
Molecular Mechanisms of Ferroptosis and Updates of Ferroptosis Studies in Cancers and Leukemia


Abstract
:1. Introduction
2. Molecular Mechanisms of Ferroptosis Induction
2.1. Ferroptosis Regulation through Phospholipid Metabolism
2.2. Iron Metabolism and Ferroptosis
2.2.1. Iron Homeostasis and the Regulation of Labile Iron Pool
2.2.2. Essential Roles of LIP in Lipid Peroxidation
2.2.3. Ferroptosis Regulation through Iron Metabolism
2.3. What Is the Direct Cause of Ferroptosis?
3. Mechanisms of Protection from Ferroptosis
3.1. The System xc−—GSH—GPX4 Axis
3.2. The Ferroptosis Suppressor Protein 1-Coenzyme Q Axis
3.3. The Dihydroorate Dehydrogenase/Glycerol-3-Phosphate Dehydrogenase 2–Mitochondrial CoQ Axis
3.4. The GTP Cyclohydrolase 1–Tetrahydrobiopterin–Dihydrofolate Reductase Axis
4. Regulation of Ferroptosis by p53
5. Ferroptosis in Leukemia
5.1. Dysregulation of Iron Homeostasis in Leukemia
5.2. Ferroptosis-Related Gene Signatures in Leukemia
5.3. Ferroptosis Induction as a Therapeutic Strategy in Leukemia
5.3.1. Inhibition of System xc−—GSH—GPX4 Axis in Leukemia
5.3.2. Ferroptosis Induction by Clinically Available Anti-Leukemia Agents
5.3.3. Ferroptosis Induction by Natural Compounds and Their Derivatives
5.3.4. Other Potential Therapeutic Strategies Inducing Ferroptosis in Leukemia
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, B.A.; El-Deiry, W.S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef]
- Leak, L.; Dixon, S.J. Surveying the landscape of emerging and understudied cell death mechanisms. Biochim. Biophys. Acta Mol. Cell Res. 2023, 1870, 119432. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Stockwell, B.R. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 2022, 185, 2401–2421. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Jiang, L.; Tsuduki, T.; Conrad, M.; Toyokuni, S. Embryonal erythropoiesis and aging exploit ferroptosis. Redox Biol. 2021, 48, 102175. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Miao, W.; Cui, Q.; Shi, B.; Zhang, J.; Qiu, H.; Zhang, L.; Han, Y. Inhibition of ferroptosis promotes megakaryocyte differentiation and platelet production. J. Cell Mol. Med. 2022, 26, 3582–3585. [Google Scholar] [CrossRef] [PubMed]
- Mazhar, M.; Din, A.U.; Ali, H.; Yang, G.; Ren, W.; Wang, L.; Fan, X.; Yang, S. Implication of ferroptosis in aging. Cell Death Discov. 2021, 7, 149. [Google Scholar] [CrossRef]
- Ma, X.; Xiao, L.; Liu, L.; Ye, L.; Su, P.; Bi, E.; Wang, Q.; Yang, M.; Qian, J.; Yi, Q. CD36-mediated ferroptosis dampens intratumoral CD8+ T cell effector function and impairs their antitumor ability. Cell Metab. 2021, 33, 1001–1012.e5. [Google Scholar] [CrossRef]
- Xu, S.; Chaudhary, O.; Rodriguez-Morales, P.; Sun, X.; Chen, D.; Zappasodi, R.; Xu, Z.; Pinto, A.F.M.; Williams, A.; Schulze, I.; et al. Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8+ T cells in tumors. Immunity 2021, 54, 1561–1577.e7. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Chen, Z.; Zhang, H.; Chen, C.; Zeng, M.; Yunis, J.; Wei, Y.; Wan, Y.; Wang, N.; Zhou, M.; et al. Selenium-GPX4 axis protects follicular helper T cells from ferroptosis. Nat. Immunol. 2021, 22, 1127–1139. [Google Scholar] [CrossRef]
- Xiong, J.; Qi, W.; Liu, J.; Zhang, Z.; Wang, Z.; Bao, J.; Wu, C.; Liang, F. Research Progress of Ferroptosis: A Bibliometrics and Visual Analysis Study. J. Healthc. Eng. 2021, 2021, 2178281. [Google Scholar] [CrossRef] [PubMed]
- Hangauer, M.J.; Viswanathan, V.S.; Ryan, M.J.; Bole, D.; Eaton, J.K.; Matov, A.; Galeas, J.; Dhruv, H.D.; Berens, M.E.; Schreiber, S.L.; et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 2017, 551, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, V.S.; Ryan, M.J.; Dhruv, H.D.; Gill, S.; Eichhoff, O.M.; Seashore-Ludlow, B.; Kaffenberger, S.D.; Eaton, J.K.; Shimada, K.; Aguirre, A.J.; et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 2017, 547, 453–457. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Y.; Wu, X.; Xu, F.; Ma, H.; Wu, M.; Xia, Y. Targeting Ferroptosis Pathway to Combat Therapy Resistance and Metastasis of Cancer. Front. Pharm. 2022, 13, 909821. [Google Scholar] [CrossRef]
- Lei, G.; Zhuang, L.; Gan, B. Targeting ferroptosis as a vulnerability in cancer. Nat. Rev. Cancer 2022, 22, 381–396. [Google Scholar] [CrossRef]
- Soula, M.; Weber, R.A.; Zilka, O.; Alwaseem, H.; La, K.; Yen, F.; Molina, H.; Garcia-Bermudez, J.; Pratt, D.A.; Birsoy, K. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat. Chem. Biol. 2020, 16, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Magtanong, L.; Mueller, G.D.; Williams, K.J.; Billmann, M.; Chan, K.; Armenta, D.A.; Pope, L.E.; Moffat, J.; Boone, C.; Myers, C.L.; et al. Context-dependent regulation of ferroptosis sensitivity. Cell Chem. Biol. 2022, 29, 1409–1418.e6. [Google Scholar] [CrossRef]
- Ball, S.; Borthakur, G. Apoptosis targeted therapies in acute myeloid leukemia: An update. Expert Rev. Hematol. 2020, 13, 1373–1386. [Google Scholar] [CrossRef]
- Maiti, A.; Carter, B.Z.; Andreeff, M.; Konopleva, M.Y. Beyond BCL-2 Inhibition in Acute Myloid Leukemia: Other Approaches to Leverage the Apoptotic Pathway. Clin. Lymphoma Myeloma Leuk. 2022, 22, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Lasica, M.; Anderson, M.A. Review of Venetoclax in CLL, AML and Multiple Myeloma. J. Pers. Med. 2021, 11, 463. [Google Scholar] [CrossRef]
- Ong, F.; Kim, K.; Konopleva, M.Y. Venetoclax resistance: Mechanistic insights and future strategies. Cancer Drug Resist. 2022, 5, 380–400. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Jiang, X. The Chemistry and Biology of Ferroptosis. Cell Chem. Biol. 2020, 27, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Conrad, M.; Pratt, D.A. The chemical basis of ferroptosis. Nat. Chem. Biol. 2019, 15, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Guillou, H.; Zadravec, D.; Martin, P.G.; Jacobsson, A. The key roles of elongases and desaturases in mammalian fatty acid metabolism: Insights from transgenic mice. Prog. Lipid Res. 2010, 49, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Nam, M.; Son, H.Y.; Hyun, K.; Jang, S.Y.; Kim, J.W.; Kim, M.W.; Jung, Y.; Jang, E.; Yoon, S.J.; et al. Polyunsaturated fatty acid biosynthesis pathway determines ferroptosis sensitivity in gastric cancer. Proc. Natl. Acad. Sci. USA 2020, 117, 32433–32442. [Google Scholar] [CrossRef]
- Yamane, D.; Hayashi, Y.; Matsumoto, M.; Nakanishi, H.; Imagawa, H.; Kohara, M.; Lemon, S.M.; Ichi, I. FADS2-dependent fatty acid desaturation dictates cellular sensitivity to ferroptosis and permissiveness for hepatitis C virus replication. Cell Chem. Biol. 2022, 29, 799–810.e794. [Google Scholar] [CrossRef]
- Dierge, E.; Debock, E.; Guilbaud, C.; Corbet, C.; Mignolet, E.; Mignard, L.; Bastien, E.; Dessy, C.; Larondelle, Y.; Feron, O. Peroxidation of n-3 and n-6 polyunsaturated fatty acids in the acidic tumor environment leads to ferroptosis-mediated anticancer effects. Cell Metab. 2021, 33, 1701–1715.e1705. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Zandkarimi, F.; Zhang, Y.; Meena, J.K.; Kim, J.; Zhuang, L.; Tyagi, S.; Ma, L.; Westbrook, T.F.; Steinberg, G.R.; et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat. Cell Biol. 2020, 22, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Olzmann, J.A.; Carvalho, P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155. [Google Scholar] [CrossRef]
- Bai, Y.; Meng, L.; Han, L.; Jia, Y.; Zhao, Y.; Gao, H.; Kang, R.; Wang, X.; Tang, D.; Dai, E. Lipid storage and lipophagy regulates ferroptosis. Biochem. Biophys. Res. Commun. 2019, 508, 997–1003. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef]
- Dixon, S.J.; Winter, G.E.; Musavi, L.S.; Lee, E.D.; Snijder, B.; Rebsamen, M.; Superti-Furga, G.; Stockwell, B.R. Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS Chem. Biol. 2015, 10, 1604–1609. [Google Scholar] [CrossRef] [PubMed]
- Tesfay, L.; Paul, B.T.; Konstorum, A.; Deng, Z.; Cox, A.O.; Lee, J.; Furdui, C.M.; Hegde, P.; Torti, F.M.; Torti, S.V. Stearoyl-CoA Desaturase 1 Protects Ovarian Cancer Cells from Ferroptotic Cell Death. Cancer Res. 2019, 79, 5355–5366. [Google Scholar] [CrossRef]
- Magtanong, L.; Ko, P.J.; To, M.; Cao, J.Y.; Forcina, G.C.; Tarangelo, A.; Ward, C.C.; Cho, K.; Patti, G.J.; Nomura, D.K.; et al. Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem. Biol. 2019, 26, 420–432.e9. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Henry, W.S.; Ricq, E.L.; Graham, E.T.; Phadnis, V.V.; Maretich, P.; Paradkar, S.; Boehnke, N.; Deik, A.A.; Reinhardt, F.; et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature 2020, 585, 603–608. [Google Scholar] [CrossRef]
- Cui, W.; Liu, D.; Gu, W.; Chu, B. Peroxisome-driven ether-linked phospholipids biosynthesis is essential for ferroptosis. Cell Death Differ. 2021, 28, 2536–2551. [Google Scholar] [CrossRef] [PubMed]
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A Red Carpet for Iron Metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef] [PubMed]
- Gammella, E.; Buratti, P.; Cairo, G.; Recalcati, S. The transferrin receptor: The cellular iron gate. Metallomics 2017, 9, 1367–1375. [Google Scholar] [CrossRef]
- Muller, S.; Sindikubwabo, F.; Caneque, T.; Lafon, A.; Versini, A.; Lombard, B.; Loew, D.; Wu, T.D.; Ginestier, C.; Charafe-Jauffret, E.; et al. CD44 regulates epigenetic plasticity by mediating iron endocytosis. Nat. Chem. 2020, 12, 929–938. [Google Scholar] [CrossRef]
- Ohgami, R.S.; Campagna, D.R.; Greer, E.L.; Antiochos, B.; McDonald, A.; Chen, J.; Sharp, J.J.; Fujiwara, Y.; Barker, J.E.; Fleming, M.D. Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat. Genet. 2005, 37, 1264–1269. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Shang, P. The significance, trafficking and determination of labile iron in cytosol, mitochondria and lysosomes. Metallomics 2018, 10, 899–916. [Google Scholar] [CrossRef]
- Hider, R.C.; Kong, X.L. Glutathione: A key component of the cytoplasmic labile iron pool. Biometals 2011, 24, 1179–1187. [Google Scholar] [CrossRef] [PubMed]
- Donovan, A.; Lima, C.A.; Pinkus, J.L.; Pinkus, G.S.; Zon, L.I.; Robine, S.; Andrews, N.C. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005, 1, 191–200. [Google Scholar] [CrossRef]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef]
- Paul, B.T.; Manz, D.H.; Torti, F.M.; Torti, S.V. Mitochondria and Iron: Current questions. Expert Rev. Hematol. 2017, 10, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Dietz, J.V.; Fox, J.L.; Khalimonchuk, O. Down the Iron Path: Mitochondrial Iron Homeostasis and Beyond. Cells 2021, 10, 2198. [Google Scholar] [CrossRef] [PubMed]
- Wolff, N.A.; Garrick, M.D.; Zhao, L.; Garrick, L.M.; Ghio, A.J.; Thevenod, F. A role for divalent metal transporter (DMT1) in mitochondrial uptake of iron and manganese. Sci. Rep. 2018, 8, 211. [Google Scholar] [CrossRef]
- Paradkar, P.N.; Zumbrennen, K.B.; Paw, B.H.; Ward, D.M.; Kaplan, J. Regulation of mitochondrial iron import through differential turnover of mitoferrin 1 and mitoferrin 2. Mol. Cell. Biol. 2009, 29, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Wang, X.; Zhang, W.; Li, H.; Zhao, W.; Sun, J.; Yang, M. Melatonin Suppresses Ferroptosis Induced by High Glucose via Activation of the Nrf2/HO-1 Signaling Pathway in Type 2 Diabetic Osteoporosis. Oxid. Med. Cell. Longev. 2020, 2020, 9067610. [Google Scholar] [CrossRef]
- Nakamura, T.; Naguro, I.; Ichijo, H. Iron homeostasis and iron-regulated ROS in cell death, senescence and human diseases. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 1398–1409. [Google Scholar] [CrossRef]
- Yin, H.; Xu, L.; Porter, N.A. Free radical lipid peroxidation: Mechanisms and analysis. Chem. Rev. 2011, 111, 5944–5972. [Google Scholar] [CrossRef]
- O’Donnell, V.B.; Aldrovandi, M.; Murphy, R.C.; Kronke, G. Enzymatically oxidized phospholipids assume center stage as essential regulators of innate immunity and cell death. Sci. Signal. 2019, 12, eaau2293. [Google Scholar] [CrossRef]
- Stoyanovsky, D.A.; Tyurina, Y.Y.; Shrivastava, I.; Bahar, I.; Tyurin, V.A.; Protchenko, O.; Jadhav, S.; Bolevich, S.B.; Kozlov, A.V.; Vladimirov, Y.A.; et al. Iron catalysis of lipid peroxidation in ferroptosis: Regulated enzymatic or random free radical reaction? Free Radic. Biol. Med. 2019, 133, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef]
- Feng, H.; Schorpp, K.; Jin, J.; Yozwiak, C.E.; Hoffstrom, B.G.; Decker, A.M.; Rajbhandari, P.; Stokes, M.E.; Bender, H.G.; Csuka, J.M.; et al. Transferrin Receptor Is a Specific Ferroptosis Marker. Cell Rep. 2020, 30, 3411–3423.e7. [Google Scholar] [CrossRef] [PubMed]
- Geng, N.; Shi, B.J.; Li, S.L.; Zhong, Z.Y.; Li, Y.C.; Xua, W.L.; Zhou, H.; Cai, J.H. Knockdown of ferroportin accelerates erastin-induced ferroptosis in neuroblastoma cells. Eur. Rev. Med. Pharm. Sci. 2018, 22, 3826–3836. [Google Scholar] [CrossRef]
- Bao, W.D.; Pang, P.; Zhou, X.T.; Hu, F.; Xiong, W.; Chen, K.; Wang, J.; Wang, F.; Xie, D.; Hu, Y.Z.; et al. Loss of ferroportin induces memory impairment by promoting ferroptosis in Alzheimer’s disease. Cell Death Differ. 2021, 28, 1548–1562. [Google Scholar] [CrossRef]
- Song, Y.; Wang, B.; Zhu, X.; Hu, J.; Sun, J.; Xuan, J.; Ge, Z. Human umbilical cord blood-derived MSCs exosome attenuate myocardial injury by inhibiting ferroptosis in acute myocardial infarction mice. Cell Biol. Toxicol. 2021, 37, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Peng, S.; Sun, Z.; Heng, X.; Zhu, X. Temozolomide Drives Ferroptosis via a DMT1-Dependent Pathway in Glioblastoma Cells. Yonsei Med. J. 2021, 62, 843–849. [Google Scholar] [CrossRef]
- Fang, X.; Cai, Z.; Wang, H.; Han, D.; Cheng, Q.; Zhang, P.; Gao, F.; Yu, Y.; Song, Z.; Wu, Q.; et al. Loss of Cardiac Ferritin H Facilitates Cardiomyopathy via Slc7a11-Mediated Ferroptosis. Circ. Res. 2020, 127, 486–501. [Google Scholar] [CrossRef]
- Brown, C.W.; Amante, J.J.; Chhoy, P.; Elaimy, A.L.; Liu, H.; Zhu, L.J.; Baer, C.E.; Dixon, S.J.; Mercurio, A.M. Prominin2 Drives Ferroptosis Resistance by Stimulating Iron Export. Dev. Cell 2019, 51, 575–586.e4. [Google Scholar] [CrossRef]
- Liu, J.; Kuang, F.; Kroemer, G.; Klionsky, D.J.; Kang, R.; Tang, D. Autophagy-Dependent Ferroptosis: Machinery and Regulation. Cell Chem. Biol. 2020, 27, 420–435. [Google Scholar] [CrossRef]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J., 3rd; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef]
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis is an autophagic cell death process. Cell Res. 2016, 26, 1021–1032. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.Y.; Park, E.; Lee, S.J.; Chung, S.W. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget 2015, 6, 24393–24403. [Google Scholar] [CrossRef]
- Fang, X.; Wang, H.; Han, D.; Xie, E.; Yang, X.; Wei, J.; Gu, S.; Gao, F.; Zhu, N.; Yin, X.; et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2672–2680. [Google Scholar] [CrossRef]
- Adedoyin, O.; Boddu, R.; Traylor, A.; Lever, J.M.; Bolisetty, S.; George, J.F.; Agarwal, A. Heme oxygenase-1 mitigates ferroptosis in renal proximal tubule cells. Am. J. Physiol. Ren. Physiol. 2018, 314, F702–F714. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Q.; Chang, S.Y.; Wu, Q.; Gou, Y.J.; Jia, L.; Cui, Y.M.; Yu, P.; Shi, Z.H.; Wu, W.S.; Gao, G.; et al. The Protective Role of Mitochondrial Ferritin on Erastin-Induced Ferroptosis. Front. Aging Neurosci. 2016, 8, 308. [Google Scholar] [CrossRef]
- Fuhrmann, D.C.; Mondorf, A.; Beifuss, J.; Jung, M.; Brune, B. Hypoxia inhibits ferritinophagy, increases mitochondrial ferritin, and protects from ferroptosis. Redox Biol. 2020, 36, 101670. [Google Scholar] [CrossRef]
- Wang, P.; Cui, Y.; Ren, Q.; Yan, B.; Zhao, Y.; Yu, P.; Gao, G.; Shi, H.; Chang, S.; Chang, Y.Z. Mitochondrial ferritin attenuates cerebral ischaemia/reperfusion injury by inhibiting ferroptosis. Cell Death Dis. 2021, 12, 447. [Google Scholar] [CrossRef]
- Wang, X.; Ma, H.; Sun, J.; Zheng, T.; Zhao, P.; Li, H.; Yang, M. Mitochondrial Ferritin Deficiency Promotes Osteoblastic Ferroptosis Via Mitophagy in Type 2 Diabetic Osteoporosis. Biol. Trace Elem. Res. 2022, 200, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Guo, M.; Shen, M.; Kong, D.; Zhang, F.; Shao, J.; Tan, S.; Wang, S.; Chen, A.; Cao, P.; et al. The BRD7-P53-SLC25A28 axis regulates ferroptosis in hepatic stellate cells. Redox Biol. 2020, 36, 101619. [Google Scholar] [CrossRef]
- Du, J.; Zhou, Y.; Li, Y.; Xia, J.; Chen, Y.; Chen, S.; Wang, X.; Sun, W.; Wang, T.; Ren, X.; et al. Identification of Frataxin as a regulator of ferroptosis. Redox Biol. 2020, 32, 101483. [Google Scholar] [CrossRef]
- Alvarez, S.W.; Sviderskiy, V.O.; Terzi, E.M.; Papagiannakopoulos, T.; Moreira, A.L.; Adams, S.; Sabatini, D.M.; Birsoy, K.; Possemato, R. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature 2017, 551, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Ikeda, M.; Ide, T.; Tadokoro, T.; Miyamoto, H.D.; Furusawa, S.; Tsutsui, Y.; Miyake, R.; Ishimaru, K.; Watanabe, M.; et al. Doxorubicin causes ferroptosis and cardiotoxicity by intercalating into mitochondrial DNA and disrupting Alas1-dependent heme synthesis. Sci. Signal. 2022, 15, eabn8017. [Google Scholar] [CrossRef]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, S.E.; Tyurina, Y.Y.; Zhao, J.; St Croix, C.M.; Dar, H.H.; Mao, G.; Tyurin, V.A.; Anthonymuthu, T.S.; Kapralov, A.A.; Amoscato, A.A.; et al. PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals. Cell 2017, 171, 628–641.e26. [Google Scholar] [CrossRef] [PubMed]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef]
- Matsushita, M.; Freigang, S.; Schneider, C.; Conrad, M.; Bornkamm, G.W.; Kopf, M. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J. Exp. Med. 2015, 212, 555–568. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Li, H.; Graham, E.T.; Deik, A.A.; Eaton, J.K.; Wang, W.; Sandoval-Gomez, G.; Clish, C.B.; Doench, J.G.; Schreiber, S.L. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Chem. Biol. 2020, 16, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Chu, B.; Kon, N.; Chen, D.; Li, T.; Liu, T.; Jiang, L.; Song, S.; Tavana, O.; Gu, W. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat. Cell Biol. 2019, 21, 579–591. [Google Scholar] [CrossRef]
- Jurnak, F. The Pivotal Role of Aldehyde Toxicity in Autism Spectrum Disorder: The Therapeutic Potential of Micronutrient Supplementation. Nutr. Metab. Insights 2015, 8, 57–77. [Google Scholar] [CrossRef]
- Jacobs, A.T.; Marnett, L.J. Systems analysis of protein modification and cellular responses induced by electrophile stress. Acc. Chem. Res. 2010, 43, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Pizzimenti, S.; Ciamporcero, E.; Daga, M.; Pettazzoni, P.; Arcaro, A.; Cetrangolo, G.; Minelli, R.; Dianzani, C.; Lepore, A.; Gentile, F.; et al. Interaction of aldehydes derived from lipid peroxidation and membrane proteins. Front. Physiol. 2013, 4, 242. [Google Scholar] [CrossRef] [PubMed]
- Castro, J.P.; Jung, T.; Grune, T.; Siems, W. 4-Hydroxynonenal (HNE) modified proteins in metabolic diseases. Free Radic. Biol. Med. 2017, 111, 309–315. [Google Scholar] [CrossRef]
- Otsuki, Y.; Yamasaki, J.; Suina, K.; Okazaki, S.; Koike, N.; Saya, H.; Nagano, O. Vasodilator oxyfedrine inhibits aldehyde metabolism and thereby sensitizes cancer cells to xCT-targeted therapy. Cancer Sci. 2020, 111, 127–136. [Google Scholar] [CrossRef]
- Okazaki, S.; Shintani, S.; Hirata, Y.; Suina, K.; Semba, T.; Yamasaki, J.; Umene, K.; Ishikawa, M.; Saya, H.; Nagano, O. Synthetic lethality of the ALDH3A1 inhibitor dyclonine and xCT inhibitors in glutathione deficiency-resistant cancer cells. Oncotarget 2018, 9, 33832–33843. [Google Scholar] [CrossRef]
- Yusuf, R.Z.; Saez, B.; Sharda, A.; van Gastel, N.; Yu, V.W.C.; Baryawno, N.; Scadden, E.W.; Acharya, S.; Chattophadhyay, S.; Huang, C.; et al. Aldehyde dehydrogenase 3a2 protects AML cells from oxidative death and the synthetic lethality of ferroptosis inducers. Blood 2020, 136, 1303–1316. [Google Scholar] [CrossRef]
- Zhu, Z.Y.; Liu, Y.D.; Gong, Y.; Jin, W.; Topchiy, E.; Turdi, S.; Gao, Y.F.; Culver, B.; Wang, S.Y.; Ge, W.; et al. Mitochondrial aldehyde dehydrogenase (ALDH2) rescues cardiac contractile dysfunction in an APP/PS1 murine model of Alzheimer’s disease via inhibition of ACSL4-dependent ferroptosis. Acta Pharm. Sin. 2022, 43, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Pedrera, L.; Espiritu, R.A.; Ros, U.; Weber, J.; Schmitt, A.; Stroh, J.; Hailfinger, S.; von Karstedt, S.; Garcia-Saez, A.J. Ferroptotic pores induce Ca(2+) fluxes and ESCRT-III activation to modulate cell death kinetics. Cell Death Differ. 2021, 28, 1644–1657. [Google Scholar] [CrossRef] [PubMed]
- Dai, E.; Meng, L.; Kang, R.; Wang, X.; Tang, D. ESCRT-III-dependent membrane repair blocks ferroptosis. Biochem. Biophys. Res. Commun. 2020, 522, 415–421. [Google Scholar] [CrossRef]
- Riegman, M.; Sagie, L.; Galed, C.; Levin, T.; Steinberg, N.; Dixon, S.J.; Wiesner, U.; Bradbury, M.S.; Niethammer, P.; Zaritsky, A.; et al. Ferroptosis occurs through an osmotic mechanism and propagates independently of cell rupture. Nat. Cell. Biol. 2020, 22, 1042–1048. [Google Scholar] [CrossRef]
- Wu, J.; Minikes, A.M.; Gao, M.; Bian, H.; Li, Y.; Stockwell, B.R.; Chen, Z.N.; Jiang, X. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature 2019, 572, 402–406. [Google Scholar] [CrossRef]
- Yang, W.H.; Ding, C.C.; Sun, T.; Rupprecht, G.; Lin, C.C.; Hsu, D.; Chi, J.T. The Hippo Pathway Effector TAZ Regulates Ferroptosis in Renal Cell Carcinoma. Cell Rep. 2019, 28, 2501–2508.e2504. [Google Scholar] [CrossRef]
- Nakamura, T.; Ogawa, M.; Kojima, K.; Takayanagi, S.; Ishihara, S.; Hattori, K.; Naguro, I.; Ichijo, H. The mitochondrial Ca2+ uptake regulator, MICU1, is involved in cold stress-induced ferroptosis. EMBO Rep. 2021, 22, e51532. [Google Scholar] [CrossRef]
- Lo, M.; Wang, Y.Z.; Gout, P.W. The xc− cystine/glutamate antiporter: A potential target for therapy of cancer and other diseases. J. Cell Physiol. 2008, 215, 593–602. [Google Scholar] [CrossRef]
- Zhang, Y.; Swanda, R.V.; Nie, L.; Liu, X.; Wang, C.; Lee, H.; Lei, G.; Mao, C.; Koppula, P.; Cheng, W.; et al. mTORC1 couples cyst(e)ine availability with GPX4 protein synthesis and ferroptosis regulation. Nat. Commun. 2021, 12, 1589. [Google Scholar] [CrossRef]
- Zhang, H.F.; Klein Geltink, R.I.; Parker, S.J.; Sorensen, P.H. Transsulfuration, minor player or crucial for cysteine homeostasis in cancer. Trends Cell. Biol. 2022, 32, 800–814. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Lin, X.; Huang, C. Activation of the reverse transsulfuration pathway through NRF2/CBS confers erastin-induced ferroptosis resistance. Br. J. Cancer 2020, 122, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Chen, X.; Jiang, L.; Lu, B.; Yuan, M.; Zhu, D.; Zhu, H.; He, Q.; Yang, B.; Ying, M. DJ-1 suppresses ferroptosis through preserving the activity of S-adenosyl homocysteine hydrolase. Nat. Commun. 2020, 11, 1251. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef]
- Koppula, P.; Zhuang, L.; Gan, B. Cystine transporter SLC7A11/xCT in cancer: Ferroptosis, nutrient dependency, and cancer therapy. Protein Cell 2021, 12, 599–620. [Google Scholar] [CrossRef]
- Zhang, Y.; Tan, H.; Daniels, J.D.; Zandkarimi, F.; Liu, H.; Brown, L.M.; Uchida, K.; O’Connor, O.A.; Stockwell, B.R. Imidazole Ketone Erastin Induces Ferroptosis and Slows Tumor Growth in a Mouse Lymphoma Model. Cell Chem. Biol. 2019, 26, 623–633.e9. [Google Scholar] [CrossRef] [PubMed]
- Badgley, M.A.; Kremer, D.M.; Maurer, H.C.; DelGiorno, K.E.; Lee, H.J.; Purohit, V.; Sagalovskiy, I.R.; Ma, A.; Kapilian, J.; Firl, C.E.M.; et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 2020, 368, 85–89. [Google Scholar] [CrossRef]
- Pardieu, B.; Pasanisi, J.; Ling, F.; Dal Bello, R.; Penneroux, J.; Su, A.; Joudinaud, R.; Chat, L.; Wu, H.C.; Duchmann, M.; et al. Cystine uptake inhibition potentiates front-line therapies in acute myeloid leukemia. Leukemia 2022, 36, 1585–1595. [Google Scholar] [CrossRef]
- Cao, J.Y.; Dixon, S.J. Mechanisms of ferroptosis. Cell Mol. Life Sci. 2016, 73, 2195–2209. [Google Scholar] [CrossRef]
- Sun, Y.; Zheng, Y.; Wang, C.; Liu, Y. Glutathione depletion induces ferroptosis, autophagy, and premature cell senescence in retinal pigment epithelial cells. Cell Death Dis. 2018, 9, 753. [Google Scholar] [CrossRef]
- Kang, Y.P.; Mockabee-Macias, A.; Jiang, C.; Falzone, A.; Prieto-Farigua, N.; Stone, E.; Harris, I.S.; DeNicola, G.M. Non-canonical Glutamate-Cysteine Ligase Activity Protects against Ferroptosis. Cell Metab. 2021, 33, 174–189.e7. [Google Scholar] [CrossRef] [PubMed]
- Ursini, F.; Maiorino, M.; Gregolin, C. The selenoenzyme phospholipid hydroperoxide glutathione peroxidase. Biochim. Biophys. Acta 1985, 839, 62–70. [Google Scholar] [CrossRef]
- Shimada, K.; Skouta, R.; Kaplan, A.; Yang, W.S.; Hayano, M.; Dixon, S.J.; Brown, L.M.; Valenzuela, C.A.; Wolpaw, A.J.; Stockwell, B.R. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 2016, 12, 497–503. [Google Scholar] [CrossRef]
- Zou, Y.; Palte, M.J.; Deik, A.A.; Li, H.; Eaton, J.K.; Wang, W.; Tseng, Y.Y.; Deasy, R.; Kost-Alimova, M.; Dancik, V.; et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat. Commun. 2019, 10, 1617. [Google Scholar] [CrossRef]
- Eaton, J.K.; Furst, L.; Ruberto, R.A.; Moosmayer, D.; Hilpmann, A.; Ryan, M.J.; Zimmermann, K.; Cai, L.L.; Niehues, M.; Badock, V.; et al. Selective covalent targeting of GPX4 using masked nitrile-oxide electrophiles. Nat. Chem. Biol. 2020, 16, 497–506. [Google Scholar] [CrossRef]
- Conrad, M.; Proneth, B. Selenium: Tracing Another Essential Element of Ferroptotic Cell Death. Cell Chem. Biol. 2020, 27, 409–419. [Google Scholar] [CrossRef]
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Porto Freitas, F.; Seibt, T.; et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 2018, 172, 409–422.e21. [Google Scholar] [CrossRef]
- Alim, I.; Caulfield, J.T.; Chen, Y.; Swarup, V.; Geschwind, D.H.; Ivanova, E.; Seravalli, J.; Ai, Y.; Sansing, L.H.; Ste Marie, E.J.; et al. Selenium Drives a Transcriptional Adaptive Program to Block Ferroptosis and Treat Stroke. Cell 2019, 177, 1262–1279.e25. [Google Scholar] [CrossRef] [PubMed]
- Savaskan, N.E.; Ufer, C.; Kuhn, H.; Borchert, A. Molecular biology of glutathione peroxidase 4: From genomic structure to developmental expression and neural function. Biol. Chem. 2007, 388, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Yoo, S.E.; Na, R.; Walter, C.A.; Richardson, A.; Ran, Q. Short form glutathione peroxidase 4 is the essential isoform required for survival and somatic mitochondrial functions. J. Biol. Chem. 2009, 284, 30836–30844. [Google Scholar] [CrossRef]
- Schneider, M.; Forster, H.; Boersma, A.; Seiler, A.; Wehnes, H.; Sinowatz, F.; Neumuller, C.; Deutsch, M.J.; Walch, A.; Hrabe de Angelis, M.; et al. Mitochondrial glutathione peroxidase 4 disruption causes male infertility. FASEB J. 2009, 23, 3233–3242. [Google Scholar] [CrossRef]
- Yant, L.J.; Ran, Q.; Rao, L.; Van Remmen, H.; Shibatani, T.; Belter, J.G.; Motta, L.; Richardson, A.; Prolla, T.A. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic. Biol. Med. 2003, 34, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Tadokoro, T.; Ikeda, M.; Ide, T.; Deguchi, H.; Ikeda, S.; Okabe, K.; Ishikita, A.; Matsushima, S.; Koumura, T.; Yamada, K.I.; et al. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight 2020, 5, e132747. [Google Scholar] [CrossRef] [PubMed]
- Kitakata, H.; Endo, J.; Matsushima, H.; Yamamoto, S.; Ikura, H.; Hirai, A.; Koh, S.; Ichihara, G.; Hiraide, T.; Moriyama, H.; et al. MITOL/MARCH5 determines the susceptibility of cardiomyocytes to doxorubicin-induced ferroptosis by regulating GSH homeostasis. J. Mol. Cell Cardiol. 2021, 161, 116–129. [Google Scholar] [CrossRef]
- Ta, N.; Qu, C.; Wu, H.; Zhang, D.; Sun, T.; Li, Y.; Wang, J.; Wang, X.; Tang, T.; Chen, Q.; et al. Mitochondrial outer membrane protein FUNDC2 promotes ferroptosis and contributes to doxorubicin-induced cardiomyopathy. Proc. Natl. Acad. Sci. USA 2022, 119, e2117396119. [Google Scholar] [CrossRef]
- To, T.L.; Cuadros, A.M.; Shah, H.; Hung, W.H.W.; Li, Y.; Kim, S.H.; Rubin, D.H.F.; Boe, R.H.; Rath, S.; Eaton, J.K.; et al. A Compendium of Genetic Modifiers of Mitochondrial Dysfunction Reveals Intra-organelle Buffering. Cell 2019, 179, 1222–1238.e1217. [Google Scholar] [CrossRef]
- Mao, C.; Liu, X.; Zhang, Y.; Lei, G.; Yan, Y.; Lee, H.; Koppula, P.; Wu, S.; Zhuang, L.; Fang, B.; et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 2021, 593, 586–590. [Google Scholar] [CrossRef]
- Wu, S.; Mao, C.; Kondiparthi, L.; Poyurovsky, M.V.; Olszewski, K.; Gan, B. A ferroptosis defense mechanism mediated by glycerol-3-phosphate dehydrogenase 2 in mitochondria. Proc. Natl. Acad. Sci. USA 2022, 119, e2121987119. [Google Scholar] [CrossRef]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hekimi, S. Understanding Ubiquinone. Trends Cell Biol. 2016, 26, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Hayano, M.; Pagano, N.C.; Stockwell, B.R. Cell-Line Selectivity Improves the Predictive Power of Pharmacogenomic Analyses and Helps Identify NADPH as Biomarker for Ferroptosis Sensitivity. Cell Chem. Biol. 2016, 23, 225–235. [Google Scholar] [CrossRef]
- Ding, C.C.; Rose, J.; Sun, T.; Wu, J.; Chen, P.H.; Lin, C.C.; Yang, W.H.; Chen, K.Y.; Lee, H.; Xu, E.; et al. MESH1 is a cytosolic NADPH phosphatase that regulates ferroptosis. Nat. Metab. 2020, 2, 270–277. [Google Scholar] [CrossRef]
- Koppula, P.; Lei, G.; Zhang, Y.; Yan, Y.; Mao, C.; Kondiparthi, L.; Shi, J.; Liu, X.; Horbath, A.; Das, M.; et al. A targetable CoQ-FSP1 axis drives ferroptosis- and radiation-resistance in KEAP1 inactive lung cancers. Nat. Commun. 2022, 13, 2206. [Google Scholar] [CrossRef]
- Venkatesh, D.; O’Brien, N.A.; Zandkarimi, F.; Tong, D.R.; Stokes, M.E.; Dunn, D.E.; Kengmana, E.S.; Aron, A.T.; Klein, A.M.; Csuka, J.M.; et al. MDM2 and MDMX promote ferroptosis by PPARalpha-mediated lipid remodeling. Genes Dev. 2020, 34, 526–543. [Google Scholar] [CrossRef]
- Mishima, E.; Ito, J.; Wu, Z.; Nakamura, T.; Wahida, A.; Doll, S.; Tonnus, W.; Nepachalovich, P.; Eggenhofer, E.; Aldrovandi, M.; et al. A non-canonical vitamin K cycle is a potent ferroptosis suppressor. Nature 2022, 608, 778–783. [Google Scholar] [CrossRef]
- Gan, B. Mitochondrial regulation of ferroptosis. J. Cell Biol. 2021, 220, e202105043. [Google Scholar] [CrossRef] [PubMed]
- Kraft, V.A.N.; Bezjian, C.T.; Pfeiffer, S.; Ringelstetter, L.; Muller, C.; Zandkarimi, F.; Merl-Pham, J.; Bao, X.; Anastasov, N.; Kossl, J.; et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent. Sci. 2020, 6, 41–53. [Google Scholar] [CrossRef]
- Levine, A.J. p53: 800 million years of evolution and 40 years of discovery. Nat. Rev. Cancer 2020, 20, 471–480. [Google Scholar] [CrossRef]
- Momand, J.; Jung, D.; Wilczynski, S.; Niland, J. The MDM2 gene amplification database. Nucleic. Acids. Res. 1998, 26, 3453–3459. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.G.; Maiti, A.; Kadia, T.M.; Vyas, P.; Majeti, R.; Wei, A.H.; Garcia-Manero, G.; Craddock, C.; Sallman, D.A.; Kantarjian, H.M. TP53-Mutated Myelodysplastic Syndrome and Acute Myeloid Leukemia: Biology, Current Therapy, and Future Directions. Cancer Discov. 2022, 12, 2516–2529. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.S.; Duong, C.P.; Haupt, S.; Montgomery, K.G.; House, C.M.; Azar, W.J.; Pearson, H.B.; Fisher, O.M.; Read, M.; Guerra, G.R.; et al. Inhibiting the system xC−/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat. Commun. 2017, 8, 14844. [Google Scholar] [CrossRef]
- Ou, Y.; Wang, S.J.; Li, D.; Chu, B.; Gu, W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc. Natl. Acad. Sci. USA 2016, 113, E6806–E6812. [Google Scholar] [CrossRef]
- Zhang, Y.; Qian, Y.; Zhang, J.; Yan, W.; Jung, Y.S.; Chen, M.; Huang, E.; Lloyd, K.; Duan, Y.; Wang, J.; et al. Ferredoxin reductase is critical for p53-dependent tumor suppression via iron regulatory protein 2. Genes Dev. 2017, 31, 1243–1256. [Google Scholar] [CrossRef]
- Hu, W.; Zhang, C.; Wu, R.; Sun, Y.; Levine, A.; Feng, Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc. Natl. Acad. Sci. USA 2010, 107, 7455–7460. [Google Scholar] [CrossRef]
- Tarangelo, A.; Magtanong, L.; Bieging-Rolett, K.T.; Li, Y.; Ye, J.; Attardi, L.D.; Dixon, S.J. p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell Rep. 2018, 22, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Chu, B.; Yang, X.; Liu, Z.; Jin, Y.; Kon, N.; Rabadan, R.; Jiang, X.; Stockwell, B.R.; Gu, W. iPLA2beta-mediated lipid detoxification controls p53-driven ferroptosis independent of GPX4. Nat. Commun. 2021, 12, 3644. [Google Scholar] [CrossRef] [PubMed]
- Kuganesan, N.; Dlamini, S.; Tillekeratne, L.M.V.; Taylor, W.R. Tumor suppressor p53 promotes ferroptosis in oxidative stress conditions independent of modulation of ferroptosis by p21, CDKs, RB, and E2F. J. Biol. Chem. 2021, 297, 101365. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Kroemer, G.; Tang, D. The tumor suppressor protein p53 and the ferroptosis network. Free Radic. Biol. Med. 2019, 133, 162–168. [Google Scholar] [CrossRef]
- Ji, H.; Wang, W.; Li, X.; Han, X.; Zhang, X.; Wang, J.; Liu, C.; Huang, L.; Gao, W. p53: A double-edged sword in tumor ferroptosis. Pharm. Res. 2022, 177, 106013. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gu, W. p53 in ferroptosis regulation: The new weapon for the old guardian. Cell Death Differ. 2022, 29, 895–910. [Google Scholar] [CrossRef]
- Armand, P.; Kim, H.T.; Rhodes, J.; Sainvil, M.M.; Cutler, C.; Ho, V.T.; Koreth, J.; Alyea, E.P.; Hearsey, D.; Neufeld, E.J.; et al. Iron overload in patients with acute leukemia or MDS undergoing myeloablative stem cell transplantation. Biol. Blood. Marrow. Transpl. 2011, 17, 852–860. [Google Scholar] [CrossRef]
- Kautz, L.; Nemeth, E. Molecular liaisons between erythropoiesis and iron metabolism. Blood 2014, 124, 479–482. [Google Scholar] [CrossRef]
- Atilla, E.; Toprak, S.K.; Demirer, T. Current Review of Iron Overload and Related Complications in Hematopoietic Stem Cell Transplantation. Turk. J. Haematol. 2017, 34, 1–9. [Google Scholar] [CrossRef]
- Wermke, M.; Eckoldt, J.; Gotze, K.S.; Klein, S.A.; Bug, G.; de Wreede, L.C.; Kramer, M.; Stolzel, F.; von Bonin, M.; Schetelig, J.; et al. Enhanced labile plasma iron and outcome in acute myeloid leukaemia and myelodysplastic syndrome after allogeneic haemopoietic cell transplantation (ALLIVE): A prospective, multicentre, observational trial. Lancet Haematol. 2018, 5, e201–e210. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.; Duarte, T.L.; Teles, M.J.; Mosteo, L.; Chacim, S.; Aguiar, E.; Pereira-Reis, J.; Oliveira, M.; Silva, A.M.N.; Goncalves, N.; et al. Loss of erythroblasts in acute myeloid leukemia causes iron redistribution with clinical implications. Blood. Adv. 2021, 5, 3102–3112. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, M.; Hu, Y.; Xing, H.; Chen, X.; Zhang, Y.; Zhu, P. Significance of CD71 expression by flow cytometry in diagnosis of acute leukemia. Leuk. Lymphoma 2014, 55, 892–898. [Google Scholar] [CrossRef] [PubMed]
- Gasparetto, M.; Pei, S.; Minhajuddin, M.; Stevens, B.; Smith, C.A.; Seligman, P. Low ferroportin expression in AML is correlated with good risk cytogenetics, improved outcomes and increased sensitivity to chemotherapy. Leuk. Res. 2019, 80, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Trujillo-Alonso, V.; Pratt, E.C.; Zong, H.; Lara-Martinez, A.; Kaittanis, C.; Rabie, M.O.; Longo, V.; Becker, M.W.; Roboz, G.J.; Grimm, J.; et al. FDA-approved ferumoxytol displays anti-leukaemia efficacy against cells with low ferroportin levels. Nat. Nanotechnol. 2019, 14, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.; Bao, J. FerrDb: A manually curated resource for regulators and markers of ferroptosis and ferroptosis-disease associations. Database 2020, 2020, baaa021. [Google Scholar] [CrossRef]
- Zhou, N.; Yuan, X.; Du, Q.; Zhang, Z.; Shi, X.; Bao, J.; Ning, Y.; Peng, L. FerrDb V2: Update of the manually curated database of ferroptosis regulators and ferroptosis-disease associations. Nucleic. Acids. Res. 2023, 51, D571–D582. [Google Scholar] [CrossRef] [PubMed]
- Handschuh, L. Not Only Mutations Matter: Molecular Picture of Acute Myeloid Leukemia Emerging from Transcriptome Studies. J. Oncol. 2019, 2019, 7239206. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Nai, G.Y.; Dai, Y.; Huang, X.J.; Xiong, M.Y.; Yao, X.Y.; Huang, Z.N.; Li, S.N.; Zhou, W.J.; Huang, Y.; et al. Dipetidyl peptidase-4 and transferrin receptor serve as prognostic biomarkers for acute myeloid leukemia. Ann. Transl. Med. 2021, 9, 1381. [Google Scholar] [CrossRef]
- Shao, R.; Wang, H.; Liu, W.; Wang, J.; Lu, S.; Tang, H.; Lu, Y. Establishment of a prognostic ferroptosis-related gene profile in acute myeloid leukaemia. J. Cell Mol. Med. 2021, 25, 10950–10960. [Google Scholar] [CrossRef]
- Huang, X.; Zhou, D.; Ye, X.; Jin, J. A novel ferroptosis-related gene signature can predict prognosis and influence immune microenvironment in acute myeloid leukemia. Bosn. J. Basic. Med. Sci. 2022, 22, 608–628. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Tian, S.; Zhang, P.; Zhang, N.; Shen, Y.; Deng, J. Construction and Validation of a Novel Ferroptosis-Related Prognostic Model for Acute Myeloid Leukemia. Front. Genet. 2021, 12, 708699. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Fu, Y.; Yang, Z.; Gao, Z.; Feng, H.; Zhou, M.; Zhang, L.; Chen, C. Comprehensive Analysis of a Ferroptosis Pattern and Associated Prognostic Signature in Acute Myeloid Leukemia. Front. Pharm. 2022, 13, 866325. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Hong, X.; Huang, X.; Jiang, X.; Jiang, H.; Huang, Y.; Wu, W.; Xue, Y.; Lin, D. Comprehensive analysis of ferroptosis-related gene signatures as a potential therapeutic target for acute myeloid leukemia: A bioinformatics analysis and experimental verification. Front. Oncol. 2022, 12, 930654. [Google Scholar] [CrossRef]
- Wang, J.; Zhuo, Z.; Wang, Y.; Yang, S.; Chen, J.; Wang, Y.; Geng, S.; Li, M.; Du, X.; Lai, P.; et al. Identification and Validation of a Prognostic Risk-Scoring Model Based on Ferroptosis-Associated Cluster in Acute Myeloid Leukemia. Front. Cell Dev. Biol. 2021, 9, 800267. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Lang, Z.; Zhan, Y.; Tao, Q.; Yu, Z.; Chen, L.; Fan, C.; Jin, Y.; Yu, K.; Zhu, B.; et al. A novel 10-gene ferroptosis-related prognostic signature in acute myeloid leukemia. Front. Oncol. 2022, 12, 1023040. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Zhang, L.; Tian, X.; Xiang, X.; Yu, Y.; Zeng, Z.; Cao, Y.; Chen, S.; Sun, A. Identification of immune subtypes of Ph-neg B-ALL with ferroptosis related genes and the potential implementation of Sorafenib. BMC Cancer 2021, 21, 1331. [Google Scholar] [CrossRef] [PubMed]
- Gong, H.; Li, H.; Yang, Q.; Zhang, G.; Liu, H.; Ma, Z.; Peng, H.; Nie, L.; Xiao, X.; Liu, J. A Ferroptosis Molecular Subtype-Related Signature for Predicting Prognosis and Response to Chemotherapy in Patients with Chronic Lymphocytic Leukemia. Biomed. Res. Int. 2022, 2022, 5646275. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.; Li, Y.; Xu, Z.; Miao, Y.; Yin, H.; Kong, Y.; Zhang, X.; Liang, J.; Xia, Y.; Wang, L.; et al. Identifying a novel ferroptosis-related prognostic score for predicting prognosis in chronic lymphocytic leukemia. Front. Immunol. 2022, 13, 962000. [Google Scholar] [CrossRef]
- Zhou, F.; Chen, B. Prognostic significance of ferroptosis-related genes and their methylation in AML. Hematology 2021, 26, 919–930. [Google Scholar] [CrossRef]
- Han, C.; Zheng, J.; Li, F.; Guo, W.; Cai, C. Novel Prognostic Signature for Acute Myeloid Leukemia: Bioinformatics Analysis of Combined CNV-Driven and Ferroptosis-Related Genes. Front. Genet. 2022, 13, 849437. [Google Scholar] [CrossRef]
- Zheng, Z.; Wu, W.; Lin, Z.; Liu, S.; Chen, Q.; Jiang, X.; Xue, Y.; Lin, D. Identification of seven novel ferroptosis-related long non-coding RNA signatures as a diagnostic biomarker for acute myeloid leukemia. BMC Med. Genom. 2021, 14, 236. [Google Scholar] [CrossRef]
- Tao, Y.; Wei, L.; You, H. Ferroptosis-related gene signature predicts the clinical outcome in pediatric acute myeloid leukemia patients and refines the 2017 ELN classification system. Front. Mol. Biosci. 2022, 9, 954524. [Google Scholar] [CrossRef]
- Yu, Y.; Xie, Y.; Cao, L.; Yang, L.; Yang, M.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol. Cell Oncol. 2015, 2, e1054549. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Chai, W.; Xie, M.; Yang, M.; Yu, Y.; Cao, L.; Yang, L. HMGB1 regulates erastin-induced ferroptosis via RAS-JNK/p38 signaling in HL-60/NRAS(Q61L) cells. Am. J. Cancer Res. 2019, 9, 730–739. [Google Scholar]
- Zhu, T.; Liu, B.; Wu, D.; Xu, G.; Fan, Y. Autophagy Regulates VDAC3 Ubiquitination by FBXW7 to Promote Erastin-Induced Ferroptosis in Acute Lymphoblastic Leukemia. Front. Cell Dev. Biol. 2021, 9, 740884. [Google Scholar] [CrossRef]
- Lalonde, M.E.; Sasseville, M.; Gelinas, A.M.; Milanese, J.S.; Beland, K.; Drouin, S.; Haddad, E.; Marcotte, R. Genome-wide CRISPR screens identify ferroptosis as a novel therapeutic vulnerability in acute lymphoblastic leukemia. Haematologica 2023, 108, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wu, W.; Chen, Q.; Zheng, Z.; Jiang, X.; Xue, Y.; Lin, D. TXNRD1: A Key Regulator Involved in the Ferroptosis of CML Cells Induced by Cysteine Depletion In Vitro. Oxid. Med. Cell Longev. 2021, 2021, 7674565. [Google Scholar] [CrossRef]
- Lv, Q.; Niu, H.; Yue, L.; Liu, J.; Yang, L.; Liu, C.; Jiang, H.; Dong, S.; Shao, Z.; Xing, L.; et al. Abnormal Ferroptosis in Myelodysplastic Syndrome. Front. Oncol. 2020, 10, 1656. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, X.; Wang, H. Combination regimen of granulocyte colony-stimulating factor and recombinant human thrombopoietin improves the curative effect on elderly patients with leukemia through inducing pyroptosis and ferroptosis of leukemia cells. Cancer Gene 2022, 29, 1742–1750. [Google Scholar] [CrossRef]
- Birsen, R.; Larrue, C.; Decroocq, J.; Johnson, N.; Guiraud, N.; Gotanegre, M.; Cantero-Aguilar, L.; Grignano, E.; Huynh, T.; Fontenay, M.; et al. APR-246 induces early cell death by ferroptosis in acute myeloid leukemia. Haematologica 2022, 107, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Bruedigam, C.; Porter, A.H.; Song, A.; Vroeg in de Wei, G.; Stoll, T.; Straube, J.; Cooper, L.T.; Cheng, G.; Kahl, V.F.; Sobinoff, A.; et al. Imetelstat-Mediated Alterations in Lipid Metabolism to Induce Ferroptosis As Therapeutic Strategy for Acute Myeloid Leukemia. Blood 2022, 140, 487–488. [Google Scholar] [CrossRef]
- Ma, H.; Liu, Y.; Miao, Z.; Cheng, S.; Zhu, Y.; Wu, Y.; Fan, X.; Yang, J.; Li, X.; Guo, L. Neratinib inhibits proliferation and promotes apoptosis of acute myeloid leukemia cells by activating autophagy-dependent ferroptosis. Drug Dev. Res. 2022, 83, 1641–1653. [Google Scholar] [CrossRef]
- Du, J.; Wang, T.; Li, Y.; Zhou, Y.; Wang, X.; Yu, X.; Ren, X.; An, Y.; Wu, Y.; Sun, W.; et al. DHA inhibits proliferation and induces ferroptosis of leukemia cells through autophagy dependent degradation of ferritin. Free Radic. Biol. Med. 2019, 131, 356–369. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.Y.; Huang, Z.X.; Chen, G.Q.; Sheng, F.; Zheng, Y.S. Typhaneoside prevents acute myeloid leukemia (AML) through suppressing proliferation and inducing ferroptosis associated with autophagy. Biochem. Biophys. Res. Commun. 2019, 516, 1265–1271. [Google Scholar] [CrossRef] [PubMed]
- Lou, S.; Hong, H.; Maihesuti, L.; Gao, H.; Zhu, Z.; Xu, L.; Tian, S.; Kai, G.; Huang, G.; Zhao, H. Inhibitory effect of hydnocarpin D on T-cell acute lymphoblastic leukemia via induction of autophagy-dependent ferroptosis. Exp. Biol. Med. Maywood 2021, 246, 1541–1553. [Google Scholar] [CrossRef]
- Catanzaro, E.; Turrini, E.; Kerre, T.; Sioen, S.; Baeyens, A.; Guerrini, A.; Abdi Bellau, M.L.; Sacchetti, G.; Paganetto, G.; Krysko, D.V.; et al. Perillaldehyde is a new ferroptosis inducer with a relevant clinical potential for acute myeloid leukemia therapy. Biomed. Pharmacother. 2022, 154, 113662. [Google Scholar] [CrossRef]
- Li, Q.; Su, R.; Bao, X.; Cao, K.; Du, Y.; Wang, N.; Wang, J.; Xing, F.; Yan, F.; Huang, K.; et al. Glycyrrhetinic acid nanoparticles combined with ferrotherapy for improved cancer immunotherapy. Acta Biomater. 2022, 144, 109–120. [Google Scholar] [CrossRef]
- Greco, G.; Schnekenburger, M.; Catanzaro, E.; Turrini, E.; Ferrini, F.; Sestili, P.; Diederich, M.; Fimognari, C. Discovery of Sulforaphane as an Inducer of Ferroptosis in U-937 Leukemia Cells: Expanding Its Anticancer Potential. Cancers 2021, 14, 76. [Google Scholar] [CrossRef]
- Lai, X.; Sun, Y.; Zhang, X.; Wang, D.; Wang, J.; Wang, H.; Zhao, Y.; Liu, X.; Xu, X.; Song, H.; et al. Honokiol Induces Ferroptosis by Upregulating HMOX1 in Acute Myeloid Leukemia Cells. Front. Pharm. 2022, 13, 897791. [Google Scholar] [CrossRef]
- Du, Y.; Bao, J.; Zhang, M.J.; Li, L.L.; Xu, X.L.; Chen, H.; Feng, Y.B.; Peng, X.Q.; Chen, F.H. Targeting ferroptosis contributes to ATPR-induced AML differentiation via ROS-autophagy-lysosomal pathway. Gene 2020, 755, 144889. [Google Scholar] [CrossRef]
- Chen, L.; Fang, W.; Liu, J.; Qi, X.; Zhao, L.; Wang, Y.; Liu, Y.; Kong, D.; Sun, X.; Li, X.; et al. Poricoic acid A (PAA) inhibits T-cell acute lymphoblastic leukemia through inducing autophagic cell death and ferroptosis. Biochem. Biophys. Res. Commun. 2022, 608, 108–115. [Google Scholar] [CrossRef]
- Du, Y.; Han, M.; Cao, K.; Li, Q.; Pang, J.; Dou, L.; Liu, S.; Shi, Z.; Yan, F.; Feng, S. Gold Nanorods Exhibit Intrinsic Therapeutic Activity via Controlling N6-Methyladenosine-Based Epitranscriptomics in Acute Myeloid Leukemia. ACS Nano 2021, 15, 17689–17704. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, X.; Liu, N.; Shi, Y.; Liu, Y.; Ouyang, L.; Tam, S.; Xiao, D.; Liu, S.; Wen, F.; et al. A Nuclear Long Non-Coding RNA LINC00618 Accelerates Ferroptosis in a Manner Dependent upon Apoptosis. Mol Ther. 2021, 29, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.H.; Huang, J.J.; Zu, P.; Liu, J.; Gao, X.; Du, J.W.; Li, Y.F. CircKDM4C upregulates P53 by sponging hsa-let-7b-5p to induce ferroptosis in acute myeloid leukemia. Environ. Toxicol. 2021, 36, 1288–1302. [Google Scholar] [CrossRef]
- Jin, L.; Tong, L. PAQR3 inhibits proliferation and aggravates ferroptosis in acute lymphoblastic leukemia through modulation Nrf2 stability. Immun. Inflamm. Dis. 2021, 9, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Pontel, L.B.; Bueno-Costa, A.; Morellato, A.E.; Carvalho Santos, J.; Roue, G.; Esteller, M. Acute lymphoblastic leukemia necessitates GSH-dependent ferroptosis defenses to overcome FSP1-epigenetic silencing. Redox Biol. 2022, 55, 102408. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, R.M.; Lazo-Langner, A. Systematic review of azacitidine regimens in myelodysplastic syndrome and acute myeloid leukemia. BMC Hematol. 2018, 18, 3. [Google Scholar] [CrossRef]
- Liu, X.; Shi, H.; Shen, J.; Li, Y.; Yan, W.; Sun, Y.; Liao, A.; Tan, Y.; Yang, W.; Wang, H. Dual Growth Factor (rhTPO + G-CSF) and Chemotherapy Combination Regimen for Elderly Patients with Acute Myeloid Leukemia: A Phase II Single-Arm Multicenter Study. Int. J. Gen. Med. 2021, 14, 6093–6099. [Google Scholar] [CrossRef]
- Fujihara, K.M.; Zhang, B.Z.; Jackson, T.D.; Ogunkola, M.O.; Nijagal, B.; Milne, J.V.; Sallman, D.A.; Ang, C.S.; Nikolic, I.; Kearney, C.J.; et al. Eprenetapopt triggers ferroptosis, inhibits NFS1 cysteine desulfurase, and synergizes with serine and glycine dietary restriction. Sci. Adv. 2022, 8, eabm9427. [Google Scholar] [CrossRef]
- Ge, C.; Zhang, S.; Mu, H.; Zheng, S.; Tan, Z.; Huang, X.; Xu, C.; Zou, J.; Zhu, Y.; Feng, D.; et al. Emerging Mechanisms and Disease Implications of Ferroptosis: Potential Applications of Natural Products. Front. Cell Dev. Biol. 2021, 9, 774957. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Trachootham, D.; Liu, J.; Chen, G.; Pelicano, H.; Garcia-Prieto, C.; Lu, W.; Burger, J.A.; Croce, C.M.; Plunkett, W.; et al. Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat. Cell Biol. 2012, 14, 276–286. [Google Scholar] [CrossRef]
- Forte, D.; Garcia-Fernandez, M.; Sanchez-Aguilera, A.; Stavropoulou, V.; Fielding, C.; Martin-Perez, D.; Lopez, J.A.; Costa, A.S.H.; Tronci, L.; Nikitopoulou, E.; et al. Bone Marrow Mesenchymal Stem Cells Support Acute Myeloid Leukemia Bioenergetics and Enhance Antioxidant Defense and Escape from Chemotherapy. Cell Metab. 2020, 32, 829–843.e829. [Google Scholar] [CrossRef] [PubMed]
- von Samson-Himmelstjerna, F.A.; Kolbrink, B.; Riebeling, T.; Kunzendorf, U.; Krautwald, S. Progress and Setbacks in Translating a Decade of Ferroptosis Research into Clinical Practice. Cells 2022, 11, 2134. [Google Scholar] [CrossRef]
- Zhao, J.; Jia, Y.; Mahmut, D.; Deik, A.A.; Jeanfavre, S.; Clish, C.B.; Sankaran, V.G. Human hematopoietic stem cell vulnerability to ferroptosis. Cell 2023, 186, 732–747.e16. [Google Scholar] [CrossRef]
- Kinowaki, Y.; Taguchi, T.; Onishi, I.; Kirimura, S.; Kitagawa, M.; Yamamoto, K. Overview of Ferroptosis and Synthetic Lethality Strategies. Int. J. Mol. Sci. 2021, 22, 9271. [Google Scholar] [CrossRef]
- Bedoui, S.; Herold, M.J.; Strasser, A. Emerging connectivity of programmed cell death pathways and its physiological implications. Nat. Rev. Mol. Cell Biol. 2020, 21, 678–695. [Google Scholar] [CrossRef] [PubMed]



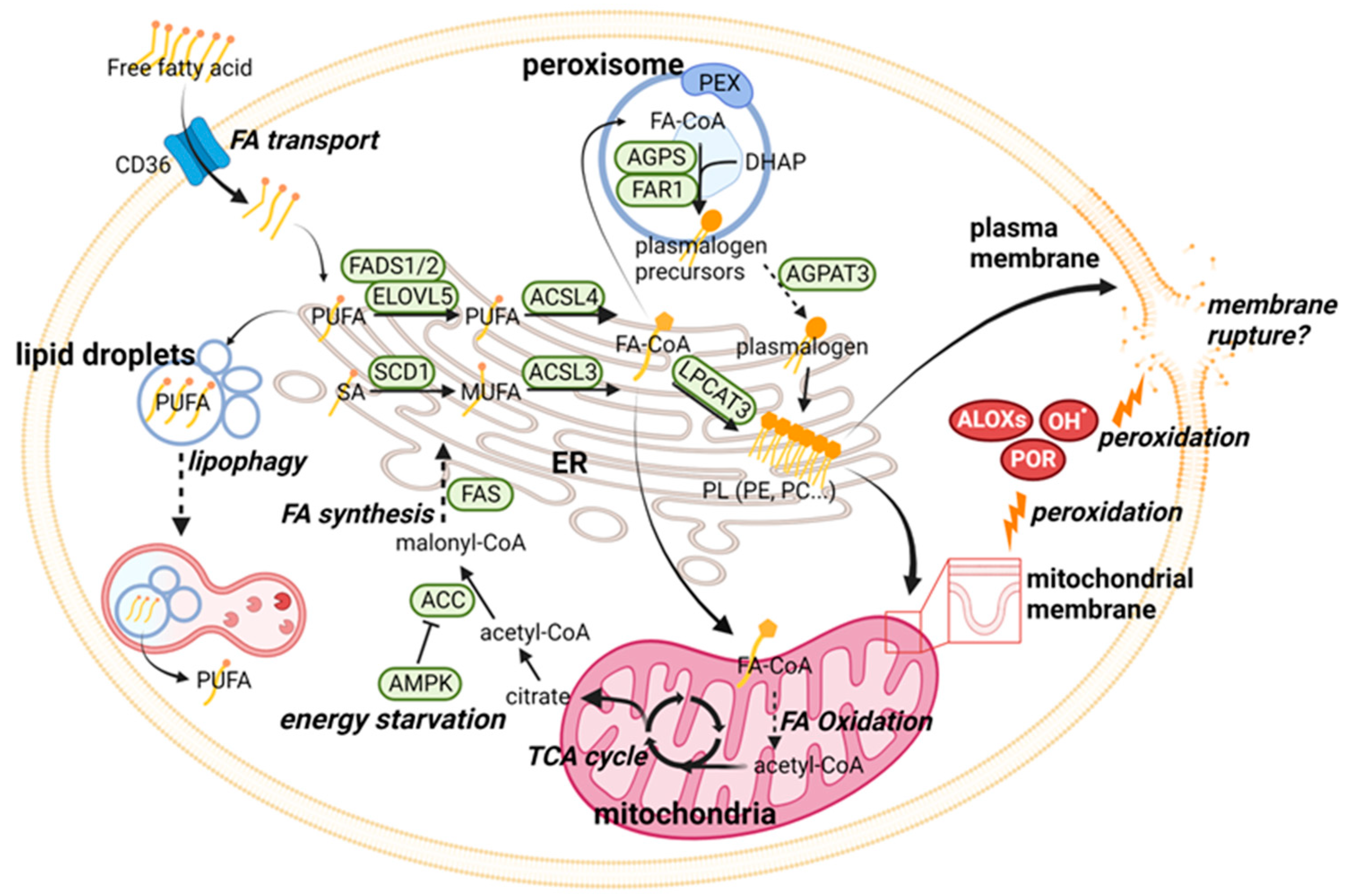{kind=link}
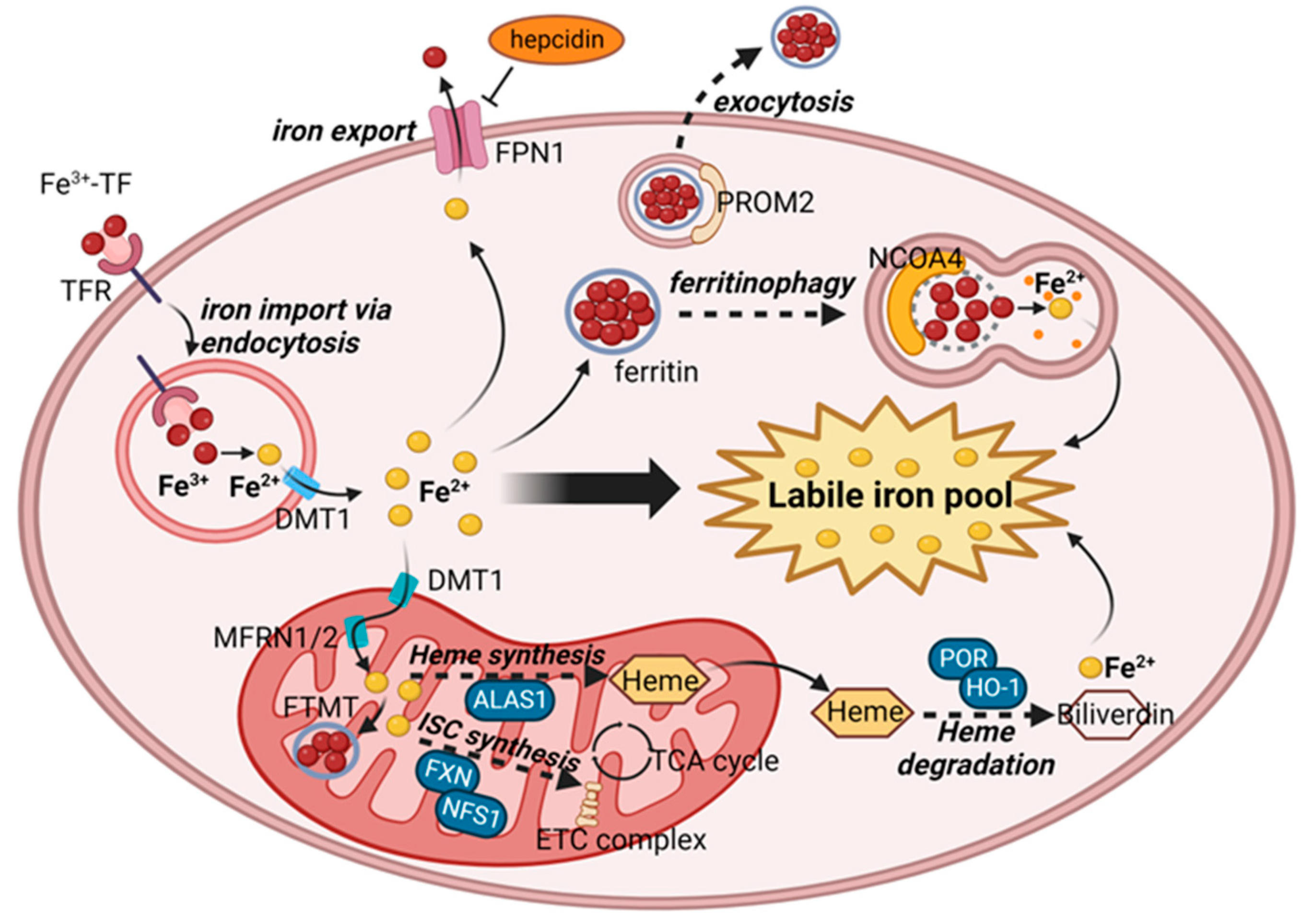{kind=link}
| Category | Pathway | Major Molecular Players |
|---|---|---|
| Phospholipid (PL) metabolism | Lipid peroxidation | ALOXs, POR, PEBP1 |
| PUFA-PL synthesis | FADS1, FADS2, ELOVL5, ACSL4, LPCAT3 | |
| MUFA-PL synthesis | SCD1, ACSL3 | |
| Plasmalogen synthesis | PEX3, PEX10, AGPS, FAR1, AGPAT3 | |
| Energy metabolism | AMPK, ACC | |
| Iron metabolism | Iron import/export | TF, TFR1, DMT1 (import)/FPN1 (export) |
| Ferritin synthesis/export | FTH1 (synthesis)/PROM2 (export) | |
| Ferritinophagy | AMPK, NCOA4 | |
| Mitochondrial iron storage | MFRN1, MFRN2, FTMT | |
| Heme synthesis/degradation | ALAS1 (synthesis)/POR, HO-1 (degradation) | |
| Iron-sulfur cluster synthesis | FXN, NFS1 | |
| Cell-cell contact | Hippo pathway | CDH1, YAP, TAZ |
| Ferroptosis protection mechanisms (Section 3) | System xc−—GSH—GPX4 axis | SLC7A11, GPX4 |
| GSH synthesis | γGCS, GSS | |
| Cyst(e)ine metabolism | CBS, SAHH | |
| Glutamine metabolism | SLC1A5, GLS2, GOT1 | |
| mTORC pathway | mTORC1 | |
| FSP1-CoQ axis | FSP1, MESH1, PPARα, MDM2, MDMX | |
| NRF2 pathway | NRF2, KEAP1 | |
| DHODH/GPD2-CoQ axis | DHODH, GPD2 | |
| GSH1-BH4-DHFR axis | BH2, BH4, DHFR |
| Models | In Vitro | In Vivo | Ferroptosis Inducers | Molecular Mechanisms of Cell Death Including Ferroptosis and Potential Combination Strategies | Ref. | ||
|---|---|---|---|---|---|---|---|
| System xc−/GSH/GPX4 inhibitors | AML | HL-60 | ● | Erastin | Mixed modes of cell death with necroptosis JNK and p38 activation Combination with AraC + daunorubicin | [184] | |
| AML | HL-60 | ● | ● | Erastin | Mixed modes of cell death with apoptosis JNK and p38 activation Nuclear translocation of HMGB1 | [185] | |
| AML | Cell lines Primary cells PDX cells | ● ● | ● | Sulfasalazine | Oxidative stress–induced cell death including ferroptosis Combination with AraC + daunorubicin | [113] | |
| ALL | Cell lines | ● | ● | Erastin, IKE | Combination with rapamycin-induced autophagy | [186] | |
| B-ALL | Cell lines PDX cells | ● ● | Erastin, sulfasalazine, RSL3, FIN56 | High sensitivity to ferroptosis due to low FSP1 expression | [187] | ||
| CML | K562 | ● | Cysteine depletion | Combination with auranofin-induced TXNRD1 inhibition | [188] | ||
| Clinically available agents | MDS | Cell lines Primary cells | ● ● | Decitabine | GSH/GPX4 inhibition–induced ferroptosis + necroptosis Combination with system xc− inhibition (erastin) | [189] | |
| AML | Cell lines | ● | ● | Thrombopoietin | Suppression of GPX4 transcription through EP300 inhibition Combination with G-CSF-induced pyroptosis | [190] | |
| AML | Cell lines Primary cells | ● ● | ● | APR-246 | GSH inhibition Combination with system xc− inhibition or GPX4 inhibition | [191] | |
| AML | Cell lines PDX cells | ● | ● ● | Imetelstat | Enhanced PUFA-phospholipid synthesis Combination with AraC + daunorubicin | [192] | |
| AML | HL-60 | ● | Neratinib | Mixed modes of cell death with apoptosis FTH1 downregulation and cellular iron accumulation Autophagy | [193] | ||
| Natural compounds and their derivatives | AML | Cell lines Primary cells | ● ● | ● | Dihydroartemisinin | Mixed modes of cell death with apoptosis AMPK-mediated ferritinophagy and increased cellular iron Mitochondrial oxidative stress and ETC inhibition | [194] |
| AML | Cell lines Primary cells | ● ● | ● | Typhaneoside | Mixed modes of cell death with apoptosis AMPK-mediated ferritinophagy and increased cellular iron Mitochondrial oxidative stress and ETC inhibition | [195] | |
| T-ALL | Cell lines | ● | Hydnocarpin D | Mixed modes of cell death with apoptosis Autophagy | [196] | ||
| AML | HL-60 Primary cells | ● ● | Perillaldehyde | GSH/GPX4 inhibition | [197] | ||
| AML | Cell lines | ● | ● | Glycyrrhetinic acid nanoparticle | GSH/GPX4 inhibition Combination with ferumoxytol or anti-PDL1 antibody | [198] | |
| AML | Cell lines | ● | Sulforaphane | GSH/GPX4 inhibition | [199] | ||
| AML | Cell lines | ● | Honokiol | Mixed modes of cell death with apoptosis HO-1 upregulation | [200] | ||
| AML | Cell lines | ● | ● | 4-Amino-2-trifluoromethyl- phenyl retinate | GSH/GPX4 inhibition NCOA4-mediated ferritinophagy NRF2 downregulation | [201] | |
| T-ALL | Cell lines Primary cells | ● ● | ● | Poricoic acid A | GSH/GPX4 inhibition–induced ferroptosis + apoptosis AMPK-mediated autophagy Mitochondrial oxidative stress and ETC inhibition | [202] | |
| Others | AML | Cell lines Primary cells MLL/AF9 mouse AML | ● ● | ● | ALDH3a2 inhibition | Alteration of lipid biosynthesis and cellular composition Combination with GPX4 inhibition Combination with AraC + daunorubicin | [96] |
| AML | Cell lines | ● | ● | GNR-CSP12 | GSH/GPX4 inhibition m6A hypomethylation and suppression of downstream genes Combination with TKIs including nilotinib or anti-PD1 antibody | [203] | |
| AML | Cell lines | ● | LINC00618 | System xc− inhibition–induced ferroptosis + apoptosis Combination with vincristine | [204] | ||
| AML | Cell lines | ● | ● | circKDM4C | GPX4 inhibition p53 upregulation Combination with system xc− inhibition (erastin) | [205] | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akiyama, H.; Carter, B.Z.; Andreeff, M.; Ishizawa, J. Molecular Mechanisms of Ferroptosis and Updates of Ferroptosis Studies in Cancers and Leukemia. Cells 2023, 12, 1128. https://doi.org/10.3390/cells12081128
Akiyama H, Carter BZ, Andreeff M, Ishizawa J. Molecular Mechanisms of Ferroptosis and Updates of Ferroptosis Studies in Cancers and Leukemia. Cells. 2023; 12(8):1128. https://doi.org/10.3390/cells12081128
Chicago/Turabian StyleAkiyama, Hiroki, Bing Z. Carter, Michael Andreeff, and Jo Ishizawa. 2023. "Molecular Mechanisms of Ferroptosis and Updates of Ferroptosis Studies in Cancers and Leukemia" Cells 12, no. 8: 1128. https://doi.org/10.3390/cells12081128