

Maternal Inflammation with Elevated Kynurenine Metabolites Is Related to the Risk of Abnormal Brain Development and Behavioral Changes in Autism Spectrum Disorder
 , , ,
, , ,
Abstract
:1. Introduction
2. Interleukin (IL)-17a as a Potential Mediator of ASD
2.1. Pro-Inflammatory Cytokines and ASD
2.2. Proinflammatory Cytokines Regulate KP Enzymes
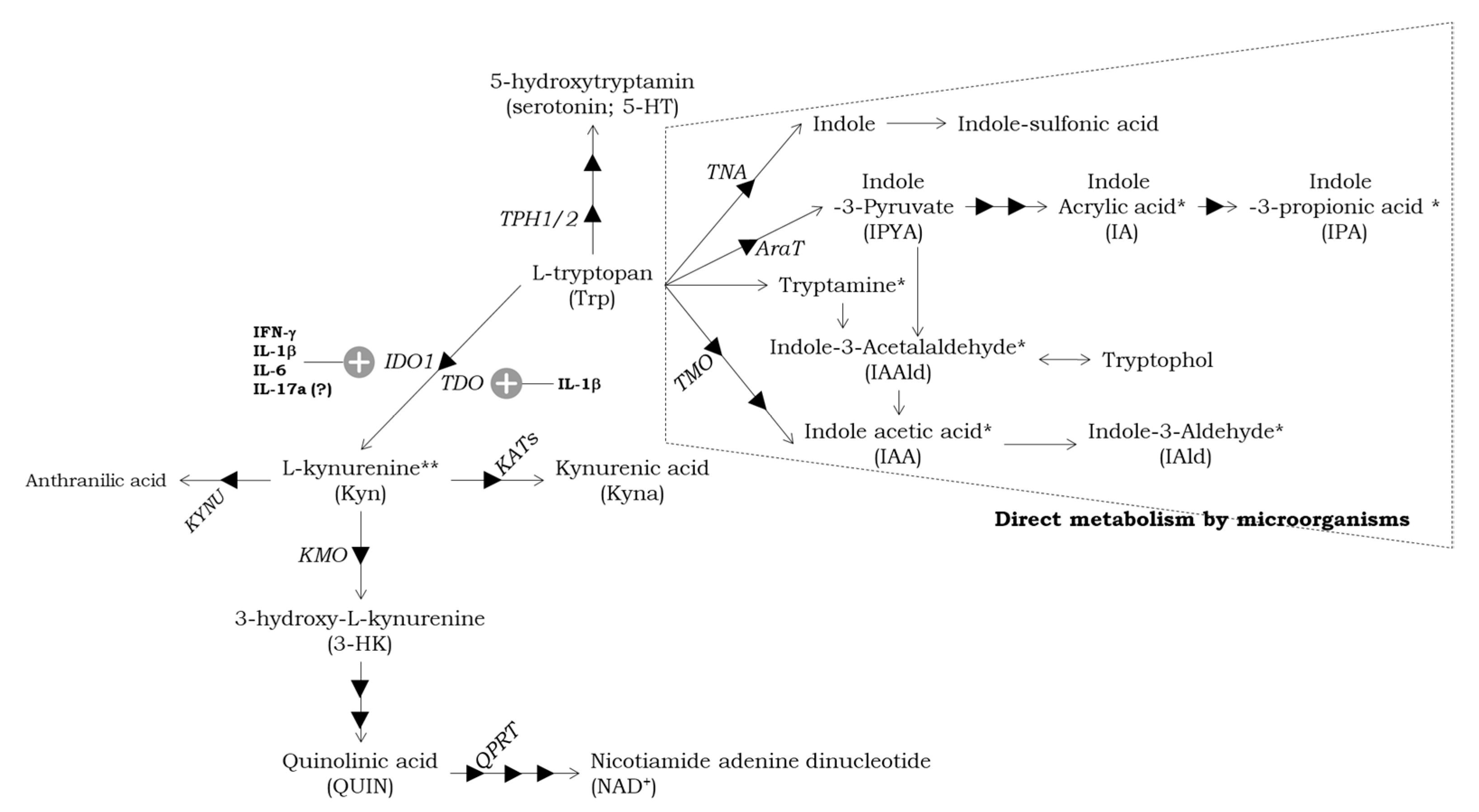
3. The Metabolism of Trp in the Intestine and the Role of Microbiota in ASD
3.1. Intestinal Trp Metabolism Pathways and Physiological Roles
3.2. The Regulation of Trp Metabolism by Gut Microbiota
3.3. Dysbiosis in ASD
4. Perinatal KP Metabolism
4.1. Neurodevelopment and KP Metabolites
4.2. Relation between Key Receptors and KP Metabolites in Developing Brain
4.3. Kynurenine Profile in ASD
5. Experimental Animal Models with Pre- and Postnatal KP Confusion to Study Neuropathological and Behavioral Deficits
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ABC | Aberrant Behavior Checklist |
| ADDM | Autism Developmental Disabilities Monitoring |
| ADI-R | Autism Diagnostic Interview-Revised |
| ADOS | Autism Diagnostic Observation Schedule |
| AhR | aryl hydrocarbon receptor |
| APCs | antigen-presenting cells |
| ASD | autism spectrum disorder |
| 5AV | 5-aminovaleric acid |
| Breg | B-regulatory cell |
| BBB | blood-brain barrier |
| CARS | Childhood Autism Rating Scale |
| CNS | central nervous system |
| DCS | D-cycloserine |
| DCX | doublecortin |
| DSM | Diagnostic and Statistical Manual of Mental Disorder |
| E | embryonic day |
| ELISA | enzyme-linked immune sorbent assay |
| E. coli | Escherichia coli |
| 15q dup | 15q11.13 duplication |
| GABA | gamma-aminobutyric acid |
| GS-MS | gas chromatography-mass spectrometry |
| GI | gastrointestinal |
| HDAC | histone deacetylase |
| HILIC | hydrophilic interaction chromatography |
| 3-HK | 3-hydroxy-L-kynurenine |
| HPLC | high performance liquid chromatography |
| 5-HT | 5-hydroxytryptamine |
| IA | indole acrylic acid |
| IAA | indole-3-acetic acid |
| IAAld | indole-3acetaaldehyde |
| IAld | indole-3-aldehyde |
| IBD | inflammatory bowel disease |
| ICD-10 | International Statistical Classification of Diseases and Related Health Problems-10 |
| ICjM | island of Calleja major |
| IDO1 | indoleamine 2,3-dioxygenase1 |
| IFN-γ | interferon-gamma |
| IL-1β | interleukin-1 beta |
| IL-6 | interleukin-6 |
| IL-17 | interleukin-17 |
| IL-21 | interlekin-21 |
| ILC3 | innate lymphocyte cells 3 |
| IPA | indole-3-propionic acid |
| KAT | kynurenine aminotransferase |
| KMO | kynurenine 3-monooxygenase |
| KP | kynurenine pathway |
| Kyn | kynurenine |
| Kyna | kynurenic acid |
| LC | locus coeruleus |
| LC-MS/MS | liquid chromatography-tandem mass spectrometry |
| LPS | lipopolysaccharide |
| LRP | low-density receptor-related protein |
| LTP | long-term potentiation |
| MIA | maternal immune activation |
| nAChR | nicotinic acetylcholine receptor |
| NMDA | N-methyl-D-aspartate |
| PDS-95 | postsynaptic density protein-95 |
| PND | postnatal day |
| poly(I:C) | polyinosinic:polycytidylic acid |
| PPI | pre-pulse inhibition |
| PV+ | parvalbumin positive |
| QPRT | quinolinic phosphoribosyltransferase |
| QUIN | quinolinic acid |
| RAADS-14 | Ritovo Autism & Asperger Diagnostic Scale-14 |
| REM | rapid eye movement |
| Ro 61-8048 | 3,4-dimethoxy-N-[4-(3-nitrophenyl)thiazol-2-yl]benzenesulfonamide 16 |
| RRS | Ruminative Response Scale |
| SCFA | short-chain fatty acid |
| SCZ | schizophrenia |
| SERT | serotonin transporter |
| SFB | segmented filamentous bacteria |
| SHP | SC-43 activated SH2 domain-containing phosphatase |
| SNP | single nucleotide polymorphism |
| SSRIs | selective serotonin reuptake inhibitors |
| STAT | signal transducers and activators of transcription |
| TCDD | 2,3,7,8-tetrachlorodibenzo-p-dioxin |
| TD | typically developing |
| TDO | tryptophan 2,3-dioxygenase |
| TGF-® | transforming growth factor-beta |
| Th | T helper |
| TMS/MS | tandem mass spectrometry |
| TNF | tumor necrosis factor |
| TPH | tryptophan hydroxylase |
| Treg | T-regulatory cell |
| Trp | tryptophan |
| UHPLC | ultra-high performance liquid chromatography |
| VGLUT | vesicular glutamate transporter |
| WSAS | Work and Social Adjustment Scale |
References
- Bryn, V.; Verkerk, R.; Skjeldal, O.H.; Saugstad, O.D.; Ormstad, H. Kynurenine Pathway in Autism Spectrum Disorders in Children. Neuropsychobiology 2017, 76, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Carpita, B.; Nardi, B.; Palego, L.; Cremone, I.M.; Massimetti, G.; Carmassi, C.; Betti, L.; Giannaccini, G.; Dell’Osso, L. Kynurenine pathway and autism spectrum phenotypes: An investigation among adults with autism spectrum disorder and their first-degree relatives. CNS Spectr. 2022, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Imamura, Y.; Kasahara, Y.; Yoshida, C.; Momono, Y.; Fang, K.; Nishiyama, T.; Sakai, D.; Konishi, Y. The Effects of Maternal Interleukin-17A on Social Behavior, Cognitive Function, and Depression-Like Behavior in Mice with Altered Kynurenine Metabolites. Int. J. Tryptophan Res. 2021, 14, 11786469211026639. [Google Scholar] [CrossRef]
- Hahn, B.; Reneski, C.H.; Pocivavsek, A.; Schwarcz, R. Prenatal kynurenine treatment in rats causes schizophrenia-like broad monitoring deficits in adulthood. Psychopharmacology 2018, 235, 651–661. [Google Scholar] [CrossRef]
- Pershing, M.L.; Bortz, D.M.; Pocivavsek, A.; Fredericks, P.J.; Jorgensen, C.V.; Vunck, S.A.; Leuner, B.; Schwarcz, R.; Bruno, J.P. Elevated levels of kynurenic acid during gestation produce neurochemical, morphological, and cognitive deficits in adulthood: Implications for schizophrenia. Neuropharmacology 2015, 90, 33–41. [Google Scholar] [CrossRef] [Green Version]
- Murphy, S.K.; Fineberg, A.M.; Maxwell, S.D.; Alloy, L.B.; Zimmermann, L.; Krigbaum, N.Y.; Cohn, B.A.; Drabick, D.A.G.; Ellman, L.M. Maternal infection and stress during pregnancy and depressive symptoms in adolescent offspring. Psychiatry Res. 2017, 257, 102–110. [Google Scholar] [CrossRef]
- Sha, Q.; Madaj, Z.; Keaton, S.; Escobar Galvis, M.L.; Smart, L.; Krzyzanowski, S.; Fazleabas, A.T.; Leach, R.; Postolache, T.T.; Achtyes, E.D.; et al. Cytokines and tryptophan metabolites can predict depressive symptoms in pregnancy. Transl. Psychiatry 2022, 12, 35. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.K.; Essa, M.M.; de Paula Martins, R.; Lovejoy, D.B.; Bilgin, A.A.; Waly, M.I.; Al-Farsi, Y.M.; Al-Sharbati, M.; Al-Shaffae, M.A.; Guillemin, G.J. Altered kynurenine pathway metabolism in autism: Implication for immune-induced glutamatergic activity. Autism Res. 2016, 9, 621–631. [Google Scholar] [CrossRef]
- Wonodi, I.; Schwarcz, R. Cortical kynurenine pathway metabolism: A novel target for cognitive enhancement in Schizophrenia. Schizophr. Bull. 2010, 36, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Sathyasaikumar, K.V.; Stachowski, E.K.; Wonodi, I.; Roberts, R.C.; Rassoulpour, A.; McMahon, R.P.; Schwarcz, R. Impaired kynurenine pathway metabolism in the prefrontal cortex of individuals with schizophrenia. Schizophr. Bull. 2011, 37, 1147–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plitman, E.; Iwata, Y.; Caravaggio, F.; Nakajima, S.; Chung, J.K.; Gerretsen, P.; Kim, J.; Takeuchi, H.; Chakravarty, M.M.; Remington, G.; et al. Kynurenic Acid in Schizophrenia: A Systematic Review and Meta-analysis. Schizophr. Bull. 2017, 43, 764–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogyu, K.; Kubo, K.; Noda, Y.; Iwata, Y.; Tsugawa, S.; Omura, Y.; Wada, M.; Tarumi, R.; Plitman, E.; Moriguchi, S.; et al. Kynurenine pathway in depression: A systematic review and meta-analysis. Neurosci. Biobehav. Rev. 2018, 90, 16–25. [Google Scholar] [CrossRef]
- Yun, Y.; Zhang, Q.; Zhao, W.; Ma, T.; Fan, H.; Bai, L.; Ma, B.; Qi, S.; Wang, Z.; An, H.; et al. Relationship between the tryptophan-kynurenine pathway and painful physical symptoms in patients with major depressive disorder. J. Psychosom. Res. 2022, 163, 111069. [Google Scholar] [CrossRef] [PubMed]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Text Revision DSM-5-TR, 5th ed.; Text Revision (DSM-5-TR); American Psychiatric Association Publishing Inc.: Washington, DC, USA, 2022. [Google Scholar]
- Maenner, M.J.; Shaw, K.A.; Bakian, A.V.; Bilder, D.A.; Durkin, M.S.; Esler, A.; Furnier, S.M.; Hallas, L.; Hall-Lande, J.; Hudson, A.; et al. Prevalence and Characteristics of Autism Spectrum Disorder Among Children Aged 8 Years—Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2018. MMWR Surveill. Summ. 2021, 70, 1–16. [Google Scholar] [CrossRef]
- Cogswell, M.E.; Coil, E.; Tian, L.H.; Tinker, S.C.; Ryerson, A.B.; Maenner, M.J.; Rice, C.E.; Peacock, G. Health Needs and Use of Services Among Children with Developmental Disabilities—United States, 2014–2018. MMWR Morb. Mortal. Wkly. Rep. 2022, 71, 453–458. [Google Scholar] [CrossRef]
- Liu, X.; Takumi, T. Genomic and genetic aspects of autism spectrum disorder. Biochem. Biophys. Res. Commun. 2014, 452, 244–253. [Google Scholar] [CrossRef] [Green Version]
- Tick, B.; Bolton, P.; Happe, F.; Rutter, M.; Rijsdijk, F. Heritability of autism spectrum disorders: A meta-analysis of twin studies. J. Child Psychol. Psychiatry 2016, 57, 585–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patterson, P.H. Maternal infection and immune involvement in autism. Trends Mol. Med. 2011, 17, 389–394. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.K.; Magnusson, C.; Gardner, R.M.; Blomstrom, A.; Newschaffer, C.J.; Burstyn, I.; Karlsson, H.; Dalman, C. Maternal hospitalization with infection during pregnancy and risk of autism spectrum disorders. Brain Behav. Immun. 2015, 44, 100–105. [Google Scholar] [CrossRef] [Green Version]
- Han, V.X.; Patel, S.; Jones, H.F.; Dale, R.C. Maternal immune activation and neuroinflammation in human neurodevelopmental disorders. Nat. Rev. Neurol. 2021, 17, 564–579. [Google Scholar] [CrossRef]
- Masi, A.; Quintana, D.S.; Glozier, N.; Lloyd, A.R.; Hickie, I.B.; Guastella, A.J. Cytokine aberrations in autism spectrum disorder: A systematic review and meta-analysis. Mol. Psychiatry 2015, 20, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Pocivavsek, A.; Thomas, M.A.; Elmer, G.I.; Bruno, J.P.; Schwarcz, R. Continuous kynurenine administration during the prenatal period, but not during adolescence, causes learning and memory deficits in adult rats. Psychopharmacology 2014, 231, 2799–2809. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Chauhan, A.; Sheikh, A.M.; Patil, S.; Chauhan, V.; Li, X.M.; Ji, L.; Brown, T.; Malik, M. Elevated immune response in the brain of autistic patients. J. Neuroimmunol. 2009, 207, 111–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, J.T.; Chana, G.; Pardo, C.A.; Achim, C.; Semendeferi, K.; Buckwalter, J.; Courchesne, E.; Everall, I.P. Microglial activation and increased microglial density observed in the dorsolateral prefrontal cortex in autism. Biol. Psychiatry 2010, 68, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Pardo, C.A.; Vargas, D.L.; Zimmerman, A.W. Immunity, neuroglia and neuroinflammation in autism. Int. Rev. Psychiatry 2005, 17, 485–495. [Google Scholar] [CrossRef] [Green Version]
- Vargas, D.L.; Nascimbene, C.; Krishnan, C.; Zimmerman, A.W.; Pardo, C.A. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol. 2005, 57, 67–81. [Google Scholar] [CrossRef]
- Wong, H.; Hoeffer, C. Maternal IL-17A in autism. Exp. Neurol. 2018, 299, 228–240. [Google Scholar] [CrossRef]
- Jiang, H.Y.; Xu, L.L.; Shao, L.; Xia, R.M.; Yu, Z.H.; Ling, Z.X.; Yang, F.; Deng, M.; Ruan, B. Maternal infection during pregnancy and risk of autism spectrum disorders: A systematic review and meta-analysis. Brain Behav. Immun. 2016, 58, 165–172. [Google Scholar] [CrossRef]
- Chen, S.W.; Zhong, X.S.; Jiang, L.N.; Zheng, X.Y.; Xiong, Y.Q.; Ma, S.J.; Qiu, M.; Huo, S.T.; Ge, J.; Chen, Q. Maternal autoimmune diseases and the risk of autism spectrum disorders in offspring: A systematic review and meta-analysis. Behav. Brain Res. 2016, 296, 61–69. [Google Scholar] [CrossRef]
- Gong, T.; Lundholm, C.; Rejno, G.; Bolte, S.; Larsson, H.; D’Onofrio, B.M.; Lichtenstein, P.; Almqvist, C. Parental asthma and risk of autism spectrum disorder in offspring: A population and family-based case-control study. Clin. Exp. Allergy 2019, 49, 883–891. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.; Zou, H.; Sheikh, A.M.; Malik, M.; Dobkin, C.; Brown, W.T.; Li, X. IL-6 is increased in the cerebellum of autistic brain and alters neural cell adhesion, migration and synaptic formation. J. Neuroinflammation 2011, 8, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, S.E.; Li, J.; Garbett, K.; Mirnics, K.; Patterson, P.H. Maternal immune activation alters fetal brain development through interleukin-6. J. Neurosci. 2007, 27, 10695–10702. [Google Scholar] [CrossRef] [Green Version]
- Rudolph, M.D.; Graham, A.M.; Feczko, E.; Miranda-Dominguez, O.; Rasmussen, J.M.; Nardos, R.; Entringer, S.; Wadhwa, P.D.; Buss, C.; Fair, D.A. Maternal IL-6 during pregnancy can be estimated from newborn brain connectivity and predicts future working memory in offspring. Nat. Neurosci. 2018, 21, 765–772. [Google Scholar] [CrossRef]
- Wilke, C.M.; Bishop, K.; Fox, D.; Zou, W. Deciphering the role of Th17 cells in human disease. Trends Immunol. 2011, 32, 603–611. [Google Scholar] [CrossRef] [Green Version]
- Yasuda, K.; Takeuchi, Y.; Hirota, K. Correction to: The pathogenicity of Th17 cells in autoimmune diseases. Semin. Immunopathol. 2019, 41, 299. [Google Scholar] [CrossRef] [Green Version]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Ivanov, I.I.; Spolski, R.; Min, R.; Shenderov, K.; Egawa, T.; Levy, D.E.; Leonard, W.J.; Littman, D.R. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat. Immunol. 2007, 8, 967–974. [Google Scholar] [CrossRef]
- Mangan, P.R.; Harrington, L.E.; O’Quinn, D.B.; Helms, W.S.; Bullard, D.C.; Elson, C.O.; Hatton, R.D.; Wahl, S.M.; Schoeb, T.R.; Weaver, C.T. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 2006, 441, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Akintunde, M.E.; Rose, M.; Krakowiak, P.; Heuer, L.; Ashwood, P.; Hansen, R.; Hertz-Picciotto, I.; Van de Water, J. Increased production of IL-17 in children with autism spectrum disorders and co-morbid asthma. J. Neuroimmunol. 2015, 286, 33–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Ayadhi, L.Y.; Mostafa, G.A. Elevated serum levels of interleukin-17A in children with autism. J. Neuroinflammation 2012, 9, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, G.B.; Yim, Y.S.; Wong, H.; Kim, S.; Kim, H.; Kim, S.V.; Hoeffer, C.A.; Littman, D.R.; Huh, J.R. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science 2016, 351, 933–939. [Google Scholar] [CrossRef] [Green Version]
- Garay, P.A.; Hsiao, E.Y.; Patterson, P.H.; McAllister, A.K. Maternal immune activation causes age- and region-specific changes in brain cytokines in offspring throughout development. Brain Behav. Immun. 2013, 31, 54–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solek, C.M.; Farooqi, N.; Verly, M.; Lim, T.K.; Ruthazer, E.S. Maternal immune activation in neurodevelopmental disorders. Dev. Dyn. 2018, 247, 588–619. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Tome, S.; Takei, Y. Intraventricular IL-17A administration activates microglia and alters their localization in the mouse embryo cerebral cortex. Mol. Brain 2020, 13, 93. [Google Scholar] [CrossRef] [PubMed]
- Gumusoglu, S.B.; Hing, B.W.Q.; Chilukuri, A.S.S.; Dewitt, J.J.; Scroggins, S.M.; Stevens, H.E. Correction: Chronic maternal interleukin-17 and autism-related cortical gene expression, neurobiology, and behavior. Neuropsychopharmacology 2020, 45, 1588. [Google Scholar] [CrossRef]
- Li, Z.; Li, K.; Zhu, L.; Kan, Q.; Yan, Y.; Kumar, P.; Xu, H.; Rostami, A.; Zhang, G.X. Inhibitory effect of IL-17 on neural stem cell proliferation and neural cell differentiation. BMC Immunol. 2013, 14, 20. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Kim, H.; Yim, Y.S.; Ha, S.; Atarashi, K.; Tan, T.G.; Longman, R.S.; Honda, K.; Littman, D.R.; Choi, G.B.; et al. Maternal gut bacteria promote neurodevelopmental abnormalities in mouse offspring. Nature 2017, 549, 528–532. [Google Scholar] [CrossRef] [Green Version]
- Honig, A.; Rieger, L.; Kapp, M.; Sutterlin, M.; Dietl, J.; Kammerer, U. Indoleamine 2,3-dioxygenase (IDO) expression in invasive extravillous trophoblast supports role of the enzyme for materno-fetal tolerance. J. Reprod. Immunol. 2004, 61, 79–86. [Google Scholar] [CrossRef]
- Sedlmayr, P.; Blaschitz, A.; Stocker, R. The role of placental tryptophan catabolism. Front. Immunol. 2014, 5, 230. [Google Scholar] [CrossRef] [Green Version]
- Munn, D.H.; Zhou, M.; Attwood, J.T.; Bondarev, I.; Conway, S.J.; Marshall, B.; Brown, C.; Mellor, A.L. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science 1998, 281, 1191–1193. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Sharma, M.D.; Baban, B.; Harding, H.P.; Zhang, Y.; Ron, D.; Mellor, A.L. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity 2005, 22, 633–642. [Google Scholar] [CrossRef] [Green Version]
- Kudo, Y.; Boyd, C.A.; Sargent, I.L.; Redman, C.W. Decreased tryptophan catabolism by placental indoleamine 2,3-dioxygenase in preeclampsia. Am. J. Obstet. Gynecol. 2003, 188, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Sung, N.; Gilman-Sachs, A.; Kwak-Kim, J. T Helper (Th) Cell Profiles in Pregnancy and Recurrent Pregnancy Losses: Th1/Th2/Th9/Th17/Th22/Tfh Cells. Front. Immunol. 2020, 11, 2025. [Google Scholar] [CrossRef]
- Wang, R.; Weng, Y.; Zhao, S.; Li, S.; Wen, X.; Huang, G. [IL-6 up-regulates indoleamine 2, 3-dioxygenase (IDO) expression in chorionic villi and decidua]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2021, 37, 158–163. [Google Scholar] [PubMed]
- Cheng, H.; Huang, Y.; Huang, G.; Chen, Z.; Tang, J.; Pan, L.; Lv, J.; Long, A.; Wang, R.; Chen, Z.; et al. Effect of the IDO Gene on Pregnancy in Mice with Recurrent Pregnancy Loss. Reprod. Sci. 2021, 28, 52–59. [Google Scholar] [CrossRef]
- Wang, R.; Zhao, S.; Chen, X.; Xiao, Z.; Wen, X.; Zhong, X.; Li, S.; Cheng, H.; Huang, G. Molecular mechanisms involved in the IL-6-mediated upregulation of indoleamine 2,3-dioxygenase 1 (IDO1) expression in the chorionic villi and decidua of women in early pregnancy. BMC Pregnancy Childbirth 2022, 22, 983. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, R.S.S.; de Sousa, J.R.; Araujo, M.T.F.; Martins Filho, A.J.; de Alcantara, B.N.; Araujo, F.M.C.; Queiroz, M.G.L.; Cruz, A.C.R.; Vasconcelos, B.H.B.; Chiang, J.O.; et al. In situ immune response and mechanisms of cell damage in central nervous system of fatal cases microcephaly by Zika virus. Sci. Rep. 2018, 8, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanai, M.; Funakoshi, H.; Takahashi, H.; Hayakawa, T.; Mizuno, S.; Matsumoto, K.; Nakamura, T. Tryptophan 2,3-dioxygenase is a key modulator of physiological neurogenesis and anxiety-related behavior in mice. Mol. Brain 2009, 2, 8. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, D.; Dvorakova, T.; Schramme, F.; Stroobant, V.; Van den Eynde, B.J. Tryptophan 2,3-Dioxygenase Expression Identified in Murine Decidual Stromal Cells Is Not Essential for Feto-Maternal Tolerance. Front. Immunol. 2020, 11, 601759. [Google Scholar] [CrossRef]
- Pilotte, L.; Larrieu, P.; Stroobant, V.; Colau, D.; Dolusic, E.; Frederick, R.; De Plaen, E.; Uyttenhove, C.; Wouters, J.; Masereel, B.; et al. Reversal of tumoral immune resistance by inhibition of tryptophan 2,3-dioxygenase. Proc. Natl. Acad. Sci. USA 2012, 109, 2497–2502. [Google Scholar] [CrossRef] [Green Version]
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478, 197–203. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, D.; Dvorakova, T.; Stroobant, V.; Bouzin, C.; Daumerie, A.; Solvay, M.; Klaessens, S.; Letellier, M.C.; Renauld, J.C.; van Baren, N.; et al. Tryptophan 2,3-Dioxygenase Expression Identified in Human Hepatocellular Carcinoma Cells and in Intratumoral Pericytes of Most Cancers. Cancer Immunol. Res. 2020, 8, 19–31. [Google Scholar] [CrossRef]
- Bent, R.; Moll, L.; Grabbe, S.; Bros, M. Interleukin-1 Beta-A Friend or Foe in Malignancies? Int. J. Mol. Sci. 2018, 19, 2155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urata, Y.; Koga, K.; Hirota, Y.; Akiyama, I.; Izumi, G.; Takamura, M.; Nagai, M.; Harada, M.; Hirata, T.; Yoshino, O.; et al. IL-1beta increases expression of tryptophan 2,3-dioxygenase and stimulates tryptophan catabolism in endometrioma stromal cells. Am. J. Reprod. Immunol. 2014, 72, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Pantouris, G.; Loudon-Griffiths, J.; Mowat, C.G. Insights into the mechanism of inhibition of tryptophan 2,3-dioxygenase by isatin derivatives. J. Enzym. Inhib. Med. Chem. 2016, 31, 70–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zador, F.; Joca, S.; Nagy-Grocz, G.; Dvoracsko, S.; Szucs, E.; Tomboly, C.; Benyhe, S.; Vecsei, L. Pro-Inflammatory Cytokines: Potential Links between the Endocannabinoid System and the Kynurenine Pathway in Depression. Int. J. Mol. Sci. 2021, 22, 5903. [Google Scholar] [CrossRef]
- Nakashima, A.; Ito, M.; Yoneda, S.; Shiozaki, A.; Hidaka, T.; Saito, S. Circulating and decidual Th17 cell levels in healthy pregnancy. Am. J. Reprod. Immunol. 2010, 63, 104–109. [Google Scholar] [CrossRef]
- Santner-Nanan, B.; Peek, M.J.; Khanam, R.; Richarts, L.; Zhu, E.; Fazekas de St Groth, B.; Nanan, R. Systemic increase in the ratio between Foxp3+ and IL-17-producing CD4+ T cells in healthy pregnancy but not in preeclampsia. J. Immunol. 2009, 183, 7023–7030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cua, D.J.; Tato, C.M. Innate IL-17-producing cells: The sentinels of the immune system. Nat. Rev. Immunol. 2010, 10, 479–489. [Google Scholar] [CrossRef]
- Romani, L.; Zelante, T.; De Luca, A.; Fallarino, F.; Puccetti, P. IL-17 and therapeutic kynurenines in pathogenic inflammation to fungi. J. Immunol. 2008, 180, 5157–5162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krause, R.; Zollner-Schwetz, I.; Salzer, H.J.; Valentin, T.; Rabensteiner, J.; Pruller, F.; Raggam, R.; Meinitzer, A.; Prattes, J.; Rinner, B.; et al. Elevated levels of interleukin 17A and kynurenine in candidemic patients, compared with levels in noncandidemic patients in the intensive care unit and those in healthy controls. J. Infect. Dis. 2015, 211, 445–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, T.W.; Clanchy, F.I.L.; Huang, Y.S.; Chiang, N.Y.; Darlington, L.G.; Williams, R.O. An integrated cytokine and kynurenine network as the basis of neuroimmune communication. Front. Neurosci. 2022, 16, 1002004. [Google Scholar] [CrossRef] [PubMed]
- Clarke, G.; Grenham, S.; Scully, P.; Fitzgerald, P.; Moloney, R.D.; Shanahan, F.; Dinan, T.G.; Cryan, J.F. The microbiome-gut-brain axis during early life regulates the hippocampal serotonergic system in a sex-dependent manner. Mol Psychiatry 2013, 18, 666–673. [Google Scholar] [CrossRef] [Green Version]
- Yano, J.M.; Yu, K.; Donaldson, G.P.; Shastri, G.G.; Ann, P.; Ma, L.; Nagler, C.R.; Ismagilov, R.F.; Mazmanian, S.K.; Hsiao, E.Y. Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell 2015, 161, 264–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zelante, T.; Iannitti, R.G.; Cunha, C.; De Luca, A.; Giovannini, G.; Pieraccini, G.; Zecchi, R.; D’Angelo, C.; Massi-Benedetti, C.; Fallarino, F.; et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 2013, 39, 372–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agus, A.; Planchais, J.; Sokol, H. Gut Microbiota Regulation of Tryptophan Metabolism in Health and Disease. Cell Host Microbe 2018, 23, 716–724. [Google Scholar] [CrossRef] [Green Version]
- Roager, H.M.; Licht, T.R. Microbial tryptophan catabolites in health and disease. Nat. Commun. 2018, 9, 3294. [Google Scholar] [CrossRef] [Green Version]
- Mawe, G.M.; Hoffman, J.M. Serotonin signalling in the gut—Functions, dysfunctions and therapeutic targets. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 473–486. [Google Scholar] [CrossRef] [Green Version]
- Terry, N.; Margolis, K.G. Serotonergic Mechanisms Regulating the GI Tract: Experimental Evidence and Therapeutic Relevance. Handb. Exp. Pharmacol. 2017, 239, 319–342. [Google Scholar] [CrossRef] [Green Version]
- Heredia, D.J.; Gershon, M.D.; Koh, S.D.; Corrigan, R.D.; Okamoto, T.; Smith, T.K. Important role of mucosal serotonin in colonic propulsion and peristaltic reflexes: In vitro analyses in mice lacking tryptophan hydroxylase 1. J. Physiol. 2013, 591, 5939–5957. [Google Scholar] [CrossRef]
- Margolis, K.G.; Li, Z.; Stevanovic, K.; Saurman, V.; Israelyan, N.; Anderson, G.M.; Snyder, I.; Veenstra-VanderWeele, J.; Blakely, R.D.; Gershon, M.D. Serotonin transporter variant drives preventable gastrointestinal abnormalities in development and function. J. Clin. Invest. 2016, 126, 2221–2235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baron, R.; Rawadi, G. Wnt signaling and the regulation of bone mass. Curr. Osteoporos. Rep. 2007, 5, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Hata, T.; Asano, Y.; Yoshihara, K.; Kimura-Todani, T.; Miyata, N.; Zhang, X.T.; Takakura, S.; Aiba, Y.; Koga, Y.; Sudo, N. Regulation of gut luminal serotonin by commensal microbiota in mice. PLoS ONE 2017, 12, e0180745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reigstad, C.S.; Salmonson, C.E.; Rainey, J.F., 3rd; Szurszewski, J.H.; Linden, D.R.; Sonnenburg, J.L.; Farrugia, G.; Kashyap, P.C. Gut microbes promote colonic serotonin production through an effect of short-chain fatty acids on enterochromaffin cells. FASEB J. 2015, 29, 1395–1403. [Google Scholar] [CrossRef] [Green Version]
- Alexeev, E.E.; Lanis, J.M.; Kao, D.J.; Campbell, E.L.; Kelly, C.J.; Battista, K.D.; Gerich, M.E.; Jenkins, B.R.; Walk, S.T.; Kominsky, D.J.; et al. Microbiota-Derived Indole Metabolites Promote Human and Murine Intestinal Homeostasis through Regulation of Interleukin-10 Receptor. Am. J. Pathol. 2018, 188, 1183–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubbard, T.D.; Murray, I.A.; Perdew, G.H. Indole and Tryptophan Metabolism: Endogenous and Dietary Routes to Ah Receptor Activation. Drug Metab. Dispos. 2015, 43, 1522–1535. [Google Scholar] [CrossRef] [Green Version]
- Wlodarska, M.; Luo, C.; Kolde, R.; d’Hennezel, E.; Annand, J.W.; Heim, C.E.; Krastel, P.; Schmitt, E.K.; Omar, A.S.; Creasey, E.A.; et al. Indoleacrylic Acid Produced by Commensal Peptostreptococcus Species Suppresses Inflammation. Cell Host Microbe 2017, 22, 25–37.e26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, X.; Gao, Y.; Yang, R. Gut Microbiota-Derived Tryptophan Metabolites Maintain Gut and Systemic Homeostasis. Cells 2022, 11, 2296. [Google Scholar] [CrossRef]
- Lamas, B.; Natividad, J.M.; Sokol, H. Aryl hydrocarbon receptor and intestinal immunity. Mucosal Immunol. 2018, 11, 1024–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockinger, B.; Di Meglio, P.; Gialitakis, M.; Duarte, J.H. The aryl hydrocarbon receptor: Multitasking in the immune system. Annu. Rev. Immunol. 2014, 32, 403–432. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.B.; Van Benschoten, A.H.; Cimermancic, P.; Donia, M.S.; Zimmermann, M.; Taketani, M.; Ishihara, A.; Kashyap, P.C.; Fraser, J.S.; Fischbach, M.A. Discovery and characterization of gut microbiota decarboxylases that can produce the neurotransmitter tryptamine. Cell Host Microbe 2014, 16, 495–503. [Google Scholar] [CrossRef] [Green Version]
- Juorio, A.V.; Paterson, I.A. Tryptamine may couple dopaminergic and serotonergic transmission in the brain. Gen. Pharmacol. 1990, 21, 613–616. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, Y.; Williams, B.B.; Battaglioli, E.J.; Whitaker, W.R.; Till, L.; Grover, M.; Linden, D.R.; Akiba, Y.; Kandimalla, K.K.; Zachos, N.C.; et al. Gut Microbiota-Produced Tryptamine Activates an Epithelial G-Protein-Coupled Receptor to Increase Colonic Secretion. Cell Host Microbe 2018, 23, 775–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galligan, J.J. Beneficial actions of microbiota-derived tryptophan metabolites. Neurogastroenterol. Motil. 2018, 30, e13283. [Google Scholar] [CrossRef] [PubMed]
- Dodd, D.; Spitzer, M.H.; Van Treuren, W.; Merrill, B.D.; Hryckowian, A.J.; Higginbottom, S.K.; Le, A.; Cowan, T.M.; Nolan, G.P.; Fischbach, M.A.; et al. A gut bacterial pathway metabolizes aromatic amino acids into nine circulating metabolites. Nature 2017, 551, 648–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Lee, J. Indole as an intercellular signal in microbial communities. FEMS Microbiol. Rev. 2010, 34, 426–444. [Google Scholar] [CrossRef] [PubMed]
- Wikoff, W.R.; Anfora, A.T.; Liu, J.; Schultz, P.G.; Lesley, S.A.; Peters, E.C.; Siuzdak, G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. USA 2009, 106, 3698–3703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukic, I.; Getselter, D.; Koren, O.; Elliott, E. Role of Tryptophan in Microbiota-Induced Depressive-Like Behavior: Evidence from Tryptophan Depletion Study. Front. Behav. Neurosci. 2019, 13, 123. [Google Scholar] [CrossRef] [Green Version]
- Dehhaghi, M.; Kazemi Shariat Panahi, H.; Guillemin, G.J. Microorganisms, Tryptophan Metabolism, and Kynurenine Pathway: A Complex Interconnected Loop Influencing Human Health Status. Int. J. Tryptophan Res. 2019, 12, 1178646919852996. [Google Scholar] [CrossRef] [Green Version]
- Cryan, J.F.; Dinan, T.G. Mind-altering microorganisms: The impact of the gut microbiota on brain and behaviour. Nat. Rev. Neurosci. 2012, 13, 701–712. [Google Scholar] [CrossRef]
- He, Y.W.; Wang, H.S.; Zeng, J.; Fang, X.; Chen, H.Y.; Du, J.; Yang, X.Y. Sodium butyrate inhibits interferon-gamma induced indoleamine 2,3-dioxygenase expression via STAT1 in nasopharyngeal carcinoma cells. Life Sci. 2013, 93, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Yu, T.; Huang, X.; Bilotta, A.J.; Xu, L.; Lu, Y.; Sun, J.; Pan, F.; Zhou, J.; Zhang, W.; et al. Intestinal microbiota-derived short-chain fatty acids regulation of immune cell IL-22 production and gut immunity. Nat. Commun. 2020, 11, 4457. [Google Scholar] [CrossRef]
- Li, G.; Lin, J.; Zhang, C.; Gao, H.; Lu, H.; Gao, X.; Zhu, R.; Li, Z.; Li, M.; Liu, Z. Microbiota metabolite butyrate constrains neutrophil functions and ameliorates mucosal inflammation in inflammatory bowel disease. Gut Microbes 2021, 13, 1968257. [Google Scholar] [CrossRef] [PubMed]
- Glauben, R.; Batra, A.; Fedke, I.; Zeitz, M.; Lehr, H.A.; Leoni, F.; Mascagni, P.; Fantuzzi, G.; Dinarello, C.A.; Siegmund, B. Histone hyperacetylation is associated with amelioration of experimental colitis in mice. J. Immunol. 2006, 176, 5015–5022. [Google Scholar] [CrossRef] [Green Version]
- Sandler, R.H.; Finegold, S.M.; Bolte, E.R.; Buchanan, C.P.; Maxwell, A.P.; Vaisanen, M.L.; Nelson, M.N.; Wexler, H.M. Short-term benefit from oral vancomycin treatment of regressive-onset autism. J. Child Neurol. 2000, 15, 429–435. [Google Scholar] [CrossRef]
- Finegold, S.M.; Molitoris, D.; Song, Y.; Liu, C.; Vaisanen, M.L.; Bolte, E.; McTeague, M.; Sandler, R.; Wexler, H.; Marlowe, E.M.; et al. Gastrointestinal microflora studies in late-onset autism. Clin. Infect. Dis. 2002, 35, S6–S16. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Liu, C.; Finegold, S.M. Real-time PCR quantitation of clostridia in feces of autistic children. Appl. Environ. Microbiol. 2004, 70, 6459–6465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parracho, H.M.; Bingham, M.O.; Gibson, G.R.; McCartney, A.L. Differences between the gut microflora of children with autistic spectrum disorders and that of healthy children. J. Med. Microbiol. 2005, 54, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Finegold, S.M.; Dowd, S.E.; Gontcharova, V.; Liu, C.; Henley, K.E.; Wolcott, R.D.; Youn, E.; Summanen, P.H.; Granpeesheh, D.; Dixon, D.; et al. Pyrosequencing study of fecal microflora of autistic and control children. Anaerobe 2010, 16, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.L.; Hornig, M.; Buie, T.; Bauman, M.L.; Cho Paik, M.; Wick, I.; Bennett, A.; Jabado, O.; Hirschberg, D.L.; Lipkin, W.I. Impaired carbohydrate digestion and transport and mucosal dysbiosis in the intestines of children with autism and gastrointestinal disturbances. PLoS ONE 2011, 6, e24585. [Google Scholar] [CrossRef] [Green Version]
- Adams, J.B.; Johansen, L.J.; Powell, L.D.; Quig, D.; Rubin, R.A. Gastrointestinal flora and gastrointestinal status in children with autism--comparisons to typical children and correlation with autism severity. BMC Gastroenterol. 2011, 11, 22. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Christophersen, C.T.; Sorich, M.J.; Gerber, J.P.; Angley, M.T.; Conlon, M.A. Increased abundance of Sutterella spp. and Ruminococcus torques in feces of children with autism spectrum disorder. Mol. Autism 2013, 4, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kushak, R.I.; Winter, H.S.; Buie, T.M.; Cox, S.B.; Phillips, C.D.; Ward, N.L. Analysis of the Duodenal Microbiome in Autistic Individuals: Association with Carbohydrate Digestion. J. Pediatr. Gastroenterol. Nutr. 2017, 64, e110–e116. [Google Scholar] [CrossRef] [PubMed]
- Luna, R.A.; Oezguen, N.; Balderas, M.; Venkatachalam, A.; Runge, J.K.; Versalovic, J.; Veenstra-VanderWeele, J.; Anderson, G.M.; Savidge, T.; Williams, K.C. Distinct Microbiome-Neuroimmune Signatures Correlate with Functional Abdominal Pain in Children with Autism Spectrum Disorder. Cell. Mol. Gastroenterol. Hepatol. 2017, 3, 218–230. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Li, J.; Wu, F.; Zheng, H.; Peng, Q.; Zhou, H. Altered composition and function of intestinal microbiota in autism spectrum disorders: A systematic review. Transl. Psychiatry 2019, 9, 43. [Google Scholar] [CrossRef] [Green Version]
- Frye, R.E.; Rose, S.; Slattery, J.; MacFabe, D.F. Gastrointestinal dysfunction in autism spectrum disorder: The role of the mitochondria and the enteric microbiome. Microb. Ecol. Health Dis. 2015, 26, 27458. [Google Scholar] [CrossRef] [PubMed]
- Argou-Cardozo, I.; Zeidan-Chulia, F. Clostridium Bacteria and Autism Spectrum Conditions: A Systematic Review and Hypothetical Contribution of Environmental Glyphosate Levels. Med. Sci. 2018, 6, 29. [Google Scholar] [CrossRef] [Green Version]
- Silva, Y.P.; Bernardi, A.; Frozza, R.L. The Role of Short-Chain Fatty Acids from Gut Microbiota in Gut-Brain Communication. Front. Endocrinol. 2020, 11, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srikantha, P.; Mohajeri, M.H. The Possible Role of the Microbiota-Gut-Brain-Axis in Autism Spectrum Disorder. Int. J. Mol. Sci. 2019, 20, 2115. [Google Scholar] [CrossRef] [Green Version]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef] [Green Version]
- Kang, D.W.; Park, J.G.; Ilhan, Z.E.; Wallstrom, G.; Labaer, J.; Adams, J.B.; Krajmalnik-Brown, R. Reduced incidence of Prevotella and other fermenters in intestinal microflora of autistic children. PLoS ONE 2013, 8, e68322. [Google Scholar] [CrossRef] [Green Version]
- Golubeva, A.V.; Joyce, S.A.; Moloney, G.; Burokas, A.; Sherwin, E.; Arboleya, S.; Flynn, I.; Khochanskiy, D.; Moya-Perez, A.; Peterson, V.; et al. Microbiota-related Changes in Bile Acid & Tryptophan Metabolism are Associated with Gastrointestinal Dysfunction in a Mouse Model of Autism. EBioMedicine 2017, 24, 166–178. [Google Scholar] [CrossRef] [Green Version]
- Kang, D.W.; Adams, J.B.; Gregory, A.C.; Borody, T.; Chittick, L.; Fasano, A.; Khoruts, A.; Geis, E.; Maldonado, J.; McDonough-Means, S.; et al. Microbiota Transfer Therapy alters gut ecosystem and improves gastrointestinal and autism symptoms: An open-label study. Microbiome 2017, 5, 10. [Google Scholar] [CrossRef]
- Hsiao, E.Y.; McBride, S.W.; Hsien, S.; Sharon, G.; Hyde, E.R.; McCue, T.; Codelli, J.A.; Chow, J.; Reisman, S.E.; Petrosino, J.F.; et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 2013, 155, 1451–1463. [Google Scholar] [CrossRef] [Green Version]
- Sharon, G.; Cruz, N.J.; Kang, D.W.; Gandal, M.J.; Wang, B.; Kim, Y.M.; Zink, E.M.; Casey, C.P.; Taylor, B.C.; Lane, C.J.; et al. Human Gut Microbiota from Autism Spectrum Disorder Promote Behavioral Symptoms in Mice. Cell 2019, 177, 1600–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Notarangelo, F.M.; Beggiato, S.; Schwarcz, R. Assessment of Prenatal Kynurenine Metabolism Using Tissue Slices: Focus on the Neosynthesis of Kynurenic Acid in Mice. Dev. Neurosci. 2019, 41, 102–111. [Google Scholar] [CrossRef]
- Notarangelo, F.M.; Pocivavsek, A. Elevated kynurenine pathway metabolism during neurodevelopment: Implications for brain and behavior. Neuropharmacology 2017, 112, 275–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Zundert, S.K.; Broekhuizen, M.; Smit, A.J.; van Rossem, L.; Mirzaian, M.; Willemsen, S.P.; Danser, A.J.; De Rijke, Y.B.; Reiss, I.K.; Merkus, D.; et al. The Role of the Kynurenine Pathway in the (Patho) physiology of Maternal Pregnancy and Fetal Outcomes: A Systematic Review. Int. J. Tryptophan Res. 2022, 15, 11786469221135545. [Google Scholar] [CrossRef]
- Khalil, O.S.; Pisar, M.; Forrest, C.M.; Vincenten, M.C.; Darlington, L.G.; Stone, T.W. Prenatal inhibition of the kynurenine pathway leads to structural changes in the hippocampus of adult rat offspring. Eur. J. Neurosci. 2014, 39, 1558–1571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pisar, M.; Forrest, C.M.; Khalil, O.S.; McNair, K.; Vincenten, M.C.; Qasem, S.; Darlington, L.G.; Stone, T.W. Modified neocortical and cerebellar protein expression and morphology in adult rats following prenatal inhibition of the kynurenine pathway. Brain Res. 2014, 1576, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Varga, D.; Heredi, J.; Kanvasi, Z.; Ruszka, M.; Kis, Z.; Ono, E.; Iwamori, N.; Iwamori, T.; Takakuwa, H.; Vecsei, L.; et al. Systemic L-Kynurenine sulfate administration disrupts object recognition memory, alters open field behavior and decreases c-Fos immunopositivity in C57Bl/6 mice. Front. Behav. Neurosci. 2015, 9, 157. [Google Scholar] [CrossRef] [Green Version]
- Perkins, M.N.; Stone, T.W. An iontophoretic investigation of the actions of convulsant kynurenines and their interaction with the endogenous excitant quinolinic acid. Brain Res. 1982, 247, 184–187. [Google Scholar] [CrossRef]
- Hilmas, C.; Pereira, E.F.; Alkondon, M.; Rassoulpour, A.; Schwarcz, R.; Albuquerque, E.X. The brain metabolite kynurenic acid inhibits alpha7 nicotinic receptor activity and increases non-alpha7 nicotinic receptor expression: Physiopathological implications. J. Neurosci. 2001, 21, 7463–7473. [Google Scholar] [CrossRef] [PubMed]
- Ceresoli-Borroni, G.; Schwarcz, R. Perinatal kynurenine pathway metabolism in the normal and asphyctic rat brain. Amino Acids 2000, 19, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Fukui, S.; Schwarcz, R.; Rapoport, S.I.; Takada, Y.; Smith, Q.R. Blood-brain barrier transport of kynurenines: Implications for brain synthesis and metabolism. J. Neurochem. 1991, 56, 2007–2017. [Google Scholar] [CrossRef]
- Goeden, N.; Notarangelo, F.M.; Pocivavsek, A.; Beggiato, S.; Bonnin, A.; Schwarcz, R. Prenatal Dynamics of Kynurenine Pathway Metabolism in Mice: Focus on Kynurenic Acid. Dev. Neurosci. 2017, 39, 519–528. [Google Scholar] [CrossRef]
- Tashiro, T.; Murakami, Y.; Mouri, A.; Imamura, Y.; Nabeshima, T.; Yamamoto, Y.; Saito, K. Kynurenine 3-monooxygenase is implicated in antidepressants-responsive depressive-like behaviors and monoaminergic dysfunctions. Behav. Brain Res. 2017, 317, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Erhardt, S.; Pocivavsek, A.; Repici, M.; Liu, X.C.; Imbeault, S.; Maddison, D.C.; Thomas, M.A.R.; Smalley, J.L.; Larsson, M.K.; Muchowski, P.J.; et al. Adaptive and Behavioral Changes in Kynurenine 3-Monooxygenase Knockout Mice: Relevance to Psychotic Disorders. Biol. Psychiatry 2017, 82, 756–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namba, T.; Ming, G.L.; Song, H.; Waga, C.; Enomoto, A.; Kaibuchi, K.; Kohsaka, S.; Uchino, S. NMDA receptor regulates migration of newly generated neurons in the adult hippocampus via Disrupted-In-Schizophrenia 1 (DISC1). J. Neurochem. 2011, 118, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Nacher, J.; McEwen, B.S. The role of N-methyl-D-asparate receptors in neurogenesis. Hippocampus 2006, 16, 267–270. [Google Scholar] [CrossRef]
- Colonnese, M.T.; Zhao, J.P.; Constantine-Paton, M. NMDA receptor currents suppress synapse formation on sprouting axons in vivo. J. Neurosci. 2005, 25, 1291–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ultanir, S.K.; Kim, J.E.; Hall, B.J.; Deerinck, T.; Ellisman, M.; Ghosh, A. Regulation of spine morphology and spine density by NMDA receptor signaling in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 19553–19558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.J.; Choi, S.Y.; Kim, E. NMDA receptor dysfunction in autism spectrum disorders. Curr. Opin. Pharmacol. 2015, 20, 8–13. [Google Scholar] [CrossRef]
- O’Roak, B.J.; Vives, L.; Girirajan, S.; Karakoc, E.; Krumm, N.; Coe, B.P.; Levy, R.; Ko, A.; Lee, C.; Smith, J.D.; et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 2012, 485, 246–250. [Google Scholar] [CrossRef] [Green Version]
- O’Roak, B.J.; Vives, L.; Fu, W.; Egertson, J.D.; Stanaway, I.B.; Phelps, I.G.; Carvill, G.; Kumar, A.; Lee, C.; Ankenman, K.; et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 2012, 338, 1619–1622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarabeux, J.; Kebir, O.; Gauthier, J.; Hamdan, F.F.; Xiong, L.; Piton, A.; Spiegelman, D.; Henrion, E.; Millet, B.; S2D Team; et al. Rare mutations in N-methyl-D-aspartate glutamate receptors in autism spectrum disorders and schizophrenia. Transl. Psychiatry 2011, 1, e55. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.; Chen, W.; Myers, S.J.; Yuan, H.; Traynelis, S.F. Human GRIN2B variants in neurodevelopmental disorders. J. Pharmacol. Sci. 2016, 132, 115–121. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Ma, Y.; Fan, R.; Yang, Z.; Li, M.D. Implication of Genes for the N-Methyl-D-Aspartate (NMDA) Receptor in Substance Addictions. Mol. Neurobiol. 2018, 55, 7567–7578. [Google Scholar] [CrossRef]
- Wink, L.K.; Minshawi, N.F.; Shaffer, R.C.; Plawecki, M.H.; Posey, D.J.; Horn, P.S.; Adams, R.; Pedapati, E.V.; Schaefer, T.L.; McDougle, C.J.; et al. d-Cycloserine enhances durability of social skills training in autism spectrum disorder. Mol. Autism 2017, 8, 2. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.F.; Chen, P.S.; Hsu, Y.T.; Lee, C.W.; Wang, T.F.; Chen, Y.J.; Lin, H.C. (D)-Cycloserine Ameliorates Autism-Like Deficits by Removing GluA2-Containing AMPA Receptors in a Valproic Acid-Induced Rat Model. Mol. Neurobiol. 2018, 55, 4811–4824. [Google Scholar] [CrossRef]
- Hosenbocus, S.; Chahal, R. Memantine: A review of possible uses in child and adolescent psychiatry. J. Can. Acad. Child Adolesc. Psychiatry 2013, 22, 166–171. [Google Scholar]
- Soorya, L.V.; Fogg, L.; Ocampo, E.; Printen, M.; Youngkin, S.; Halpern, D.; Kolevzon, A.; Lee, S.; Grodberg, D.; Anagnostou, E. Neurocognitive Outcomes from Memantine: A Pilot, Double-Blind, Placebo-Controlled Trial in Children with Autism Spectrum Disorder. J. Child Adolesc. Psychopharmacol. 2021, 31, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Budreck, E.C.; Kwon, O.B.; Jung, J.H.; Baudouin, S.; Thommen, A.; Kim, H.S.; Fukazawa, Y.; Harada, H.; Tabuchi, K.; Shigemoto, R.; et al. Neuroligin-1 controls synaptic abundance of NMDA-type glutamate receptors through extracellular coupling. Proc. Natl. Acad. Sci. USA 2013, 110, 725–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berkel, S.; Marshall, C.R.; Weiss, B.; Howe, J.; Roeth, R.; Moog, U.; Endris, V.; Roberts, W.; Szatmari, P.; Pinto, D.; et al. Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nat. Genet. 2010, 42, 489–491. [Google Scholar] [CrossRef] [PubMed]
- Won, H.; Lee, H.R.; Gee, H.Y.; Mah, W.; Kim, J.I.; Lee, J.; Ha, S.; Chung, C.; Jung, E.S.; Cho, Y.S.; et al. Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature 2012, 486, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Silverman, J.L.; Smith, D.G.; Rizzo, S.J.; Karras, M.N.; Turner, S.M.; Tolu, S.S.; Bryce, D.K.; Smith, D.L.; Fonseca, K.; Ring, R.H.; et al. Negative allosteric modulation of the mGluR5 receptor reduces repetitive behaviors and rescues social deficits in mouse models of autism. Sci. Transl. Med. 2012, 4, 131ra151. [Google Scholar] [CrossRef] [Green Version]
- Benson, A.D.; Burket, J.A.; Deutsch, S.I. Balb/c mice treated with D-cycloserine arouse increased social interest in conspecifics. Brain Res. Bull. 2013, 99, 95–99. [Google Scholar] [CrossRef]
- Auerbach, B.D.; Osterweil, E.K.; Bear, M.F. Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature 2011, 480, 63–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albuquerque, E.X.; Pereira, E.F.; Alkondon, M.; Rogers, S.W. Mammalian nicotinic acetylcholine receptors: From structure to function. Physiol. Rev. 2009, 89, 73–120. [Google Scholar] [CrossRef] [Green Version]
- Bouzat, C.; Lasala, M.; Nielsen, B.E.; Corradi, J.; Esandi, M.D.C. Molecular function of alpha7 nicotinic receptors as drug targets. J. Physiol. 2018, 596, 1847–1861. [Google Scholar] [CrossRef] [Green Version]
- Broide, R.S.; Winzer-Serhan, U.H.; Chen, Y.; Leslie, F.M. Distribution of alpha7 Nicotinic Acetylcholine Receptor Subunit mRNA in the Developing Mouse. Front. Neuroanat. 2019, 13, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eissa, N.; Al-Houqani, M.; Sadeq, A.; Ojha, S.K.; Sasse, A.; Sadek, B. Current Enlightenment About Etiology and Pharmacological Treatment of Autism Spectrum Disorder. Front. Neurosci. 2018, 12, 304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.L.; Adams, C.E.; Stevens, K.E.; Chow, K.H.; Freedman, R.; Patterson, P.H. The interaction between maternal immune activation and alpha 7 nicotinic acetylcholine receptor in regulating behaviors in the offspring. Brain Behav. Immun. 2015, 46, 192–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McTighe, S.M.; Neal, S.J.; Lin, Q.; Hughes, Z.A.; Smith, D.G. The BTBR mouse model of autism spectrum disorders has learning and attentional impairments and alterations in acetylcholine and kynurenic acid in prefrontal cortex. PLoS ONE 2013, 8, e62189. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, R.F.; Tran, M.B.; Hogenkamp, D.J.; Ayala, N.L.; Johnstone, T.; Dunnigan, A.J.; Gee, T.K.; Gee, K.W. Allosteric modulation of nicotinic and GABA(A) receptor subtypes differentially modify autism-like behaviors in the BTBR mouse model. Neuropharmacology 2017, 126, 38–47. [Google Scholar] [CrossRef]
- Mahmood, H.M.; Aldhalaan, H.M.; Alshammari, T.K.; Alqasem, M.A.; Alshammari, M.A.; Albekairi, N.A.; AlSharari, S.D. The Role of Nicotinic Receptors in the Attenuation of Autism-Related Behaviors in a Murine BTBR T + tf/J Autistic Model. Autism Res. 2020, 13, 1311–1334. [Google Scholar] [CrossRef]
- Ghaleiha, A.; Ghyasvand, M.; Mohammadi, M.R.; Farokhnia, M.; Yadegari, N.; Tabrizi, M.; Hajiaghaee, R.; Yekehtaz, H.; Akhondzadeh, S. Galantamine efficacy and tolerability as an augmentative therapy in autistic children: A randomized, double-blind, placebo-controlled trial. J. Psychopharmacol. 2014, 28, 677–685. [Google Scholar] [CrossRef]
- Buckley, A.W.; Sassower, K.; Rodriguez, A.J.; Jennison, K.; Wingert, K.; Buckley, J.; Thurm, A.; Sato, S.; Swedo, S. An open label trial of donepezil for enhancement of rapid eye movement sleep in young children with autism spectrum disorders. J. Child Adolesc. Psychopharmacol. 2011, 21, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, S.J.; Lalande, M. Neurodevelopmental disorders involving genomic imprinting at human chromosome 15q11-q13. Neurobiol. Dis. 2010, 39, 13–20. [Google Scholar] [CrossRef]
- Ziats, M.N.; Goin-Kochel, R.P.; Berry, L.N.; Ali, M.; Ge, J.; Guffey, D.; Rosenfeld, J.A.; Bader, P.; Gambello, M.J.; Wolf, V.; et al. The complex behavioral phenotype of 15q13.3 microdeletion syndrome. Genet. Med. 2016, 18, 1111–1118. [Google Scholar] [CrossRef] [Green Version]
- Yasui, D.H.; Scoles, H.A.; Horike, S.; Meguro-Horike, M.; Dunaway, K.W.; Schroeder, D.I.; Lasalle, J.M. 15q11.2-13.3 chromatin analysis reveals epigenetic regulation of CHRNA7 with deficiencies in Rett and autism brain. Hum. Mol. Genet. 2011, 20, 4311–4323. [Google Scholar] [CrossRef] [PubMed]
- Stone, T.W. Neuropharmacology of quinolinic and kynurenic acids. Pharmacol. Rev. 1993, 45, 309–379. [Google Scholar]
- Alkondon, M.; Pereira, E.F.; Eisenberg, H.M.; Kajii, Y.; Schwarcz, R.; Albuquerque, E.X. Age dependency of inhibition of alpha7 nicotinic receptors and tonically active N-methyl-D-aspartate receptors by endogenously produced kynurenic acid in the brain. J. Pharmacol. Exp. Ther. 2011, 337, 572–582. [Google Scholar] [CrossRef] [Green Version]
- Alkondon, M.; Pereira, E.F.; Albuquerque, E.X. Endogenous activation of nAChRs and NMDA receptors contributes to the excitability of CA1 stratum radiatum interneurons in rat hippocampal slices: Effects of kynurenic acid. Biochem. Pharmacol. 2011, 82, 842–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albuquerque, E.X.; Schwarcz, R. Kynurenic acid as an antagonist of alpha7 nicotinic acetylcholine receptors in the brain: Facts and challenges. Biochem. Pharmacol. 2013, 85, 1027–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tribollet, E.; Bertrand, D.; Marguerat, A.; Raggenbass, M. Comparative distribution of nicotinic receptor subtypes during development, adulthood and aging: An autoradiographic study in the rat brain. Neuroscience 2004, 124, 405–420. [Google Scholar] [CrossRef]
- Shytle, R.D.; Mori, T.; Townsend, K.; Vendrame, M.; Sun, N.; Zeng, J.; Ehrhart, J.; Silver, A.A.; Sanberg, P.R.; Tan, J. Cholinergic modulation of microglial activation by alpha 7 nicotinic receptors. J. Neurochem. 2004, 89, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Papouin, T.; Dunphy, J.M.; Tolman, M.; Dineley, K.T.; Haydon, P.G. Septal Cholinergic Neuromodulation Tunes the Astrocyte-Dependent Gating of Hippocampal NMDA Receptors to Wakefulness. Neuron 2017, 94, 840–854. [Google Scholar] [CrossRef] [Green Version]
- Petrov, K.A.; Girard, E.; Nikitashina, A.D.; Colasante, C.; Bernard, V.; Nurullin, L.; Leroy, J.; Samigullin, D.; Colak, O.; Nikolsky, E.; et al. Schwann cells sense and control acetylcholine spillover at the neuromuscular junction by alpha7 nicotinic receptors and butyrylcholinesterase. J. Neurosci. 2014, 34, 11870–11883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, G.; Vijayaraghavan, S. Nicotinic receptor signaling in nonexcitable cells. J. Neurobiol. 2002, 53, 524–534. [Google Scholar] [CrossRef] [PubMed]
- Konradsson-Geuken, A.; Wu, H.Q.; Gash, C.R.; Alexander, K.S.; Campbell, A.; Sozeri, Y.; Pellicciari, R.; Schwarcz, R.; Bruno, J.P. Cortical kynurenic acid bi-directionally modulates prefrontal glutamate levels as assessed by microdialysis and rapid electrochemistry. Neuroscience 2010, 169, 1848–1859. [Google Scholar] [CrossRef] [Green Version]
- Potter, M.C.; Elmer, G.I.; Bergeron, R.; Albuquerque, E.X.; Guidetti, P.; Wu, H.Q.; Schwarcz, R. Reduction of endogenous kynurenic acid formation enhances extracellular glutamate, hippocampal plasticity, and cognitive behavior. Neuropsychopharmacology 2010, 35, 1734–1742. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.Q.; Pereira, E.F.; Bruno, J.P.; Pellicciari, R.; Albuquerque, E.X.; Schwarcz, R. The astrocyte-derived alpha7 nicotinic receptor antagonist kynurenic acid controls extracellular glutamate levels in the prefrontal cortex. J. Mol. Neurosci. 2010, 40, 204–210. [Google Scholar] [CrossRef] [Green Version]
- Beggiato, S.; Antonelli, T.; Tomasini, M.C.; Tanganelli, S.; Fuxe, K.; Schwarcz, R.; Ferraro, L. Kynurenic acid, by targeting alpha7 nicotinic acetylcholine receptors, modulates extracellular GABA levels in the rat striatum in vivo. Eur. J. Neurosci. 2013, 37, 1470–1477. [Google Scholar] [CrossRef]
- Rassoulpour, A.; Wu, H.Q.; Ferre, S.; Schwarcz, R. Nanomolar concentrations of kynurenic acid reduce extracellular dopamine levels in the striatum. J. Neurochem. 2005, 93, 762–765. [Google Scholar] [CrossRef]
- Zmarowski, A.; Wu, H.Q.; Brooks, J.M.; Potter, M.C.; Pellicciari, R.; Schwarcz, R.; Bruno, J.P. Astrocyte-derived kynurenic acid modulates basal and evoked cortical acetylcholine release. Eur. J. Neurosci. 2009, 29, 529–538. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, J.; Berg, D.K. Role of endogenous nicotinic signaling in guiding neuronal development. Biochem. Pharmacol. 2007, 74, 1112–1119. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Otsu, Y.; Vasuta, C.; Nawa, H.; Murphy, T.H. Action-potential-independent GABAergic tone mediated by nicotinic stimulation of immature striatal miniature synaptic transmission. J. Neurophysiol. 2007, 98, 581–593. [Google Scholar] [CrossRef]
- Lozada, A.F.; Wang, X.; Gounko, N.V.; Massey, K.A.; Duan, J.; Liu, Z.; Berg, D.K. Glutamatergic synapse formation is promoted by alpha7-containing nicotinic acetylcholine receptors. J. Neurosci. 2012, 32, 7651–7661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhulkifle, H.; Agouni, A.; Zeidan, A.; Al-Kuwari, M.S.; Parray, A.; Tolefat, M.; Korashy, H.M. Influence of the Aryl Hydrocarbon Receptor Activating Environmental Pollutants on Autism Spectrum Disorder. Int. J. Mol. Sci. 2021, 22, 9258. [Google Scholar] [CrossRef] [PubMed]
- Schultz, R.; Suominen, J.; Varre, T.; Hakovirta, H.; Parvinen, M.; Toppari, J.; Pelto-Huikko, M. Expression of aryl hydrocarbon receptor and aryl hydrocarbon receptor nuclear translocator messenger ribonucleic acids and proteins in rat and human testis. Endocrinology 2003, 144, 767–776. [Google Scholar] [CrossRef]
- Kimura, E.; Kubo, K.I.; Endo, T.; Nakajima, K.; Kakeyama, M.; Tohyama, C. Excessive activation of AhR signaling disrupts neuronal migration in the hippocampal CA1 region in the developing mouse. J. Toxicol. Sci. 2017, 42, 25–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, E.; Kohda, M.; Maekawa, F.; Fujii-Kuriyama, Y.; Tohyama, C. Correction to: Neurons expressing the aryl hydrocarbon receptor in the locus coeruleus and island of Calleja major are novel targets of dioxin in the mouse brain. Histochem. Cell. Biol. 2021, 156, 293. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Huang, L.; Jin, J.Y.; Jolly, S.; Zang, Y.; Wu, H.; Yan, L.; Pi, W.; Li, L.; Mellor, A.L.; et al. IDO Immune Status after Chemoradiation May Predict Survival in Lung Cancer Patients. Cancer Res. 2018, 78, 809–816. [Google Scholar] [CrossRef] [Green Version]
- DiNatale, B.C.; Murray, I.A.; Schroeder, J.C.; Flaveny, C.A.; Lahoti, T.S.; Laurenzana, E.M.; Omiecinski, C.J.; Perdew, G.H. Kynurenic acid is a potent endogenous aryl hydrocarbon receptor ligand that synergistically induces interleukin-6 in the presence of inflammatory signaling. Toxicol. Sci. 2010, 115, 89–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Lara, L.; Perez-Severiano, F.; Gonzalez-Esquivel, D.; Elizondo, G.; Segovia, J. Absence of aryl hydrocarbon receptors increases endogenous kynurenic acid levels and protects mouse brain against excitotoxic insult and oxidative stress. J. Neurosci. Res. 2015, 93, 1423–1433. [Google Scholar] [CrossRef]
- Gomez-Fernandez, A.; de la Torre-Aguilar, M.J.; Gil-Campos, M.; Flores-Rojas, K.; Cruz-Rico, M.D.; Martin-Borreguero, P.; Perez-Navero, J.L. Children with Autism Spectrum Disorder with Regression Exhibit a Different Profile in Plasma Cytokines and Adhesion Molecules Compared to Children Without Such Regression. Front. Pediatr. 2018, 6, 264. [Google Scholar] [CrossRef] [Green Version]
- Yoshimura, Y.; Kikuchi, M.; Saito, D.N.; Hirosawa, T.; Takahashi, T.; Munesue, T.; Kosaka, H.; Naito, N.; Ouchi, Y.; Minabe, Y. Markers for the central serotonin system correlate to verbal ability and paralinguistic social voice processing in autism spectrum disorder. Sci. Rep. 2020, 10, 14558. [Google Scholar] [CrossRef]
- Muller, C.L.; Anacker, A.M.J.; Veenstra-VanderWeele, J. The serotonin system in autism spectrum disorder: From biomarker to animal models. Neuroscience 2016, 321, 24–41. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.D.; Notarangelo, F.M.; Wang, J.Z.; Schwarcz, R. Kynurenic acid and 3-hydroxykynurenine production from D-kynurenine in mice. Brain Res. 2012, 1455, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez Esquivel, D.; Ramirez-Ortega, D.; Pineda, B.; Castro, N.; Rios, C.; Perez de la Cruz, V. Kynurenine pathway metabolites and enzymes involved in redox reactions. Neuropharmacology 2017, 112, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Maddison, D.C.; Giorgini, F. The kynurenine pathway and neurodegenerative disease. Semin. Cell Dev. Biol. 2015, 40, 134–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenabi, E.; Karami, M.; Khazaei, S.; Bashirian, S. The association between preeclampsia and autism spectrum disorders among children: A meta-analysis. Korean J. Pediatr. 2019, 62, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Lampi, K.M.; Lehtonen, L.; Tran, P.L.; Suominen, A.; Lehti, V.; Banerjee, P.N.; Gissler, M.; Brown, A.S.; Sourander, A. Risk of autism spectrum disorders in low birth weight and small for gestational age infants. J. Pediatr. 2012, 161, 830–836. [Google Scholar] [CrossRef] [Green Version]
- Crump, C.; Sundquist, J.; Sundquist, K. Preterm or Early Term Birth and Risk of Autism. Pediatrics 2021, 148, e2020032300. [Google Scholar] [CrossRef] [PubMed]
- Ormstad, H.; Bryn, V.; Verkerk, R.; Skjeldal, O.H.; Halvorsen, B.; Saugstad, O.D.; Isaksen, J.; Maes, M. Serum Tryptophan, Tryptophan Catabolites and Brain-derived Neurotrophic Factor in Subgroups of Youngsters with Autism Spectrum Disorders. CNS Neurol. Disord. Drug Targets 2018, 17, 626–639. [Google Scholar] [CrossRef] [Green Version]
- Bilgic, A.; Abusoglu, S.; Sadic Celikkol, C.; Oflaz, M.B.; Akca, O.F.; Sivrikaya, A.; Baysal, T.; Unlu, A. Altered kynurenine pathway metabolite levels in toddlers and preschool children with autism spectrum disorder. Int. J. Neurosci. 2022, 132, 826–834. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Qian-Qian, L.; Cong, Y.; Xiao-Bing, Z.; Hong-Zhu, D. Reduction of essential amino acid levels and sex-specific alterations in serum amino acid concentration profiles in children with autism spectrum disorder. Psychiatry Res. 2021, 297, 113675. [Google Scholar] [CrossRef] [PubMed]
- Tu, W.J.; Chen, H.; He, J. Application of LC-MS/MS analysis of plasma amino acids profiles in children with autism. J. Clin. Biochem. Nutr. 2012, 51, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Naushad, S.M.; Jain, J.M.; Prasad, C.K.; Naik, U.; Akella, R.R. Autistic children exhibit distinct plasma amino acid profile. Indian J. Biochem. Biophys. 2013, 50, 474–478. [Google Scholar]
- Chen, W.X.; Chen, Y.R.; Peng, M.Z.; Liu, X.; Cai, Y.N.; Huang, Z.F.; Yang, S.Y.; Huang, J.Y.; Wang, R.H.; Yi, P.; et al. Plasma Amino Acid Profile in Children with Autism Spectrum Disorder in Southern China: Analysis of 110 Cases. J. Autism Dev. Disord. 2023. [Google Scholar] [CrossRef]
- Adams, J.B.; Audhya, T.; McDonough-Means, S.; Rubin, R.A.; Quig, D.; Geis, E.; Gehn, E.; Loresto, M.; Mitchell, J.; Atwood, S.; et al. Nutritional and metabolic status of children with autism vs. neurotypical children, and the association with autism severity. Nutr. Metab. 2011, 8, 34. [Google Scholar] [CrossRef] [Green Version]
- Noto, A.; Fanos, V.; Barberini, L.; Grapov, D.; Fattuoni, C.; Zaffanello, M.; Casanova, A.; Fenu, G.; De Giacomo, A.; De Angelis, M.; et al. The urinary metabolomics profile of an Italian autistic children population and their unaffected siblings. J. Matern.-Fetal Neonatal Med. 2014, 27, 46–52. [Google Scholar] [CrossRef]
- Kaluzna-Czaplinska, J.; Zurawicz, E.; Struck, W.; Markuszewski, M. Identification of organic acids as potential biomarkers in the urine of autistic children using gas chromatography/mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2014, 966, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Gevi, F.; Zolla, L.; Gabriele, S.; Persico, A.M. Urinary metabolomics of young Italian autistic children supports abnormal tryptophan and purine metabolism. Mol. Autism 2016, 7, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timperio, A.M.; Gevi, F.; Cucinotta, F.; Ricciardello, A.; Turriziani, L.; Scattoni, M.L.; Persico, A.M. Urinary Untargeted Metabolic Profile Differentiates Children with Autism from Their Unaffected Siblings. Metabolites 2022, 12, 797. [Google Scholar] [CrossRef] [PubMed]
- Harutyunyan, A.A.; Harutyunyan, H.A.; Yenkoyan, K.B. Novel Probable Glance at Inflammatory Scenario Development in Autistic Pathology. Front. Psychiatry 2021, 12, 788779. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, A.W.; Jyonouchi, H.; Comi, A.M.; Connors, S.L.; Milstien, S.; Varsou, A.; Heyes, M.P. Cerebrospinal fluid and serum markers of inflammation in autism. Pediatr. Neurol. 2005, 33, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.J.; Liu, C.L.; Sang, B.; Zhu, X.M.; Du, Y.J. The combined role of serotonin and interleukin-6 as biomarker for autism. Neuroscience 2015, 284, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Gabriele, S.; Sacco, R.; Persico, A.M. Blood serotonin levels in autism spectrum disorder: A systematic review and meta-analysis. Eur. Neuropsychopharmacol. 2014, 24, 919–929. [Google Scholar] [CrossRef]
- Nakai, N.; Nagano, M.; Saitow, F.; Watanabe, Y.; Kawamura, Y.; Kawamoto, A.; Tamada, K.; Mizuma, H.; Onoe, H.; Watanabe, Y.; et al. Serotonin rebalances cortical tuning and behavior linked to autism symptoms in 15q11-13 CNV mice. Sci. Adv. 2017, 3, e1603001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rind, H.B.; Russo, A.F.; Whittemore, S.R. Developmental regulation of tryptophan hydroxylase messenger RNA expression and enzyme activity in the raphe and its target fields. Neuroscience 2000, 101, 665–677. [Google Scholar] [CrossRef]
- Yang, S.Y.; Yoo, H.J.; Cho, I.H.; Park, M.; Kim, S.A. Association with tryptophan hydroxylase 2 gene polymorphisms and autism spectrum disorders in Korean families. Neurosci. Res. 2012, 73, 333–336. [Google Scholar] [CrossRef]
- Rodriguez-Gomez, D.A.; Garcia-Guaqueta, D.P.; Charry-Sanchez, J.D.; Sarquis-Buitrago, E.; Blanco, M.; Velez-van-Meerbeke, A.; Talero-Gutierrez, C. A systematic review of common genetic variation and biological pathways in autism spectrum disorder. BMC Neurosci. 2021, 22, 60. [Google Scholar] [CrossRef]
- Saitow, F.; Takumi, T.; Suzuki, H. Change in serotonergic modulation contributes to the synaptic imbalance of neuronal circuit at the prefrontal cortex in the 15q11-13 duplication mouse model of autism. Neuropharmacology 2020, 165, 107931. [Google Scholar] [CrossRef] [PubMed]
- Takumi, T.; Tamada, K.; Hatanaka, F.; Nakai, N.; Bolton, P.F. Behavioral neuroscience of autism. Neurosci. Biobehav. Rev. 2020, 110, 60–76. [Google Scholar] [CrossRef]
- Lauder, J.M.; Krebs, H. Serotonin as a differentiation signal in early neurogenesis. Dev. Neurosci. 1978, 1, 15–30. [Google Scholar] [CrossRef]
- Salter, M.; Knowles, R.G.; Pogson, C.I. How does displacement of albumin-bound tryptophan cause sustained increases in the free tryptophan concentration in plasma and 5-hydroxytryptamine synthesis in brain? Biochem. J. 1989, 262, 365–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishizawa, S.; Benkelfat, C.; Young, S.N.; Leyton, M.; Mzengeza, S.; de Montigny, C.; Blier, P.; Diksic, M. Differences between males and females in rates of serotonin synthesis in human brain. Proc. Natl. Acad. Sci. USA 1997, 94, 5308–5313. [Google Scholar] [CrossRef] [Green Version]
- Mendelsohn, D.; Riedel, W.J.; Sambeth, A. Effects of acute tryptophan depletion on memory, attention and executive functions: A systematic review. Neurosci. Biobehav. Rev. 2009, 33, 926–952. [Google Scholar] [CrossRef]
- Robinson, N.; Bergen, S.E. Environmental Risk Factors for Schizophrenia and Bipolar Disorder and Their Relationship to Genetic Risk: Current Knowledge and Future Directions. Front. Genet. 2021, 12, 686666. [Google Scholar] [CrossRef] [PubMed]
- Lyall, K.; Schmidt, R.J.; Hertz-Picciotto, I. Maternal lifestyle and environmental risk factors for autism spectrum disorders. Int. J. Epidemiol. 2014, 43, 443–464. [Google Scholar] [CrossRef] [Green Version]
- Notarangelo, F.M.; Schwarcz, R. Restraint Stress during Pregnancy Rapidly Raises Kynurenic Acid Levels in Mouse Placenta and Fetal Brain. Dev. Neurosci. 2016, 38, 458–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Notarangelo, F.M.; Schwarcz, R. A single prenatal lipopolysaccharide injection has acute, but not long-lasting, effects on cerebral kynurenine pathway metabolism in mice. Eur. J. Neurosci. 2021, 54, 5968–5981. [Google Scholar] [CrossRef]
- Holloway, T.; Moreno, J.L.; Umali, A.; Rayannavar, V.; Hodes, G.E.; Russo, S.J.; Gonzalez-Maeso, J. Prenatal stress induces schizophrenia-like alterations of serotonin 2A and metabotropic glutamate 2 receptors in the adult offspring: Role of maternal immune system. J. Neurosci. 2013, 33, 1088–1098. [Google Scholar] [CrossRef] [PubMed]
- MacDowell, K.S.; Munarriz-Cuezva, E.; Meana, J.J.; Leza, J.C.; Ortega, J.E. Paliperidone Reversion of Maternal Immune Activation-Induced Changes on Brain Serotonin and Kynurenine Pathways. Front. Pharmacol. 2021, 12, 682602. [Google Scholar] [CrossRef] [PubMed]
- Canetta, S.; Bolkan, S.; Padilla-Coreano, N.; Song, L.J.; Sahn, R.; Harrison, N.L.; Gordon, J.A.; Brown, A.; Kellendonk, C. Maternal immune activation leads to selective functional deficits in offspring parvalbumin interneurons. Mol. Psychiatry 2016, 21, 956–968. [Google Scholar] [CrossRef] [PubMed]
- Khalil, O.S.; Forrest, C.M.; Pisar, M.; Smith, R.A.; Darlington, L.G.; Stone, T.W. Prenatal activation of maternal TLR3 receptors by viral-mimetic poly(I:C) modifies GluN2B expression in embryos and sonic hedgehog in offspring in the absence of kynurenine pathway activation. Immunopharmacol. Immunotoxicol. 2013, 35, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Zavitsanou, K.; Lim, C.K.; Purves-Tyson, T.; Karl, T.; Kassiou, M.; Banister, S.D.; Guillemin, G.J.; Weickert, C.S. Effect of maternal immune activation on the kynurenine pathway in preadolescent rat offspring and on MK801-induced hyperlocomotion in adulthood: Amelioration by COX-2 inhibition. Brain Behav. Immun. 2014, 41, 173–181. [Google Scholar] [CrossRef]
- Forrest, C.M.; Kennedy, P.G.; Rodgers, J.; Dalton, R.N.; Turner, C.; Darlington, L.G.; Cobb, S.R.; Stone, T.W. Kynurenine pathway metabolism following prenatal KMO inhibition and in Mecp2(+/−) mice, using liquid chromatography-tandem mass spectrometry. Neurochem. Int. 2016, 100, 110–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forrest, C.M.; Khalil, O.S.; Pisar, M.; Darlington, L.G.; Stone, T.W. Prenatal inhibition of the tryptophan-kynurenine pathway alters synaptic plasticity and protein expression in the rat hippocampus. Brain Res. 2013, 1504, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Forrest, C.M.; Khalil, O.S.; Pisar, M.; McNair, K.; Kornisiuk, E.; Snitcofsky, M.; Gonzalez, N.; Jerusalinsky, D.; Darlington, L.G.; Stone, T.W. Changes in synaptic transmission and protein expression in the brains of adult offspring after prenatal inhibition of the kynurenine pathway. Neuroscience 2013, 254, 241–259. [Google Scholar] [CrossRef] [Green Version]
- Forrest, C.M.; McNair, K.; Pisar, M.; Khalil, O.S.; Darlington, L.G.; Stone, T.W. Altered hippocampal plasticity by prenatal kynurenine administration, kynurenine-3-monoxygenase (KMO) deletion or galantamine. Neuroscience 2015, 310, 91–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pershing, M.L.; Phenis, D.; Valentini, V.; Pocivavsek, A.; Lindquist, D.H.; Schwarcz, R.; Bruno, J.P. Prenatal kynurenine exposure in rats: Age-dependent changes in NMDA receptor expression and conditioned fear responding. Psychopharmacology 2016, 233, 3725–3735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rentschler, K.M.; Baratta, A.M.; Ditty, A.L.; Wagner, N.T.J.; Wright, C.J.; Milosavljevic, S.; Mong, J.A.; Pocivavsek, A. Prenatal Kynurenine Elevation Elicits Sex-Dependent Changes in Sleep and Arousal During Adulthood: Implications for Psychotic Disorders. Schizophr. Bull. 2021, 47, 1320–1330. [Google Scholar] [CrossRef]
- Wright, C.J.; Rentschler, K.M.; Wagner, N.T.J.; Lewis, A.M.; Beggiato, S.; Pocivavsek, A. Time of Day-Dependent Alterations in Hippocampal Kynurenic Acid, Glutamate, and GABA in Adult Rats Exposed to Elevated Kynurenic Acid During Neurodevelopment. Front. Psychiatry 2021, 12, 734984. [Google Scholar] [CrossRef] [PubMed]
- Pocivavsek, A.; Wu, H.Q.; Elmer, G.I.; Bruno, J.P.; Schwarcz, R. Pre- and postnatal exposure to kynurenine causes cognitive deficits in adulthood. Eur. J. Neurosci. 2012, 35, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Alexander, K.S.; Pocivavsek, A.; Wu, H.Q.; Pershing, M.L.; Schwarcz, R.; Bruno, J.P. Early developmental elevations of brain kynurenic acid impair cognitive flexibility in adults: Reversal with galantamine. Neuroscience 2013, 238, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Iaccarino, H.F.; Suckow, R.F.; Xie, S.; Bucci, D.J. The effect of transient increases in kynurenic acid and quinolinic acid levels early in life on behavior in adulthood: Implications for schizophrenia. Schizophr. Res. 2013, 150, 392–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.C.; Holtze, M.; Powell, S.B.; Terrando, N.; Larsson, M.K.; Persson, A.; Olsson, S.K.; Orhan, F.; Kegel, M.; Asp, L.; et al. Behavioral disturbances in adult mice following neonatal virus infection or kynurenine treatment--role of brain kynurenic acid. Brain Behav. Immun. 2014, 36, 80–89. [Google Scholar] [CrossRef] [Green Version]
- Tufvesson-Alm, M.; Schwieler, L.; Schwarcz, R.; Goiny, M.; Erhardt, S.; Engberg, G. Importance of kynurenine 3-monooxygenase for spontaneous firing and pharmacological responses of midbrain dopamine neurons: Relevance for schizophrenia. Neuropharmacology 2018, 138, 130–139. [Google Scholar] [CrossRef]
- Alkondon, M.; Pereira, E.F.; Yu, P.; Arruda, E.Z.; Almeida, L.E.; Guidetti, P.; Fawcett, W.P.; Sapko, M.T.; Randall, W.R.; Schwarcz, R.; et al. Targeted deletion of the kynurenine aminotransferase ii gene reveals a critical role of endogenous kynurenic acid in the regulation of synaptic transmission via alpha7 nicotinic receptors in the hippocampus. J. Neurosci. 2004, 24, 4635–4648. [Google Scholar] [CrossRef] [Green Version]
- Sapko, M.T.; Guidetti, P.; Yu, P.; Tagle, D.A.; Pellicciari, R.; Schwarcz, R. Endogenous kynurenate controls the vulnerability of striatal neurons to quinolinate: Implications for Huntington’s disease. Exp. Neurol. 2006, 197, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Meyza, K.Z.; Blanchard, D.C. The BTBR mouse model of idiopathic autism—Current view on mechanisms. Neurosci. Biobehav. Rev. 2017, 76, 99–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akaba, Y.; Shiohama, T.; Komaki, Y.; Seki, F.; Ortug, A.; Sawada, D.; Uchida, W.; Kamagata, K.; Shimoji, K.; Aoki, S.; et al. Comprehensive Volumetric Analysis of Mecp2-Null Mouse Model for Rett Syndrome by T2-Weighted 3D Magnetic Resonance Imaging. Front. Neurosci. 2022, 16, 885335. [Google Scholar] [CrossRef] [PubMed]
- Almulla, A.F.; Vasupanrajit, A.; Tunvirachaisakul, C.; Al-Hakeim, H.K.; Solmi, M.; Verkerk, R.; Maes, M. The tryptophan catabolite or kynurenine pathway in schizophrenia: Meta-analysis reveals dissociations between central, serum, and plasma compartments. Mol. Psychiatry 2022, 27, 3679–3691. [Google Scholar] [CrossRef] [PubMed]
- Giorgini, F.; Huang, S.Y.; Sathyasaikumar, K.V.; Notarangelo, F.M.; Thomas, M.A.; Tararina, M.; Wu, H.Q.; Schwarcz, R.; Muchowski, P.J. Targeted deletion of kynurenine 3-monooxygenase in mice: A new tool for studying kynurenine pathway metabolism in periphery and brain. J. Biol. Chem. 2013, 288, 36554–36566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, A.K.; Wing, E.E.; Banks, W.A.; Dantzer, R. Leucine competes with kynurenine for blood-to-brain transport and prevents lipopolysaccharide-induced depression-like behavior in mice. Mol. Psychiatry 2019, 24, 1523–1532. [Google Scholar] [CrossRef]
- Amori, L.; Wu, H.Q.; Marinozzi, M.; Pellicciari, R.; Guidetti, P.; Schwarcz, R. Specific inhibition of kynurenate synthesis enhances extracellular dopamine levels in the rodent striatum. Neuroscience 2009, 159, 196–203. [Google Scholar] [CrossRef] [Green Version]
- Linderholm, K.R.; Alm, M.T.; Larsson, M.K.; Olsson, S.K.; Goiny, M.; Hajos, M.; Erhardt, S.; Engberg, G. Inhibition of kynurenine aminotransferase II reduces activity of midbrain dopamine neurons. Neuropharmacology 2016, 102, 42–47. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Age (Mean ± SD, Range) | Country | Specimen | Diagnostic Criteria | Detection Method | Observation | Ref. |
|---|---|---|---|---|---|---|
| 11.2 ± 2.02 y | Norway | Serum | ADI-R, ADOS, ICD-10 | HPLC | Kyna (⇈), QUIN (⇈) | [1] [207] |
| 8.47 ± 2.36 y, 3-10 y | Australia | DSM-IV, CARS | UHPLC | Kyn (⇈), QUIN (⇈) | [8] | |
| 35.5 ± 9.9 m | Turkey | DSM-5, CARS | HPLC | 3-HK (⇈), Kyna (⇈) | [208] | |
| 42.86 ± 11.03 m | China | DSM-IV, ADI-R, ADOS | TMS/MS | Trp (⇊) | [209] | |
| 3.46 ±0.56 y, 2–6 y | China | Plasma | DSM-IV | LC-MS/MS | Trp (⇊) | [210] |
| 4.4 ± 1.7 y | India | DSM-IV, ABC | Reverse-phase HPLC | Trp (⇊) | [211] | |
| 3.22 ± 1.18 y, 1–14 y | China | DSM-5, ADI-R, ADOS | LC-MS/MS | Trp (⇊) | [212] | |
| 10.0 ± 3.1 y, 5–16 y | USA | DSM-IV | LC-MS/MS | Trp (⇊) | [213] | |
| 27.75 ± 6.97 y | Italy | DSM-5,RAADS-14,RRS, WSAS | ELISA | Trp (⇊), Kyna (⇊) | [2] | |
| 4–16 y | Italy | Urine | ADOS | GC-MS | Trp (⇈) | [214] |
| 4–10 y | Poland | DSM-IV | GC-MS | Trp (⇊) | [215] | |
| 4.83 ± 2.40 y, 3–7 y | Italy | DSM-IV, ADI-R, ADOS, CARS | HILIC-UHPLC | Kyn (⇊), Kyna (⇊), QUIN (⇈), 5-HT (⇊) | [216] | |
| 7.06 ± 0.96 y | Italy | DSM-IV, DSM-5, ADI-R, ADOS | UHPLC | Kyn (⇊) | [217] | |
| 3–6 y | Armenia | ADI-R, ADOS, DSM-IV | LC-MS/MS | QUIN (⇈) | [218] | |
| 6.1 y, 33 m-10 y | USA | CSF | DSM-IV, ADI-R | GC-MS | QUIN (⇊) | [219] |
| Study Model | Species | Changes of Kyn Metabolites in Offspring | Behavioral Changes | Effect on Brain | Ref. |
|---|---|---|---|---|---|
| Poly(I:C) i.p. at E9.0–9.5 | Mice | Trp (⇊), Kyn (⇊), QUIN (⇈), KMO (⇈), 5-HT (⇊), QUIN/Kyna ratio (⇈) after PND80 | Impaired working memory Increased anxiety-like behavior Impaired attentional set shifting after PND80 | 5-HT2A (⇈), SERT (⇈), mGlu2 (⇊) Functional GABAergic transmission (⇊) Decreased PV+ interneuron transmission after PND80 | [236] [237] [238] |
| Poly(I:C) i.p. at E14, 16, 18 | Rat | Kyn (↑) 5 h after injection | GluN2A (↓), GluN2B (⇈), DCX (⇊) 5 h after injection | [239] | |
| Poly(I:C) i.p. at E9.0–9.5 | Mice | Trp (⇊), Kyn (⇊), QUIN (⇈), KMO (⇈), 5-HT (⇊), QUIN/Kyna ratio (⇈) after PND80 | Impaired working memory Increased anxiety-like behavior Impaired attentional set shifting after PND80 | 5-HT2A (⇈), SERT (⇈), mGlu2 (⇊) Functional GABAergic transmission (⇊) Decreased PV+ interneuron transmission after PND80 | [240] |
| KMO inhibitor i.p. at E14, 16,18 | Mice | Kyn (⇈), Kyna (⇈) 5 and 24 h after injection | [241] | ||
| KMO inhibitor i.p. at E14, 16, 18 | Rat | Kyn (⇈), Kyna (⇈) 5 and 24 h after injection | Does not affect the initial acquisition nor the subsequent memory consolidation at PND60 | GluN2A (⇈), GluN2B (⇈), PSD-95 (⇈) Increased LTP at PND21 GluN2A (⇊), GluN2B (↓), PSD-95 (↓), VGLUT-1(⇈), VGLUT-2 (⇈), Decreased LTP and basal spine densities in CA1 at PND60 | [130] [242] [243] |
| IL-17a high expression at E12.5-PND0 Kyn i.p. at E12.5–19 | Mice | Kyn (⇈), Kyna (⇈) Kyn/Trp ratio (⇈) at E18.5 TRP (⇊), Kyn/Trp ratio (⇈) at PND77 | Impaired cognitive function Lower social behavior and social cognition Depressive-like behavior at PND 56-70 | [3] | |
| Kyn i.p. at E14,16,18 | Rat | GluN1 (↑), GluN2A (↑), GluN2B (↑) Decreased LPT | [244] | ||
| Fed high Kyn diet at E15–22 | Rat | Kyn (⇈), Kyna (⇈) at E22 Kyna (⇈) at PND56 | Impaired spatial learning, reference memory, and contextual memory Impaired attentional set-shifting at PND 56-85 Reduced rapid eye movement sleep at PND85 | mGluR2 (⇊) at E21 mChrna7 (⇊) at PND2 Decreased dendritic spine density at PND56-80 mGrin1 (⇊), mGrin2a (⇊) at PND70 Time- and sex-dependent alterations in the levels of GABA and glutamate at PND56 | [5] [23] [245] [246] [247] |
| Fed high Kyn diet at E15–PND21 | Rat | Kyn (⇈), Kyna (⇈), 3-HK (⇈) at PND21 | Impaired spatial learning, reference memory, and contextual memory Impaired attentional set-shifting at PND56 | Decreased extracellular glutamate | [248] [249] |
| Kyn i.p. at PND7–10 | Rat | Kyna (⇈), QUIN (⇈) at PND10 Kyna (↑), QUIN (↑) at PND70 | Impaired social behavior Decreased locomotor activity | [250] | |
| Kyn i.p. at PND7–16 | Mice | Kyna (⇈) at PND16 | Decreased amphetamine-induced locomotor activity Impaired PPI, working memory | Enhanced amphetamine-induced dopamine release | [251] |
| KMO gene- deficit | Mice | Kyn (⇈), Kyna (⇈) | Impaired contextual memory Decreased social interaction Increased anxiety- and depressive-like behavior Increased amphetamine-induced locomotor activity | Increased spontaneous ventral tegmental area dopamine neuron activity Decreased LPT | [138] [139] [244] [252] |
| KAT II gene-deficit | Mice | Kyna (⇊), QUIN(↓) at PND14 and 21 No significant changes at PND60 | Increased performance in object exploration and recognition tasks, passive avoidance test, spatial discrimination test, and spontaneous locomotor activity at PND21 | Increased LPT Increased extracellular glutamate concentrations and endogenous α7nAChR activity | [183] [253] [254] |
| BTBR T+Itpr3tf/J | Mice | Kyna (⇈) | Display autism-relevant behaviors | Absence of the corpus Severely reduced hippocampal commissure | [165] [255] |
| Mecp2+/− | Mice | No significant changes | Exhibit hindlimb clasping and uneven breathing Model of human Rett Syndrome | Reduced in global and all local volumes in the brain | [241] [256] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murakami, Y.; Imamura, Y.; Kasahara, Y.; Yoshida, C.; Momono, Y.; Fang, K.; Sakai, D.; Konishi, Y.; Nishiyama, T. Maternal Inflammation with Elevated Kynurenine Metabolites Is Related to the Risk of Abnormal Brain Development and Behavioral Changes in Autism Spectrum Disorder. Cells 2023, 12, 1087. https://doi.org/10.3390/cells12071087
Murakami Y, Imamura Y, Kasahara Y, Yoshida C, Momono Y, Fang K, Sakai D, Konishi Y, Nishiyama T. Maternal Inflammation with Elevated Kynurenine Metabolites Is Related to the Risk of Abnormal Brain Development and Behavioral Changes in Autism Spectrum Disorder. Cells. 2023; 12(7):1087. https://doi.org/10.3390/cells12071087
Chicago/Turabian StyleMurakami, Yuki, Yukio Imamura, Yoshiyuki Kasahara, Chihiro Yoshida, Yuta Momono, Ke Fang, Daisuke Sakai, Yukuo Konishi, and Toshimasa Nishiyama. 2023. "Maternal Inflammation with Elevated Kynurenine Metabolites Is Related to the Risk of Abnormal Brain Development and Behavioral Changes in Autism Spectrum Disorder" Cells 12, no. 7: 1087. https://doi.org/10.3390/cells12071087