

Prolonged Primary Rhinovirus Infection of Human Nasal Epithelial Cells Diminishes the Viral Load of Secondary Influenza H3N2 Infection via the Antiviral State Mediated by RIG-I and Interferon-Stimulated Genes

 , , and
, , and 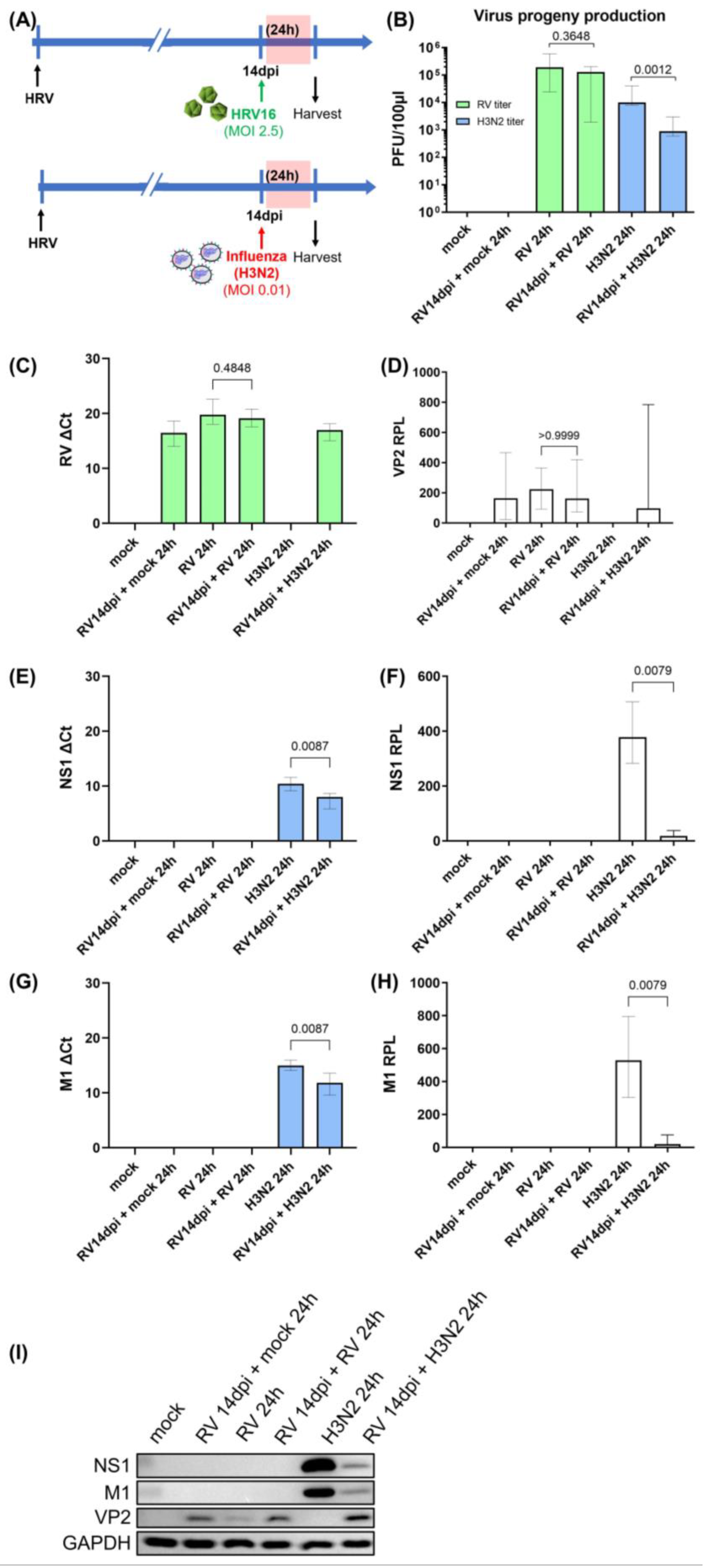{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Derivation of hNESPCs and In Vitro Differentiation of hNECs
2.2. HRV16 and H3N2 Inoculation into Fully Differentiated hNECs and Viral Plaque Assay
2.3. Cell Viability Assay
2.4. RNA Extraction and Quantitative Real-Time PCR for Host Immune Markers and HRV Viral Load
2.5. H3N2 RT-PCR Amplification and Quantitative Real-Time PCR for H3N2 Viral Load
2.6. Western Blotting
2.7. Statistical Analyses
3. Results
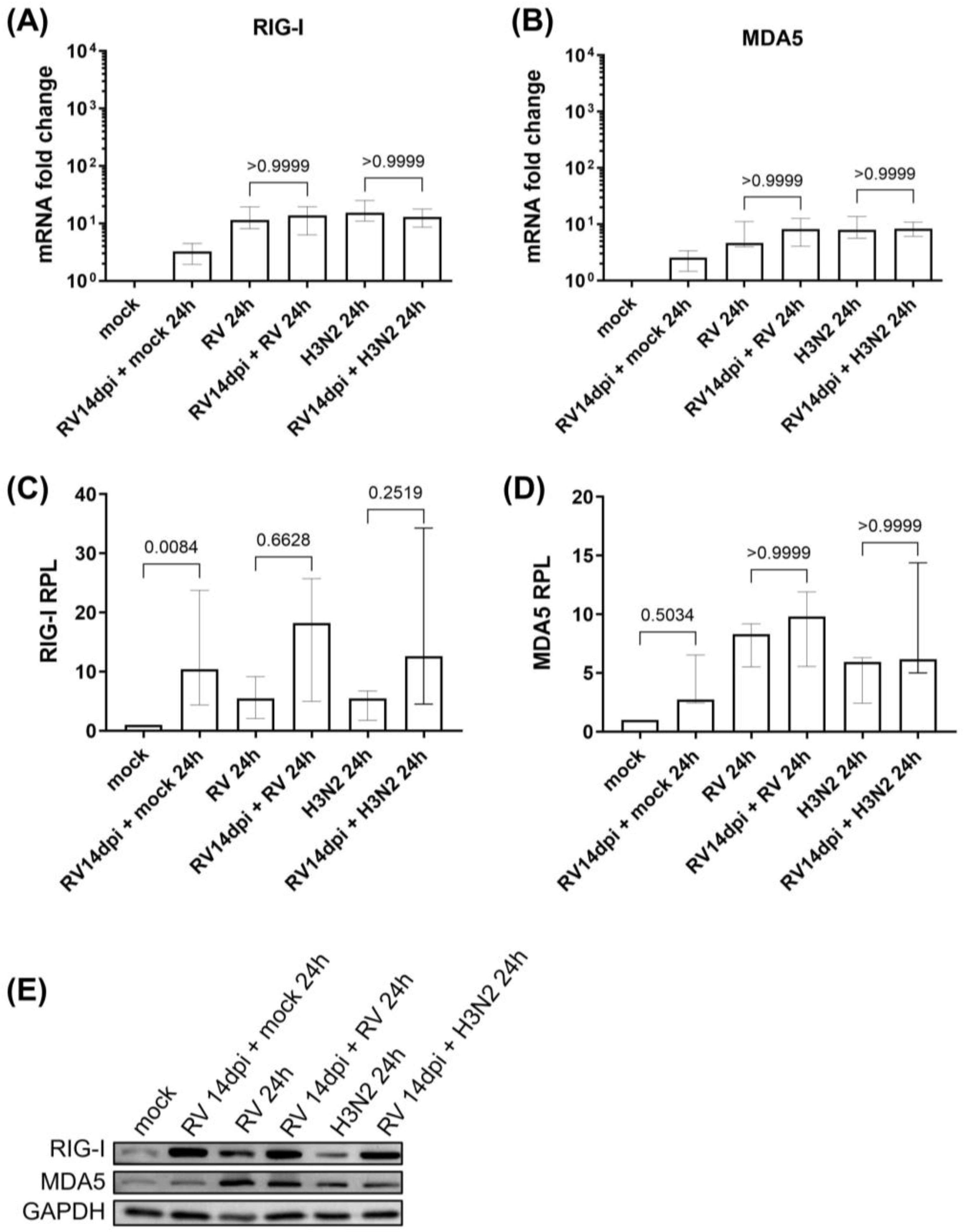
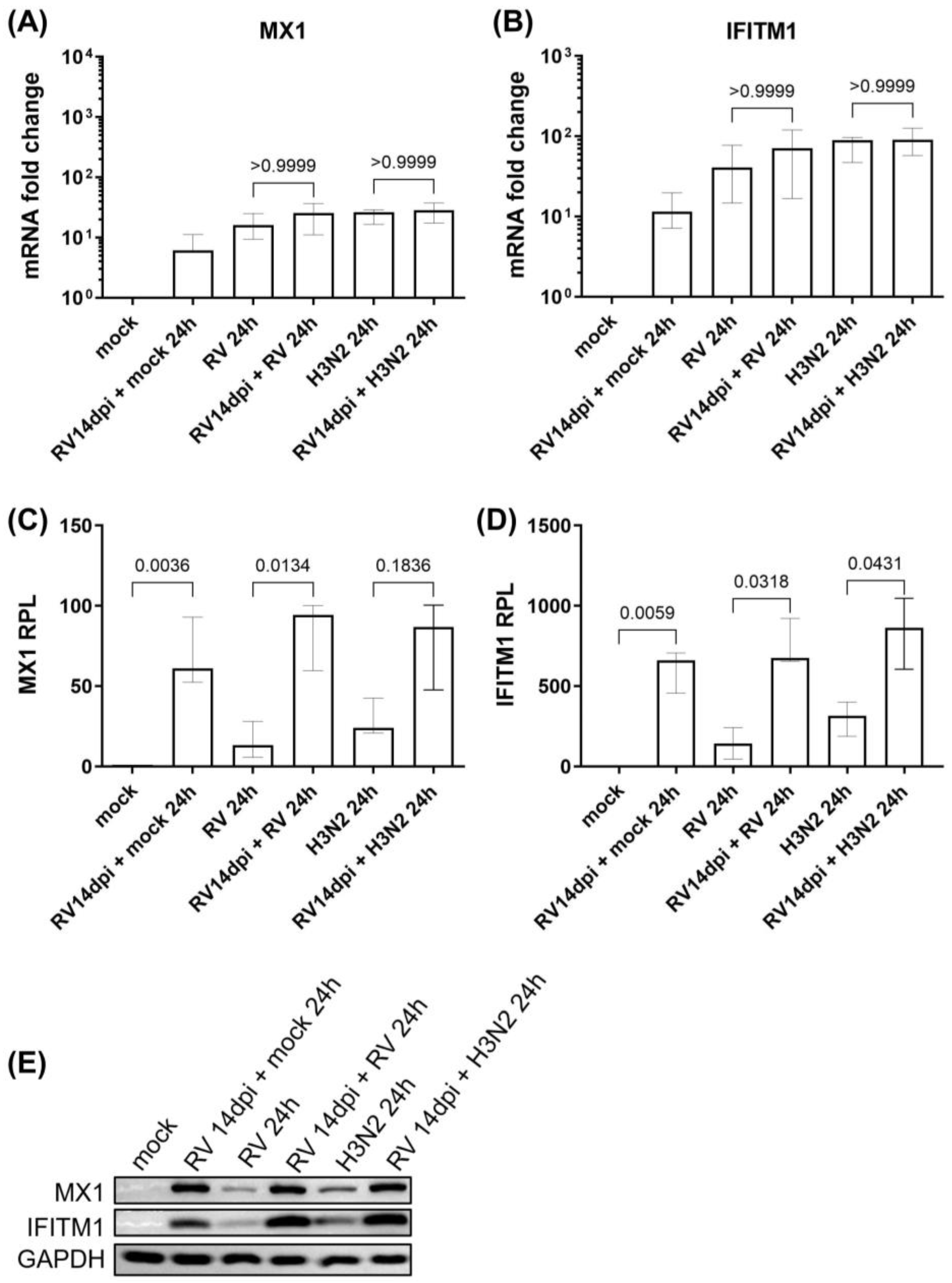
3.1. Viral Replication Dynamics and Activation of Innate Immune Responses during Secondary H3N2 Infection and HRV-A16 Re-Infection in hNECs
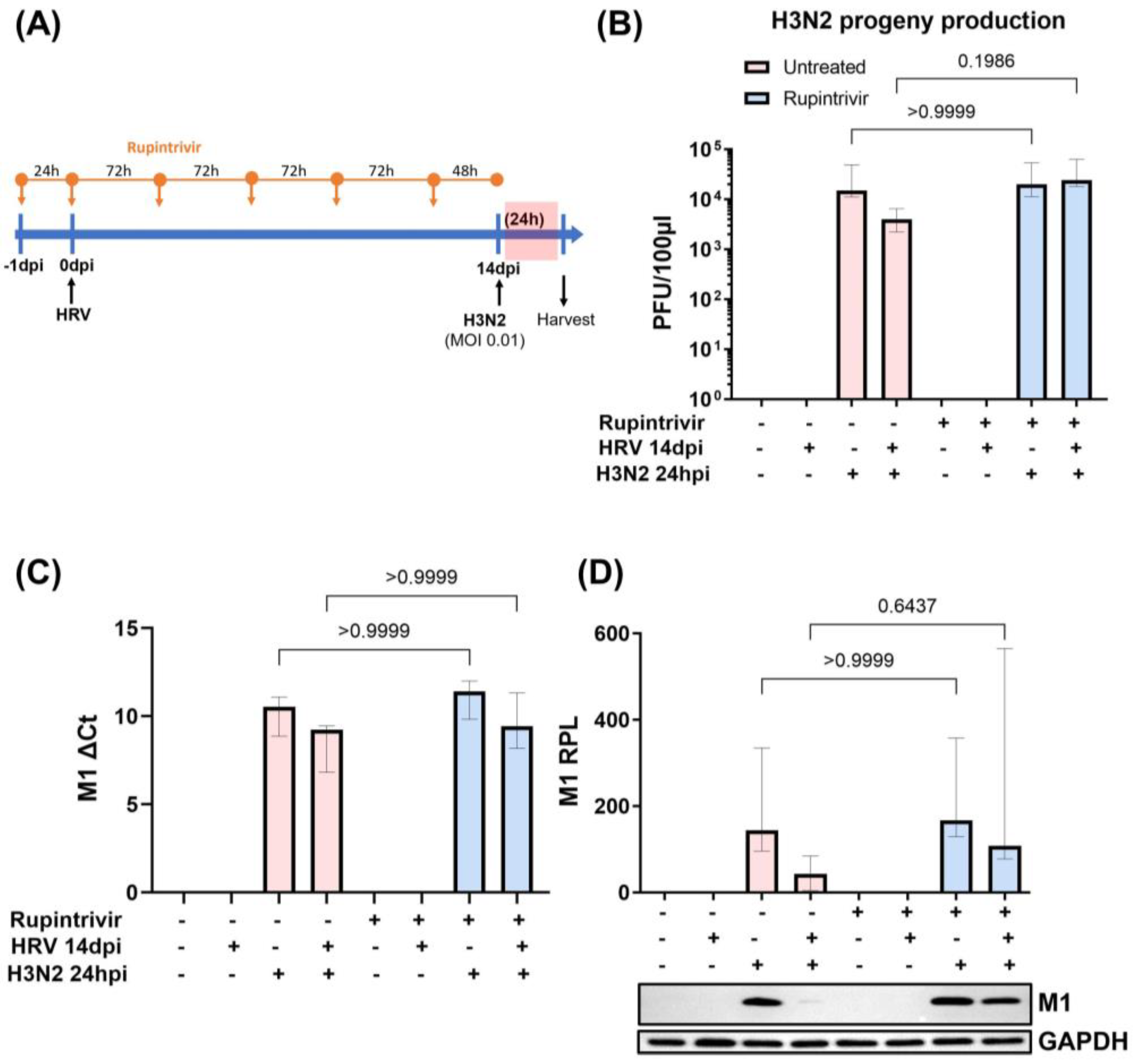
3.2. Viral Replication Dynamics and Activation of Innate Immune Responses after Longer Rupintrivir Treatment Prior to Secondary H3N2 Infection of hNECs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zlateva, K.T.; de Vries, J.J.C.; Coenjaerts, F.E.J.; van Loon, A.M.; Verheij, T.; Little, P.; Butler, C.C.; Goossens, H.; Ieven, M.; Claas, E.C.J. Prolonged shedding of rhinovirus and re-infection in adults with respiratory tract illness. Eur. Respir. J. 2014, 44, 169–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamborn, I.T.; Jing, H.; Zhang, Y.; Drutman, S.B.; Abbott, J.K.; Munir, S.; Bade, S.; Murdock, H.M.; Santos, C.P.; Brock, L.G.; et al. Recurrent rhinovirus infections in a child with inherited MDA5 deficiency. J. Exp. Med. 2017, 214, 1949–1972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelmann, I.; Mordacq, C.; Gosset, P.; Tillie-Leblond, I.; Dewilde, A.; Thumerelle, C.; Pouessel, G.; Deschildre, A. Rhinovirus and asthma: Reinfection, not persistence. Am. J. Respir. Crit. Care Med. 2013, 188, 1165–1167. [Google Scholar] [CrossRef]
- Linsuwanon, P.; Payungporn, S.; Samransamruajkit, R.; Theamboonlers, A.; Poovorawan, Y. Recurrent human rhinovirus infections in infants with refractory wheezing. Emerg. Infect. Dis. 2009, 15, 978–980. [Google Scholar] [CrossRef] [PubMed]
- Jackson, D.J.; Gangnon, R.E.; Evans, M.D.; Roberg, K.A.; Anderson, E.L.; Pappas, T.E.; Printz, M.C.; Lee, W.M.; Shult, P.A.; Reisdorf, E.; et al. Wheezing rhinovirus illnesses in early life predict asthma development in high-risk children. Am. J. Respir. Crit. Care Med. 2008, 178, 667–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusel, M.M.; de Klerk, N.H.; Holt, P.G.; Kebadze, T.; Johnston, S.L.; Sly, P.D. Role of respiratory viruses in acute upper and lower respiratory tract illness in the first year of life: A birth cohort study. Pediatr. Infect. Dis. J. 2006, 25, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Lemanske, R.F., Jr.; Jackson, D.J.; Gangnon, R.E.; Evans, M.D.; Li, Z.; Shult, P.A.; Kirk, C.J.; Reisdorf, E.; Roberg, K.A.; Anderson, E.L.; et al. Rhinovirus illnesses during infancy predict subsequent childhood wheezing. J. Allergy Clin. Immunol. 2005, 116, 571–577. [Google Scholar] [CrossRef]
- Wishaupt, J.O.; van der Ploeg, T.; de Groot, R.; Versteegh, F.G.A.; Hartwig, N.G. Single- and multiple viral respiratory infections in children: Disease and management cannot be related to a specific pathogen. BMC Infect. Dis. 2017, 17, 62. [Google Scholar] [CrossRef] [Green Version]
- Adam, K.; Pangesti, K.N.; Setiawaty, V. Multiple viral infection detected from influenza-like illness cases in Indonesia. BioMed Res. Int. 2017, 2017, 9541619. [Google Scholar] [CrossRef] [Green Version]
- Krammer, F.; Smith, G.J.D.; Fouchier, R.A.M.; Peiris, M.; Kedzierska, K.; Doherty, P.C.; Palese, P.; Shaw, M.L.; Treanor, J.; Webster, R.G.; et al. Influenza. Nat. Rev. Dis. Primers 2018, 4, 3. [Google Scholar] [CrossRef]
- Ånestad, G.; Nordbø, S.A. Virus interference. Did rhinoviruses activity hamper the progress of the 2009 influenza A (H1N1) pandemic in Norway? Med. Hypotheses 2011, 77, 1132–1134. [Google Scholar] [CrossRef] [PubMed]
- Linde, A.; Rotzén-Ostlund, M.; Zweygberg-Wirgart, B.; Rubinova, S.; Brytting, M. Does viral interference affect spread of influenza? Euro Surveill. 2009, 14, 19354. [Google Scholar] [CrossRef] [PubMed]
- Casalegno, J.S.; Ottmann, M.; Duchamp, M.B.; Escuret, V.; Billaud, G.; Frobert, E.; Morfin, F.; Lina, B. Rhinoviruses delayed the circulation of the pandemic influenza A (H1N1) 2009 virus in France. Clin. Microbiol. Infect. 2010, 16, 326–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.; Song, Z.; Li, Y.; Zhang, J.; Wang, X.L. Possible interference between seasonal epidemics of influenza and other respiratory viruses in Hong Kong, 2014–2017. BMC Infect. Dis. 2017, 17, 772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, A.; Mihaylova, V.T.; Landry, M.L.; Foxman, E.F. Interference between rhinovirus and influenza A virus: A clinical data analysis and experimental infection study. Lancet Microbe 2020, 1, e254–e262. [Google Scholar] [CrossRef] [PubMed]
- Nickbakhsh, S.; Mair, C.; Matthews, L.; Reeve, R.; Johnson Paul, C.D.; Thorburn, F.; von Wissmann, B.; Reynolds, A.; McMenamin, J.; Gunson Rory, N.; et al. Virus–virus interactions impact the population dynamics of influenza and the common cold. Proc. Natl. Acad. Sci. USA 2019, 116, 27142–27150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, K.P.; Chong, M.K.C.; Chan, K.Y.Y.; Pun, J.C.S.; Tsun, J.G.S.; Chow, S.M.W.; Ng, C.S.H.; Wang, M.H.T.; Chan, P.K.S.; Li, A.M.; et al. Suppression of influenza virus infection by rhinovirus interference—At the population, individual and cellular levels. Curr. Res. Microb. Sci. 2022, 3, 100147. [Google Scholar] [PubMed]
- Peltola, V.; Waris, M.; Kainulainen, L.; Kero, J.; Ruuskanen, O. Virus shedding after human rhinovirus infection in children, adults and patients with hypogammaglobulinaemia. Clin. Microbiol. Infect. 2013, 19, E322–E327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sansone, M.; Andersson, M.; Brittain-Long, R.; Andersson, L.M.; Olofsson, S.; Westin, J.; Lindh, M. Rhinovirus infections in western Sweden: A four-year molecular epidemiology study comparing local and globally appearing types. Eur. J. Clin. Microbiol. Infect. Dis. 2013, 32, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, E.C.; Pedersen, A.B.; Fenton, A.; Petchey, O.L. The nature and consequences of coinfection in humans. J. Infect. 2011, 63, 200–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajput, C.; Han, M.; Ishikawa, T.; Lei, J.; Jazaeri, S.; Bentley, J.K.; Hershenson, M.B. Early-life heterologous rhinovirus infections induce an exaggerated asthma-like phenotype. J. Allergy Clin. Immunol. 2020, 146, 571–582.e3. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.J.; Ijezie, E.C.; Balemba, O.B.; Miura, T.A. Attenuation of influenza A virus disease severity by viral coinfection in a mouse model. J. Virol. 2018, 92, e00881-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, N.; Sharma, S.; Barua, S.; Tripathi, B.N.; Rouse, B.T. Virological and immunological cutcomes of coinfections. Clin. Microbiol. Rev. 2018, 31, e00111-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, H.H.; Andiappan, A.K.; Duan, K.; Lum, J.; Liu, J.; Tan, K.S.; Howland, S.; Lee, B.; Ong, Y.K.; Thong, M.; et al. Transcriptomics of rhinovirus persistence reveals sustained expression of RIG-I and interferon-stimulated genes in nasal epithelial cells in vitro. Allergy 2022, 77, 2778–2793. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Tan, K.S.; Li, C.; Tran, T.; Chao, S.S.; Sugrue, R.J.; Shi, L.; Chow, V.T.; Wang, D.Y. Human nasal epithelial cells derived from multiple subjects exhibit differential responses to H3N2 influenza virus infection in vitro. J. Allergy Clin. Immunol. 2016, 138, 276–281.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, K.S.; Ong, H.H.; Yan, Y.; Liu, J.; Li, C.; Ong, Y.K.; Thong, K.T.; Choi, H.W.; Wang, D.Y.; Chow, V.T. In vitro model of fully differentiated human nasal epithelial cells infected with rhinovirus reveals epithelium-initiated immune responses. J. Infect. Dis. 2018, 217, 906–915. [Google Scholar] [CrossRef] [Green Version]
- Geib, T.; Sauder, C.; Venturelli, S.; Hässler, C.; Staeheli, P.; Schwemmle, M. Selective virus resistance conferred by expression of Borna disease virus nucleocapsid components. J. Virol. 2003, 77, 4283–4290. [Google Scholar] [CrossRef] [Green Version]
- Brindley, M.A.; Zhang, B.; Montelaro, R.C.; Maury, W. An equine infectious anemia virus variant superinfects cells through novel receptor interactions. J. Virol. 2008, 82, 9425–9432. [Google Scholar] [CrossRef] [Green Version]
- Zebovitz, E.; Brown, A. Interference among group A arboviruses. J. Virol. 1968, 2, 1283–1289. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Wei, T.; Abbott, C.M.; Harrich, D. The unexpected roles of eukaryotic translation elongation factors in RNA virus replication and pathogenesis. Microbiol. Mol. Biol. Rev. 2013, 77, 253–266. [Google Scholar] [CrossRef] [Green Version]
- Gomila, R.C.; Martin, G.W.; Gehrke, L. NF90 binds the dengue virus RNA 3′ terminus and is a positive regulator of dengue virus replication. PLoS ONE 2011, 6, e16687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salas-Benito, J.S.; De Nova-Ocampo, M. Viral interference and persistence in mosquito-borne flaviviruses. J. Immunol. Res. 2015, 2015, 873404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galligan, C.L.; Murooka, T.T.; Rahbar, R.; Baig, E.; Majchrzak-Kita, B.; Fish, E.N. Interferons and viruses: Signaling for supremacy. Immunol. Res. 2006, 35, 27–40. [Google Scholar] [CrossRef] [PubMed]
- García, M.A.; Meurs, E.F.; Esteban, M. The dsRNA protein kinase PKR: Virus and cell control. Biochimie 2007, 89, 799–811. [Google Scholar] [CrossRef]
- Schoggins, J.W.; Rice, C.M. Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol. 2011, 1, 519–525. [Google Scholar] [CrossRef]
- Yang, E.; Li, M.M.H. All about the RNA: Interferon-stimulated genes that interfere with viral RNA processes. Front. Immunol. 2020, 11, 605024. [Google Scholar] [CrossRef]
- Wong, M.-T.; Chen, S.S.L. Emerging roles of interferon-stimulated genes in the innate immune response to hepatitis C virus infection. Cell. Mol. Immunol. 2016, 13, 11–35. [Google Scholar] [CrossRef] [Green Version]
- Schoggins, J.W. Interferon-stimulated genes: What do they all do? Annu. Rev. Virol. 2019, 6, 567–584. [Google Scholar] [CrossRef]
- Kamau, E.; Onyango, C.O.; Otieno, G.P.; Kiyuka, P.K.; Agoti, C.N.; Medley, G.F.; Cane, P.A.; Nokes, D.J.; Munywoki, P.K. An intensive, active surveillance reveals continuous invasion and high diversity of rhinovirus in households. J. Infect. Dis. 2019, 219, 1049–1057. [Google Scholar] [CrossRef]
- Alper, C.M.; Doyle, W.J.; Skoner, D.P.; Buchman, C.A.; Cohen, S.; Gwaltney, J.M. Prechallenge antibodies moderate disease expression in adults experimentally exposed to rhinovirus strain hanks. Clin. Infect. Dis. 1998, 27, 119–128. [Google Scholar] [CrossRef] [Green Version]
- Heymann, P.W.; Nguyen, H.-T.; Steinke, J.W.; Turner, R.B.; Woodfolk, J.A.; Platts-Mills, T.A.E.; Martin, L.; He, H.; Biagini Myers, J.; Lindsey, M.; et al. Rhinovirus infection results in stronger and more persistent genomic dysregulation: Evidence for altered innate immune response in asthmatics at baseline, early in infection, and during convalescence. PLoS ONE 2017, 12, e0178096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravi, A.; Chang, M.; van de Pol, M.; Yang, S.; Aliprantis, A.; Thornton, B.; Carayannopoulos, L.N.; Bautmans, A.; Robberechts, M.; De Lepeleire, I.; et al. Rhinovirus-16 induced temporal interferon responses in nasal epithelium links with viral clearance and symptoms. Clin. Exp. Allergy 2019, 49, 1587–1597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravi, A.; Koster, J.; Dijkhuis, A.; Bal, S.M.; Sabogal Piñeros, Y.S.; Bonta, P.I.; Majoor, C.J.; Sterk, P.J.; Lutter, R. Interferon-induced epithelial response to rhinovirus 16 in asthma relates to inflammation and FEV1. J. Allergy Clin. Immunol. 2019, 143, 442–447.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoneyama, M.; Kikuchi, M.; Matsumoto, K.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Foy, E.; Loo, Y.M.; Gale, M., Jr.; Akira, S.; et al. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol. 2005, 175, 2851–2858. [Google Scholar] [CrossRef] [Green Version]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef]
- Kato, H.; Takeuchi, O.; Mikamo-Satoh, E.; Hirai, R.; Kawai, T.; Matsushita, K.; Hiiragi, A.; Dermody, T.S.; Fujita, T.; Akira, S. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 2008, 205, 1601–1610. [Google Scholar] [CrossRef] [Green Version]
- Cui, S.; Eisenächer, K.; Kirchhofer, A.; Brzózka, K.; Lammens, A.; Lammens, K.; Fujita, T.; Conzelmann, K.K.; Krug, A.; Hopfner, K.P. The C-terminal regulatory domain is the RNA 5′-triphosphate sensor of RIG-I. Mol. Cell 2008, 29, 169–179. [Google Scholar] [CrossRef] [Green Version]
- Schlee, M.; Roth, A.; Hornung, V.; Hagmann, C.A.; Wimmenauer, V.; Barchet, W.; Coch, C.; Janke, M.; Mihailovic, A.; Wardle, G.; et al. Recognition of 5′ triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity 2009, 31, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ludwig, J.; Schuberth, C.; Goldeck, M.; Schlee, M.; Li, H.; Juranek, S.; Sheng, G.; Micura, R.; Tuschl, T.; et al. Structural and functional insights into 5′-ppp RNA pattern recognition by the innate immune receptor RIG-I. Nat. Struct. Mol. Biol. 2010, 17, 781–787. [Google Scholar] [CrossRef] [Green Version]
- Feng, Q.; Hato, S.V.; Langereis, M.A.; Zoll, J.; Virgen-Slane, R.; Peisley, A.; Hur, S.; Semler, B.L.; van Rij, R.P.; van Kuppeveld, F.J. MDA5 detects the double-stranded RNA replicative form in picornavirus-infected cells. Cell Rep. 2012, 2, 1187–1196. [Google Scholar] [CrossRef] [Green Version]
- Slater, L.; Bartlett, N.W.; Haas, J.J.; Zhu, J.; Message, S.D.; Walton, R.P.; Sykes, A.; Dahdaleh, S.; Clarke, D.L.; Belvisi, M.G.; et al. Co-ordinated role of TLR3, RIG-I and MDA5 in the innate response to rhinovirus in bronchial epithelium. PLoS Pathog. 2010, 6, e1001178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warner, S.M.; Wiehler, S.; Michi, A.N.; Proud, D. Rhinovirus replication and innate immunity in highly differentiated human airway epithelial cells. Respir. Res. 2019, 20, 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pallansch, M.A.; Kew, O.M.; Palmenberg, A.C.; Golini, F.; Wimmer, E.; Rueckert, R.R. Picornaviral VPg sequences are contained in the replicase precursor. J. Virol. 1980, 35, 414–419. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.K.; Kim, H.E.; Park, E.B.; Lee, J.; Kim, K.H.; Lim, K.; Yum, S.; Lee, Y.H.; Kang, S.J.; Lee, J.H.; et al. Structural features of influenza A virus panhandle RNA enabling the activation of RIG-I independently of 5′-triphosphate. Nucleic Acids Res. 2016, 44, 8407–8416. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Sediri, H.; Felgenhauer, U.; Binzen, I.; Bänfer, S.; Jacob, R.; Brunotte, L.; García-Sastre, A.; Schmid-Burgk, J.L.; Schmidt, T.; et al. Influenza virus adaptation PB2-627K modulates nucleocapsid inhibition by the pathogen sensor RIG-I. Cell Host Microbe 2015, 17, 309–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opitz, B.; Rejaibi, A.; Dauber, B.; Eckhard, J.; Vinzing, M.; Schmeck, B.; Hippenstiel, S.; Suttorp, N.; Wolff, T. IFNβ induction by influenza A virus is mediated by RIG-I which is regulated by the viral NS1 protein. Cell. Microbiol. 2007, 9, 930–938. [Google Scholar] [CrossRef]
- Mibayashi, M.; Martínez-Sobrido, L.; Loo, Y.M.; Cárdenas, W.B.; Gale, M., Jr.; García-Sastre, A. Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J. Virol. 2007, 81, 514–524. [Google Scholar] [CrossRef] [Green Version]
- Anastasina, M.; Le May, N.; Bugai, A.; Fu, Y.; Söderholm, S.; Gaelings, L.; Ohman, T.; Tynell, J.; Kyttänen, S.; Barboric, M.; et al. Influenza virus NS1 protein binds cellular DNA to block transcription of antiviral genes. Biochim. Biophys. Acta 2016, 1859, 1440–1448. [Google Scholar] [CrossRef] [Green Version]
- Coch, C.; Stümpel, J.P.; Lilien-Waldau, V.; Wohlleber, D.; Kümmerer, B.M.; Bekeredjian-Ding, I.; Kochs, G.; Garbi, N.; Herberhold, S.; Schuberth-Wagner, C.; et al. RIG-I activation protects and rescues from lethal influenza virus infection and bacterial superinfection. Mol. Ther. 2017, 25, 2093–2103. [Google Scholar] [CrossRef] [Green Version]
- Pavlovic, J.; Haller, O.; Staeheli, P. Human and mouse Mx proteins inhibit different steps of the influenza virus multiplication cycle. J. Virol. 1992, 66, 2564–2569. [Google Scholar] [CrossRef] [Green Version]
- Haller, O.; Kochs, G. Human MxA protein: An interferon-induced dynamin-like GTPase with broad antiviral activity. J. Interferon Cytokine Res. 2011, 31, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Killip, M.J.; Staeheli, P.; Randall, R.E.; Jackson, D. The human interferon-induced MxA protein inhibits early stages of influenza A virus infection by retaining the incoming viral genome in the cytoplasm. J. Virol. 2013, 87, 13053–13058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, J.M.; Gilbert, C.S.; Stark, G.R.; Kerr, I.M. Differential regulation of interferon-induced mRNAs and c-myc mRNA by alpha- and gamma-interferons. Eur. J. Biochem. 1985, 153, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Evans, S.S.; Lee, D.B.; Han, T.; Tomasi, T.B.; Evans, R.L. Monoclonal antibody to the interferon-inducible protein Leu-13 triggers aggregation and inhibits proliferation of leukemic B cells. Blood 1990, 76, 2583–2593. [Google Scholar] [CrossRef] [Green Version]
- Desai, T.M.; Marin, M.; Chin, C.R.; Savidis, G.; Brass, A.L.; Melikyan, G.B. IFITM3 restricts influenza A virus entry by blocking the formation of fusion pores following virus-endosome hemifusion. PLoS Pathog. 2014, 10, e1004048. [Google Scholar] [CrossRef] [Green Version]
- Seipelt, J.; Gurané, A.; Bergmann, E.; James, M.; Sommergruber, W.; Fita, I.; Skern, T. The structures of picornaviral proteinases. Virus Res. 1999, 62, 159–168. [Google Scholar] [CrossRef]
- Pathak, H.B.; Oh, H.S.; Goodfellow, I.G.; Arnold, J.J.; Cameron, C.E. Picornavirus genome replication: Roles of precursor proteins and rate-limiting steps in oriI-dependent VPg uridylylation. J. Biol. Chem. 2008, 283, 30677–30688. [Google Scholar] [CrossRef] [Green Version]
- Pathak, H.B.; Arnold, J.J.; Wiegand, P.N.; Hargittai, M.R.; Cameron, C.E. Picornavirus genome replication: Assembly and organization of the VPg uridylylation ribonucleoprotein (initiation) complex. J. Biol. Chem. 2007, 282, 16202–16213. [Google Scholar] [CrossRef] [Green Version]
- Gamarnik, A.V.; Andino, R. Switch from translation to RNA replication in a positive-stranded RNA virus. Genes Dev. 1998, 12, 2293–2304. [Google Scholar] [CrossRef] [Green Version]
- Weng, K.F.; Li, M.L.; Hung, C.T.; Shih, S.R. Enterovirus 71 3C protease cleaves a novel target CstF-64 and inhibits cellular polyadenylation. PLoS Pathog. 2009, 5, e1000593. [Google Scholar] [CrossRef] [Green Version]
- Lozano, G.; Martínez-Salas, E. Structural insights into viral IRES-dependent translation mechanisms. Curr. Opin. Virol. 2015, 12, 113–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghildyal, R.; Jordan, B.; Li, D.; Dagher, H.; Bardin, P.G.; Gern, J.E.; Jans, D.A. Rhinovirus 3C protease can localize in the nucleus and alter active and passive nucleocytoplasmic transport. J. Virol. 2009, 83, 7349–7352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dragovich, P.S.; Webber, S.E.; Babine, R.E.; Fuhrman, S.A.; Patick, A.K.; Matthews, D.A.; Reich, S.H.; Marakovits, J.T.; Prins, T.J.; Zhou, R.; et al. Structure-based design, synthesis, and biological evaluation of irreversible human rhinovirus 3C protease inhibitors. 2. Peptide structure-activity studies. J. Med. Chem. 1998, 41, 2819–2834. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Li, B.; Yu, Y.; Iyer, S.S.; Sun, M.; Cheng, G. Positive feedback regulation of type I interferon by the interferon-stimulated gene STING. EMBO Rep. 2015, 16, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Touzot, M.; Soumelis, V.; Asselah, T. A dive into the complexity of type I interferon antiviral functions. J. Hepatol. 2012, 56, 726–728. [Google Scholar] [CrossRef] [Green Version]
- Michalska, A.; Blaszczyk, K.; Wesoly, J.; Bluyssen, H.A.R. A positive feedback amplifier circuit that regulates interferon (IFN)-stimulated gene expression and controls type I and type II IFN responses. Front. Immunol. 2018, 9, 1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheon, H.; Holvey-Bates, E.G.; Schoggins, J.W.; Forster, S.; Hertzog, P.; Imanaka, N.; Rice, C.M.; Jackson, M.W.; Junk, D.J.; Stark, G.R. IFNβ-dependent increases in STAT1, STAT2, and IRF9 mediate resistance to viruses and DNA damage. EMBO J. 2013, 32, 2751–2763. [Google Scholar] [CrossRef] [Green Version]
- Morrow, A.N.; Schmeisser, H.; Tsuno, T.; Zoon, K.C. A novel role for IFN-stimulated gene factor 3II in IFN-γ signaling and induction of antiviral activity in human cells. J. Immunol. 2011, 186, 1685–1693. [Google Scholar] [CrossRef] [Green Version]
- Yamauchi, S.; Takeuchi, K.; Chihara, K.; Honjoh, C.; Kato, Y.; Yoshiki, H.; Hotta, H.; Sada, K. STAT1 is essential for the inhibition of hepatitis C virus replication by interferon-λ but not by interferon-α. Sci. Rep. 2016, 6, 38336. [Google Scholar] [CrossRef] [Green Version]
- Kandhaya-Pillai, R.; Miro-Mur, F.; Alijotas-Reig, J.; Tchkonia, T.; Kirkland, J.L.; Schwartz, S. TNFα-senescence initiates a STAT-dependent positive feedback loop, leading to a sustained interferon signature, DNA damage, and cytokine secretion. Aging 2017, 9, 2411–2435. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.J.; Hwang, S.Y.; Imaizumi, T.; Yoo, J.Y. Negative feedback regulation of RIG-I-mediated antiviral signaling by interferon-induced ISG15 conjugation. J. Virol. 2008, 82, 1474–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, K.S.; Teo, D.E.T.; Tan, K.S.; Toh, G.A.; Ong, H.H.; Lim, C.K.; Lay, K.; Au, B.V.; Lew, T.S.; Chu, J.J.H.; et al. Enteroviral 3C protease activates the human NLRP1 inflammasome in airway epithelia. Science 2020, 370, eaay2002. [Google Scholar] [CrossRef] [PubMed]
- Essaidi-Laziosi, M.; Alvarez, C.; Puhach, O.; Sattonnet-Roche, P.; Torriani, G.; Tapparel, C.; Kaiser, L.; Eckerle, I. Sequential infections with rhinovirus and influenza modulate the replicative capacity of SARS-CoV-2 in the upper respiratory tract. Emerg. Microbes Infect. 2022, 11, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Luukkainen, A.; Puan, K.J.; Yusof, N.; Lee, B.; Tan, K.S.; Liu, J.; Yan, Y.; Toppila-Salmi, S.; Renkonen, R.; Chow, V.T.; et al. A co-culture model of PBMC and stem cell derived human nasal epithelium reveals rapid activation of NK and innate T cells upon influenza A virus infection of the nasal epithelium. Front. Immunol. 2018, 9, 2514. [Google Scholar] [CrossRef] [Green Version]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ong, H.H.; Liu, J.; Oo, Y.; Thong, M.; Wang, D.Y.; Chow, V.T. Prolonged Primary Rhinovirus Infection of Human Nasal Epithelial Cells Diminishes the Viral Load of Secondary Influenza H3N2 Infection via the Antiviral State Mediated by RIG-I and Interferon-Stimulated Genes. Cells 2023, 12, 1152. https://doi.org/10.3390/cells12081152
Ong HH, Liu J, Oo Y, Thong M, Wang DY, Chow VT. Prolonged Primary Rhinovirus Infection of Human Nasal Epithelial Cells Diminishes the Viral Load of Secondary Influenza H3N2 Infection via the Antiviral State Mediated by RIG-I and Interferon-Stimulated Genes. Cells. 2023; 12(8):1152. https://doi.org/10.3390/cells12081152
Chicago/Turabian StyleOng, Hsiao Hui, Jing Liu, Yukei Oo, Mark Thong, De Yun Wang, and Vincent T. Chow. 2023. "Prolonged Primary Rhinovirus Infection of Human Nasal Epithelial Cells Diminishes the Viral Load of Secondary Influenza H3N2 Infection via the Antiviral State Mediated by RIG-I and Interferon-Stimulated Genes" Cells 12, no. 8: 1152. https://doi.org/10.3390/cells12081152