenetetrazol- and NMDLA-induced seizures and increase kynurenic acid concentrations in the brain. Brain Res. Bull. 1992, 28, 233–238. [Google Scholar] [CrossRef]
- Miller, J.M.; MacGarvey, U.; Beal, M.F. The effect of peripheral loading with kynurenine and probenecid on extracellular striatal kynurenic acid concentrations. Neurosci. Lett. 1992, 146, 115–118. [Google Scholar] [CrossRef]
- Moroni, F.; Russi, P.; Lombardi, G.; Beni, M.; Carlà, V. Presence of kynurenic acid in the mammalian brain. J. Neurochem. 1988, 51, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Németh, H.; Robotka, H.; Kis, Z.; Rózsa, E.; Janáky, T.; Somlai, C.; Marosi, M.; Farkas, T.; Toldi, J.; Vécsei, L. Kynurenine administered together with probenecid markedly inhibits pentylenetetrazol-induced seizures. An electrophysiological and behavioural study. Neuropharmacology 2004, 47, 916–925. [Google Scholar] [CrossRef]
- Robotka, H.; Németh, H.; Somlai, C.; Vécsei, L.; Toldi, J. Systemically administered glucosamine-kynurenic acid, but not pure kynurenic acid, is effective in decreasing the evoked activity in area CA1 of the rat hippocampus. Eur. J. Pharmacol. 2005, 513, 75–80. [Google Scholar] [CrossRef]
- Guillemin, G.J.; Smith, D.G.; Smythe, G.A.; Armati, P.J.; Brew, G.J. Expression of the kynurenine pathway enzymes in human microglia and macrophages. In Developments in Tryptophan and Serotonin Metabolism; Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2003; Volume 527, pp. 105–112. [Google Scholar] [CrossRef]
- Guillemin, G.J.; Kerr, S.J.; Smythe, G.A.; Smith, D.G.; Kapoor, V.; Armati, P.J.; Croitoru, J.; Brew, B.J. Kynurenine pathway metabolism in human astrocytes: A paradox for neuronal protection. J. Neurochem. 2001, 78, 842–853. [Google Scholar] [CrossRef]
- Perkins, M.; Stone, T. An iontophoretic investigation of the actions of convulsant kynurenines and their interaction with the endogenous excitant quinolinic acid. Brain Res. 1982, 247, 184–187. [Google Scholar] [CrossRef]
- Prescott, C.; Weeks, A.M.; Staley, K.J.; Partin, K.M. Kynurenic acid has a dual action on AMPA receptor responses. Neurosci. Lett. 2006, 402, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Rózsa, É.; Robotka, H.; Vécsei, L.; Toldi, J. The Janus-face kynurenic acid. J. Neural Transm. 2008, 115, 1087–1091. [Google Scholar] [CrossRef] [PubMed]
- Yaksh, T.L. Behavioral and autonomic correlates of the tactile evoked allodynia produced by spinal glycine inhibition: Effects of modulatory receptor systems and excitatory amino acid antagonists. Pain 1989, 37, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Chapman, V.; Dickenson, A. The combination of NMDA antagonism and morphine produces profound antinociception in the rat dorsal horn. Brain Res. 1992, 573, 321–323. [Google Scholar] [CrossRef]
- Schneider, S.P.; Perl, E.R. Synaptic mediation from cutaneous mechanical nociceptors. J. Neurophysiol. 1994, 72, 612–621. [Google Scholar] [CrossRef]
- Chapman, V.; Dickenson, A. Time-related roles of excitatory amino acid receptors during persistent noxiously evoked responses of rat dorsal horn neurones. Brain Res. 1995, 703, 45–50. [Google Scholar] [CrossRef]
- Christoph, T.; Reißmüller, E.; Schiene, K.; Englberger, W.; Chizh, B.A. Antiallodynic effects of NMDA glycineB antagonists in neuropathic pain: Possible peripheral mechanisms. Brain Res. 2005, 1048, 218–227. [Google Scholar] [CrossRef]
- Ma, J.-Y.; Zhao, Z.-Q. The involvement of glia in long-term plasticity in the spinal dorsal horn of the rat. Neuroreport 2002, 13, 1781–1784. [Google Scholar] [CrossRef]
- Edwards, S.R.; Mather, L.E.; Lin, Y.; Power, I.; Cousins, M.J. Glutamate and Kynurenate in the Rat Central Nervous System Following Treatments with Tail Ischaemia or Diclofenac. J. Pharm. Pharmacol. 2000, 52, 59–66. [Google Scholar] [CrossRef]
- Csáti, A.; Edvinsson, L.; Vécsei, L.; Toldi, J.; Fulop, F.; Tajti, J.; Warfvinge, K. Kynurenic acid modulates experimentally induced inflammation in the trigeminal ganglion. J. Headache Pain 2015, 16, 99. [Google Scholar] [CrossRef]
- Tuka, B.; Nyári, A.; Cseh, E.K.; Körtési, T.; Veréb, D.; Tömösi, F.; Kecskeméti, G.; Janáky, T.; Tajti, J.; Vécsei, L. Clinical relevance of depressed kynurenine pathway in episodic migraine patients: Potential prognostic markers in the peripheral plasma during the interictal period. J. Headache Pain 2021, 22, 60. [Google Scholar] [CrossRef] [PubMed]
- Körtési, T.; Spekker, E.; Vécsei, L. Exploring the Tryptophan Metabolic Pathways in Migraine-Related Mechanisms. Cells 2022, 11, 3795. [Google Scholar] [CrossRef] [PubMed]
- Schwarcz, R.; Bruno, J.P.; Muchowski, P.J.; Wu, H.-Q. Kynurenines in the mammalian brain: When physiology meets pathology. Nat. Rev. Neurosci. 2012, 13, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Cseh, E.K.; Veres, G.; Körtési, T.; Polyák, H.; Nánási, N.; Tajti, J.; Párdutz, Á.; Klivényi, P.; Vécsei, L.; Zádori, D. Neurotransmitter and tryptophan metabolite concentration changes in the complete Freund’s adjuvant model of orofacial pain. J. Headache Pain 2020, 21, 35. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Vecsei, L.; Ashina, M. The L-kynurenine signalling pathway in trigeminal pain processing: A potential therapeutic target in migraine? Cephalalgia 2011, 31, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Knyihár-Csillik, E.; Toldi, J.; Krisztin-Péva, B.; Chadaide, Z.; Németh, H.; Fenyő, R.; Vécsei, L. Prevention of electrical stimulation-induced increase of c-fos immunoreaction in the caudal trigeminal nucleus by kynurenine combined with probenecid. Neurosci. Lett. 2007, 418, 122–126. [Google Scholar] [CrossRef]
- Heyliger, S.O.; Goodman, C.B.; Ngong, J.M.; Soliman, K.F. The analgesic effects of tryptophan and its metabolites in the rat. Pharmacol. Res. 1998, 38, 243–250. [Google Scholar] [CrossRef]
- Chauvel, V.; Vamos, E.; Pardutz, A.; Vecsei, L.; Schoenen, J.; Multon, S. Effect of systemic kynurenine on cortical spreading depression and its modulation by sex hormones in rat. Exp. Neurol. 2012, 236, 207–214. [Google Scholar] [CrossRef]
- Oláh, G.; Herédi, J.; Menyhárt, Á.; Czinege Zsolt Nagy, D.; Fuzik, J.; Kocsis, K.; Knapp, L.; Krucsó, E.; Gellért, L.; Kis, Z.; et al. Unexpected effects of peripherally administered kynurenic acid on cortical spreading depression and related blood–brain barrier permeability. Drug Des. Dev. Ther. 2013, 7, 981–987. [Google Scholar] [CrossRef]
- Zádori, D.; Veres, G.; Szalárdy, L.; Klivényi, P.; Fülöp, F.; Toldi, J.; Vécsei, L. Inhibitors of the kynurenine pathway as neurotherapeutics: A patent review (2012–2015). Expert Opin. Ther. Patents 2016, 26, 815–832. [Google Scholar] [CrossRef]
- Tajti, J.; Szok, D.; Csáti, A.; Szabó, Á.; Tanaka, M.; Vécsei, L. Exploring Novel Therapeutic Targets in the Common Pathogenic Factors in Migraine and Neuropathic Pain. Int. J. Mol. Sci. 2023, 24, 4114. [Google Scholar] [CrossRef] [PubMed]
- Boros, F.A.; Vécsei, L. Progress in the development of kynurenine and quinoline-3-carboxamide-derived drugs. Expert Opin. Investig. Drugs 2020, 29, 1223–1247. [Google Scholar] [CrossRef] [PubMed]
- Kemp, J.A.; Foster, A.C.; Leeson, P.D.; Priestley, T.; Tridgett, R.; Iversen, L.L.; Woodruff, G.N. 7-Chlorokynurenic acid is a selective antagonist at the glycine modulatory site of the N-methyl-D-aspartate receptor complex. Proc. Natl. Acad. Sci. USA 1988, 85, 6547–6550. [Google Scholar] [CrossRef] [PubMed]
- Wallace, M.; White, A.; Grako, K.A.; Lane, R.; Cato, A.; Snodgrass, H.R. Randomized, double-blind, placebo-controlled, dose-escalation study: Investigation of the safety, pharmacokinetics, and antihyperalgesic activity of L-4-chlorokynurenine in healthy volunteers. Scand. J. Pain 2017, 17, 243–251. [Google Scholar] [CrossRef]
- Ciapała, K.; Pawlik, K.; Ciechanowska, A.; Mika, J.; Rojewska, E. Effect of pharmacological modulation of the kynurenine pathway on pain-related behavior and opioid analgesia in a mouse model of neuropathic pain. Toxicol. Appl. Pharmacol. 2023, 461, 116382. [Google Scholar] [CrossRef]
- Sheng, J.A.; Bales, N.J.; Myers, S.A.; Bautista, A.I.; Roueinfar, M.; Hale, T.M.; Handa, R.J. The Hypothalamic-Pituitary-Adrenal Axis: Development, Programming Actions of Hormones, and Maternal-Fetal Interactions. Front. Behav. Neurosci. 2021, 14, 601939. [Google Scholar] [CrossRef]
- Zouikr, I.; Karshikoff, B. Lifetime Modulation of the Pain System via Neuroimmune and Neuroendocrine Interactions. Front. Immunol. 2017, 8, 276. [Google Scholar] [CrossRef]
- Smith, S.M.; Vale, W.W. The role of the hypothalamic-pituitary-adrenal axis in neuroendocrine responses to stress. Dialogues Clin. Neurosci. 2006, 8, 383–395. [Google Scholar] [CrossRef]
- Herman, J.P.; McKlveen, J.M.; Ghosal, S.; Kopp, B.; Wulsin, A.; Makinson, R.; Scheimann, J.; Myers, B. Regulation of the Hypothalamic-Pituitary-Adrenocortical Stress Response. Compr. Physiol. 2016, 6, 603–621. [Google Scholar] [CrossRef]
- Adam, E.K.; Kumari, M. Assessing salivary cortisol in large-scale, epidemiological research. Psychoneuroendocrinology 2009, 34, 1423–1436. [Google Scholar] [CrossRef]
- Adam, E.K.; Quinn, M.E.; Tavernier, R.; McQuillan, M.T.; Dahlke, K.A.; Gilbert, K.E. Diurnal cortisol slopes and mental and physical health outcomes: A systematic review and meta-analysis. Psychoneuroendocrinology 2017, 83, 25–41. [Google Scholar] [CrossRef] [PubMed]
- Gjerstad, J.K.; Lightman, S.L.; Spiga, F. Role of glucocorticoid negative feedback in the regulation of HPA axis pulsatility. Stress 2018, 21, 403–416. [Google Scholar] [CrossRef]
- De Kloet, E.R.; Vreugdenhil, E.; Oitzl, M.S.; Joëls, M. Brain corticosteroid receptor balance in health and disease. Endocr. Rev. 1998, 19, 269–301. [Google Scholar] [CrossRef] [PubMed]
- Rhen, T.; Cidlowski, J.A. Antiinflammatory action of glucocorticoids—New mechanisms for old drugs. N. Engl. J. Med. 2005, 353, 1711–1723. [Google Scholar] [CrossRef] [PubMed]
- Pace, T.W.; Hu, F.; Miller, A.H. Cytokine-effects on glucocorticoid receptor function: Relevance to glucocorticoid resistance and the pathophysiology and treatment of major depression. Brain, Behav. Immun. 2007, 21, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Pariante, C.M.; Miller, A.H. Glucocorticoid receptors in major depression: Relevance to pathophysiology and treatment. Biol. Psychiatry 2001, 49, 391–404. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.H.; Maletic, V.; Raison, C.L. Inflammation and Its Discontents: The Role of Cytokines in the Pathophysiology of Major Depression. Biol. Psychiatry 2009, 65, 732–741. [Google Scholar] [CrossRef]
- Cavadini, G.; Petrzilka, S.; Kohler, P.; Jud, C.; Tobler, I.; Birchler, T.; Fontana, A. TNF-α suppresses the expression of clock genes by interfering with E-box-mediated transcription. Proc. Natl. Acad. Sci. USA 2007, 104, 12843–12848. [Google Scholar] [CrossRef] [PubMed]
- Ohdo, S.; Koyanagi, S.; Suyama, H.; Higuchi, S.; Aramaki, H. Changing the dosing schedule minimizes the disruptive effects of interferon on clock function. Nat. Med. 2001, 7, 356–360. [Google Scholar] [CrossRef]
- Knoerl, R.; Giobbie-Hurder, A.; Berfield, J.; Berry, D.; Meyerhardt, J.A.; Wright, A.A.; Ligibel, J.A. Exploring Daily Salivary Cortisol Patterns as Biomarkers of Chronic Chemotherapy-Induced Peripheral Neuropathy Pain. Oncol. Nurs. Forum 2022, 49, 207–211. [Google Scholar] [CrossRef]
- Raison, C.L.; Borisov, A.S.; Woolwine, B.J.; Massung, B.; Vogt, G.; Miller, A.H. Interferon-α effects on diurnal hypothalamic–pituitary–adrenal axis activity: Relationship with proinflammatory cytokines and behavior. Mol. Psychiatry 2008, 15, 535–547. [Google Scholar] [CrossRef] [PubMed]
- Miller, G.E.; Chen, E.; Zhou, E.S. If it goes up, must it come down? Chronic stress and the hypothalamic-pituitary-adrenocortical axis in humans. Psychol. Bull. 2007, 133, 25–45. [Google Scholar] [CrossRef] [PubMed]
- Van Uum, S.H.M.; Sauvé, B.; Fraser, L.A.; Morley-Forster, P.; Paul, T.L.; Koren, G. Elevated content of cortisol in hair of patients with severe chronic pain: A novel biomarker for stress. Stress 2008, 11, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Begum, N.; Taylor, J.R.; Brown, C.; Rajan, J.; Keevil, B.; Pye, E.; Rainey, T.; Jones, A. Morning and evening salivary cortisol levels in patients with chronic widespread pain and those at high risk. Eur. J. Pain 2021, 26, 197–206. [Google Scholar] [CrossRef]
- Danesch, U.; Hashimoto, S.; Renkawitz, R.; Schütz, G. Transcriptional regulation of the tryptophan oxygenase gene in rat liver by glucocorticoids. J. Biol. Chem. 1983, 258, 4750–4753. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Niimi, S.; Nawa, K.; Noda, C.; Ichihara, A.; Takagi, Y.; Anai, M.; Sakaki, Y. Multihormonal regulation of transcription of the tryptophan 2,3-dioxygenase gene in primary cultures of adult rat hepatocytes with special reference to the presence of a transcriptional protein mediating the action of glucocorticoids. J. Biol. Chem. 1987, 262, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Hirota, T.; Hirota, K.; Sanno, Y.; Tanaka, T. A new glucocorticoid receptor species: Relation to induction of tryptophan dioxygenase by glucocorticoids. Endocrinology 1985, 117, 1788–1795. [Google Scholar] [CrossRef]
- Hirota, T.; Hirota, K.; Sanno, Y.; Tanaka, T. Appearance of New Hepatic Glucocorticoid Binding Proteins under Various Stressful Conditions: Relation to Endogenous Glucocorticoid Secretion1. J. Biochem. 1985, 97, 1371–1376. [Google Scholar] [CrossRef] [PubMed]
- Green, A.; Woods, H.; Joseph, M. Tryptophan metabolism in the isolated perfused liver of the rat: Effects of tryptophan concentration, hydrocortisone and allopurinol on tryptophan pyrrolase activity and kynurenine formation. Br. J. Pharmacol. 1976, 57, 103–114. [Google Scholar] [CrossRef]
- Altman, K.; Greengard, O. Correlation of kynurenine excretion with liver tryptophan pyrrolase levels in disease and after hydrocortisone induction. J. Clin. Investig. 1966, 45, 1527–1534. [Google Scholar] [CrossRef]
- Sorgdrager, F.; Doornbos, B.; Penninx, B.; de Jonge, P.; Kema, I. The association between the hypothalamic pituitary adrenal axis and tryptophan metabolism in persons with recurrent major depressive disorder and healthy controls. J. Affect. Disord. 2017, 222, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Vulto, A.; van Faassen, M.; Kerstens, M.N.; van Beek, A.P. Susceptibility to Adrenal Crisis Is Associated with Differences in Cortisol Excretion in Patients with Secondary Adrenal Insufficiency. Front. Endocrinol. 2022, 13, 849188. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.D.; Edlund, M.J.; Steffick, D.; Unützer, J. Regular use of prescribed opioids: Association with common psychiatric disorders. Pain 2005, 119, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Catley, D.; Kaell, A.T.; Kirschbaum, C.; Stone, A.A. A naturalistic evaluation of cortisol secretion in persons with fibromyalgia and rheumatoid arthritis. Arthritis Care Res. 2000, 13, 51–61. [Google Scholar] [CrossRef]
- McLean, S.A.; Williams, D.A.; Harris, R.E.; Kop, W.J.; Groner, K.H.; Ambrose, K.; Lyden, A.K.; Gracely, R.H.; Crofford, L.J.; Geisser, M.E.; et al. Momentary relationship between cortisol secretion and symptoms in patients with fibromyalgia. Arthritis Rheum. 2005, 52, 3660–3669. [Google Scholar] [CrossRef]
- Riva, R.; Mork, P.J.; Westgaard, R.H.; Lundberg, U. Comparison of the cortisol awakening response in women with shoulder and neck pain and women with fibromyalgia. Psychoneuroendocrinology 2012, 37, 299–306. [Google Scholar] [CrossRef]
- Riva, R.; Mork, P.J.; Westgaard, R.H.; Rø, M.; Lundberg, U. Fibromyalgia syndrome is associated with hypocortisolism. Int. J. Behav. Med. 2010, 17, 223–233. [Google Scholar] [CrossRef]
- Úbeda-D’ocasar, E.; Díaz-Benito, V.J.; Gallego-Sendarrubias, G.M.; Valera-Calero, J.A.; Vicario-Merino, Á.; Hervás-Pérez, J.P. Pain and Cortisol in Patients with Fibromyalgia: Systematic Review and Meta-Analysis. Diagnostics 2020, 10, 922. [Google Scholar] [CrossRef]
- Wingenfeld, K.; Nutzinger, D.; Kauth, J.; Hellhammer, D.H.; Lautenbacher, S. Salivary cortisol release and hypothalamic Pituitary adrenal axis feedback sensitivity in fibromyalgia is associated with depression but not with pain. J. Pain 2010, 11, 1195–1202. [Google Scholar] [CrossRef]
- Brooks, A.K.; Lawson, M.A.; Smith, R.A.; Janda, T.M.; Kelley, K.W.; McCusker, R.H. Interactions between inflammatory mediators and corticosteroids regulate transcription of genes within the Kynurenine Pathway in the mouse hippocampus. J. Neuroinflammation 2016, 13, 98. [Google Scholar] [CrossRef]
- Huizenga, N.A.T.M.; Koper, J.W.; de Lange, P.; Pols, H.A.P.; Stolk, R.P.; Grobbee, D.E.; de Jong, F.H.; Lamberts, S.W.J. Interperson Variability but Intraperson Stability of Baseline Plasma Cortisol Concentrations, and Its Relation to Feedback Sensitivity of the Hypothalamo-Pituitary-Adrenal Axis to a Low Dose of Dexamethasone in Elderly Individuals. J. Clin. Endocrinol. Metab. 1998, 83, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Van Rossum, E.F.C.; Lamberts, S.W.J. Polymorphisms in the Glucocorticoid Receptor Gene and Their Associations with Metabolic Parameters and Body Composition. Recent Prog. Horm. Res. 2004, 59, 333–357. [Google Scholar] [CrossRef] [PubMed]
- Soichot, M.; Vaast, A.; Vignau, J.; Guillemin, G.J.; Lhermitte, M.; Broly, F.; Allorge, D. Characterization of functional polymorphisms and glucocorticoid-responsive elements in the promoter of TDO2, a candidate gene for ethanol-induced behavioural disorders. Alcohol Alcohol. 2013, 48, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Kassel, O.; Herrlich, P. Crosstalk between the glucocorticoid receptor and other transcription factors: Molecular aspects. Mol. Cell. Endocrinol. 2007, 275, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Schoneveld, O.J.; Gaemers, I.C.; Lamers, W.H. Mechanisms of glucocorticoid signalling. Biochim. Biophys. Acta (BBA) Gene Struct. Expr. 2004, 1680, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Meier, T.B.; Drevets, W.C.; Wurfel, B.E.; Ford, B.N.; Morris, H.M.; Victor, T.A.; Bodurka, J.; Teague, T.K.; Dantzer, R.; Savitz, J. Relationship between neurotoxic kynurenine metabolites and reductions in right medial prefrontal cortical thickness in major depressive disorder. Brain, Behav. Immun. 2015, 53, 39–48. [Google Scholar] [CrossRef]
- Taleb, O.; Maammar, M.; Klein, C.; Maitre, M.; Mensah-Nyagan, A.G. A Role for Xanthurenic Acid in the Control of Brain Dopaminergic Activity. Int. J. Mol. Sci. 2021, 22, 6974. [Google Scholar] [CrossRef]
- Groven, N.; Reitan, S.K.; Fors, E.A.; Guzey, I.C. Kynurenine metabolites and ratios differ between Chronic Fatigue Syndrome, Fibromyalgia, and healthy controls. Psychoneuroendocrinology 2021, 131, 105287. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jovanovic, F.; Jovanovic, V.; Knezevic, N.N. Glucocorticoid Hormones as Modulators of the Kynurenine Pathway in Chronic Pain Conditions. Cells 2023, 12, 1178. https://doi.org/10.3390/cells12081178
Jovanovic F, Jovanovic V, Knezevic NN. Glucocorticoid Hormones as Modulators of the Kynurenine Pathway in Chronic Pain Conditions. Cells. 2023; 12(8):1178. https://doi.org/10.3390/cells12081178
Chicago/Turabian StyleJovanovic, Filip, Visnja Jovanovic, and Nebojsa Nick Knezevic. 2023. "Glucocorticoid Hormones as Modulators of the Kynurenine Pathway in Chronic Pain Conditions" Cells 12, no. 8: 1178. https://doi.org/10.3390/cells12081178