
The Role of WNT Pathway Mutations in Cancer Development and an Overview of Therapeutic Options

{kind=link}
{kind=link}
{kind=link}
{kind=link}
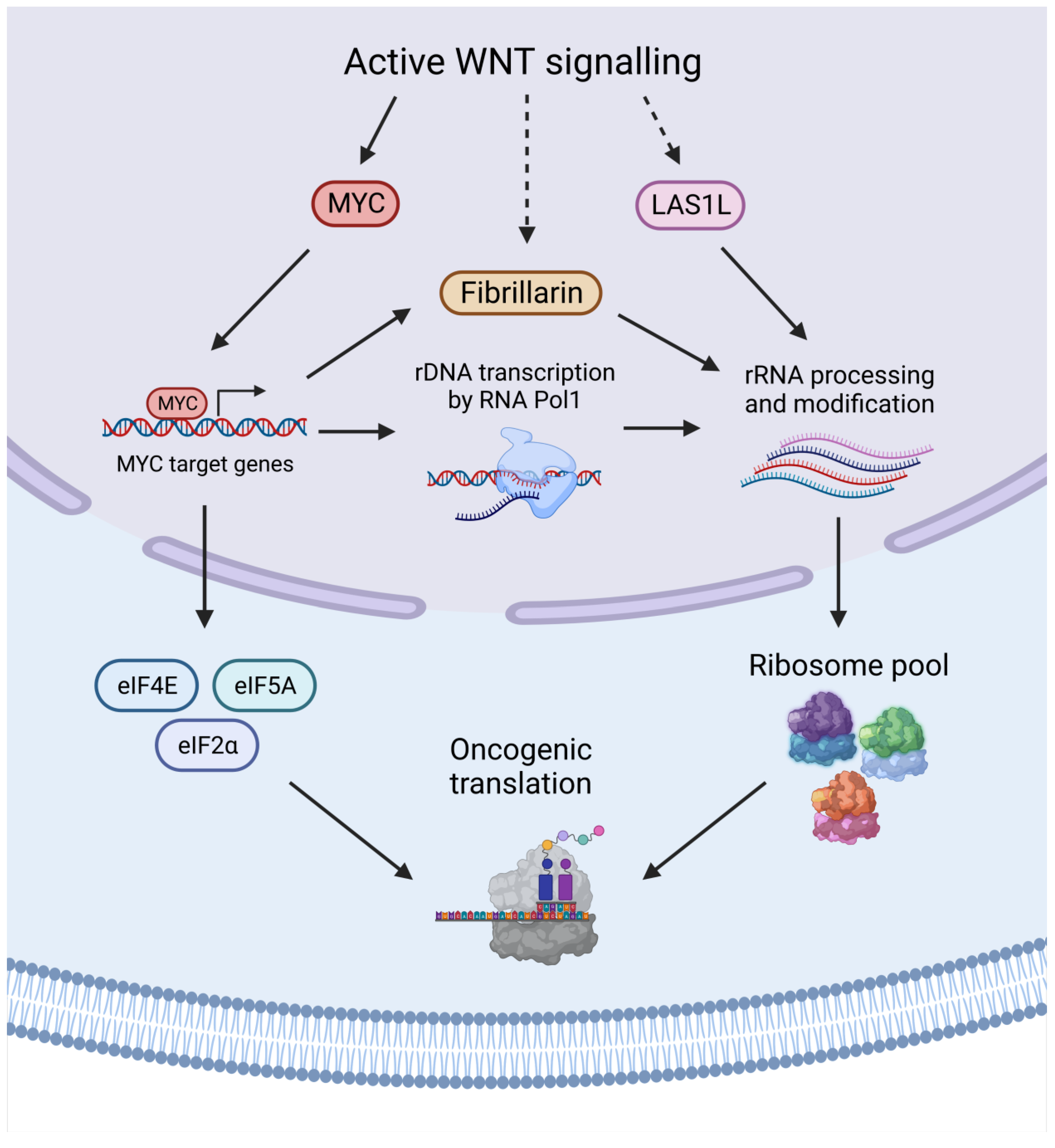{kind=link}
Abstract
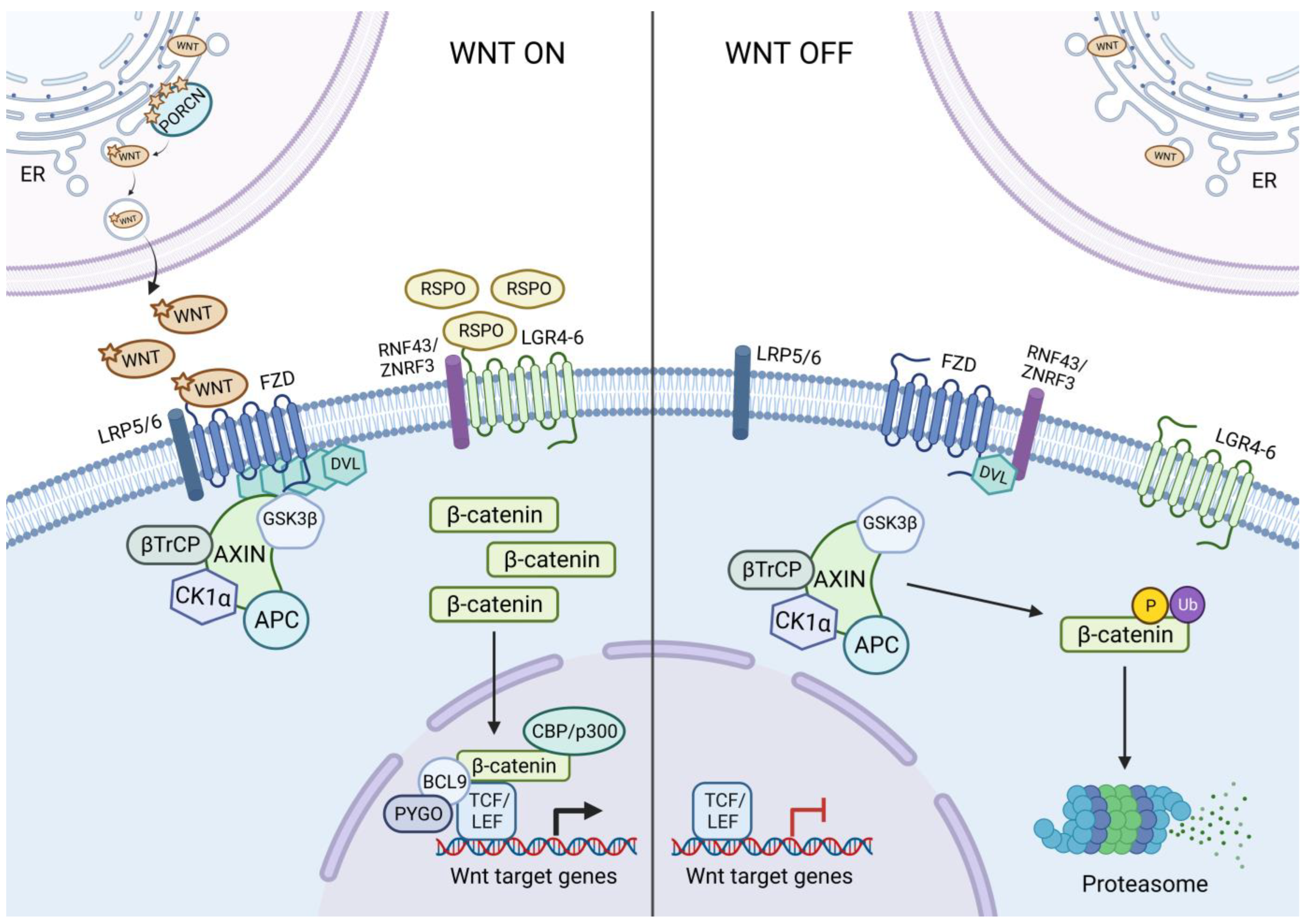
:1. Introduction
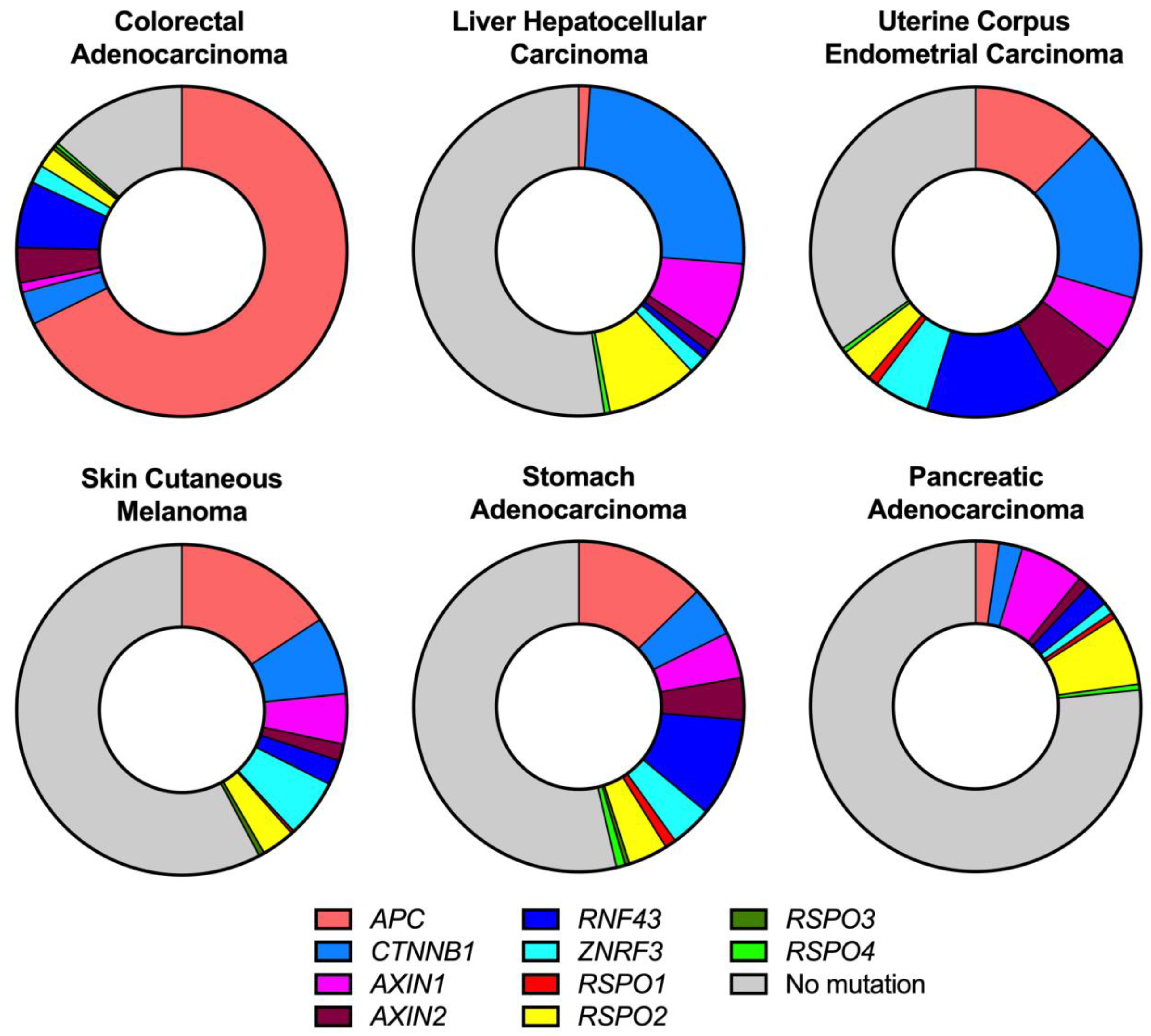
2. Wnt Activating Mutations in Cancer

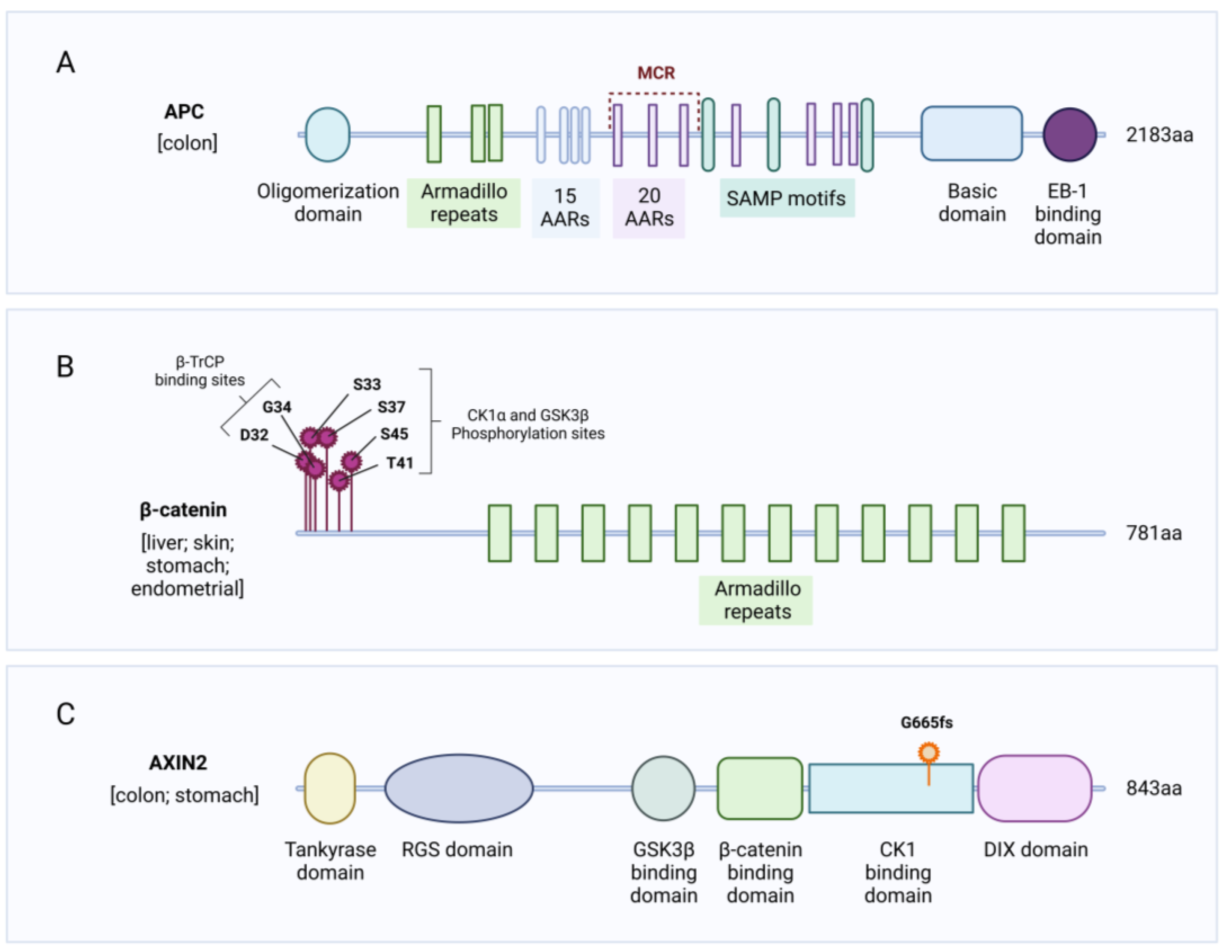
2.1. APC Mutations
2.2. CTNNB1 Mutations
2.3. AXIN Mutations
2.4. RNF43/ZNRF3 Mutations
2.5. RSPO Mutations
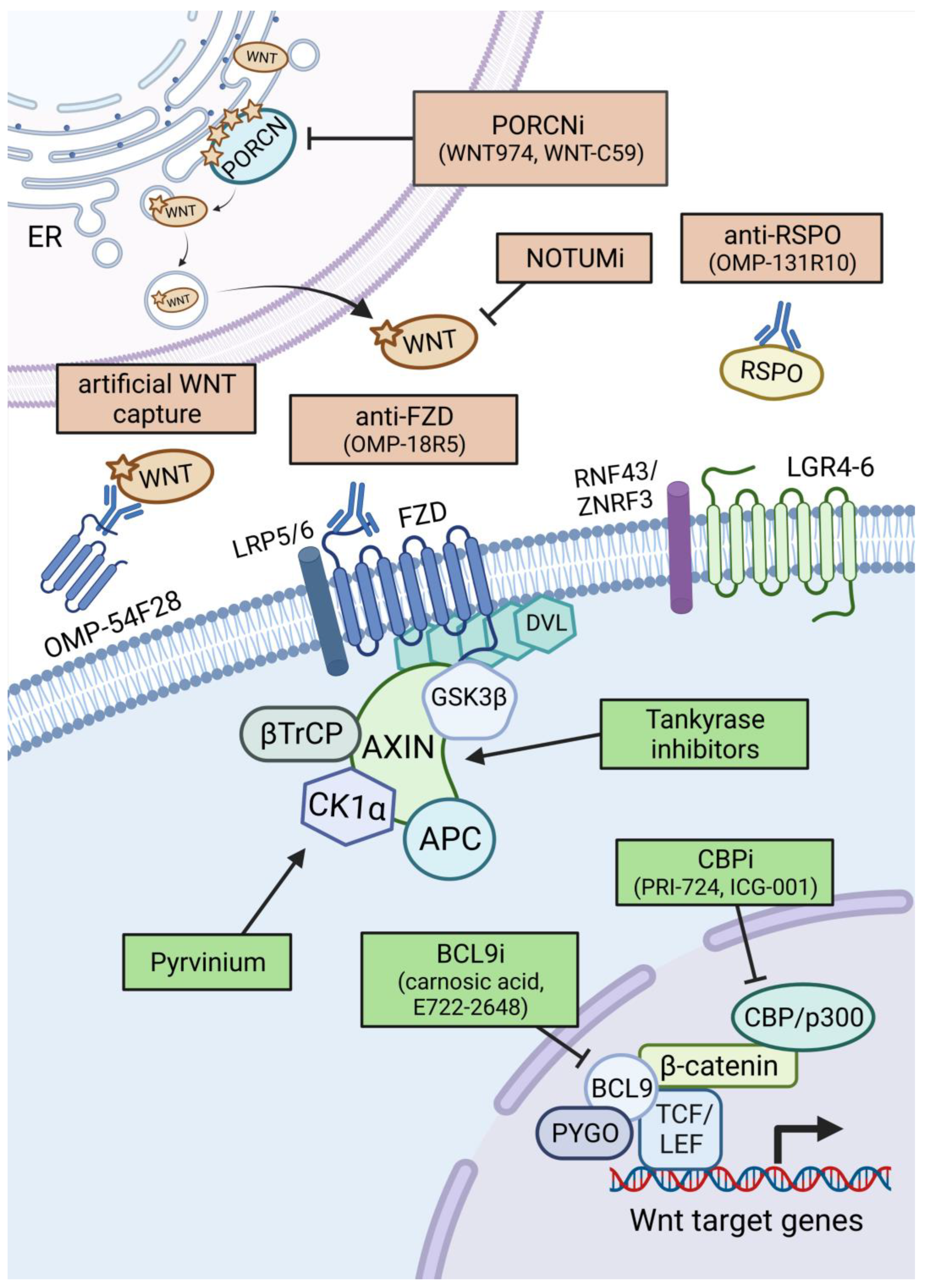
3. Therapeutic Options against Aberrant WNT Signalling in Cancer
3.1. WNT Ligand/Receptor-Based Therapies
3.1.1. Porcupine Inhibitors
3.1.2. WNT Ligand and Receptor Targeting Strategies
3.1.3. Other Extracellular Targeting Strategies
3.2. β-Catenin Targeting Therapies
3.2.1. Targeting β-Catenin in the Destruction Complex
3.2.2. Targeting β-Catenin-Mediated Transcription
4. A Therapeutic Option beyond WNT Signalling
4.1. The Key Players in Translation and Their Links to WNT Signalling
4.2. Are Ribosomes Altered in Cancer?
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nusslein-Volhard, C.; Wieschaus, E. Mutations Affecting Sgement Number and Polarity in Drosophila. Nature 1980, 287, 795–801. [Google Scholar] [CrossRef]
- Nusse, R.; Varmus, H.E. Many Tumors Induced by the Mouse Mammary Tumor Virus Contain a Provirus Integrated in the Same Region of the Host Genome. Cell 1982, 31, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Rijsewijk, F.; Schuermann, M.; Wagenaar, E.; Parren, L.; Weigel, D.; Nusse, R. The Drosophila Homolog of the Mouse Mammary Oncogene Int-1 Is Identical to the Segment Polarity Gene Wingless. Cell 1987, 50, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Jackstadt, R.; Hodder, M.C.; Sansom, O.J. WNT and β-Catenin in Cancer: Genes and Therapy. Annu. Rev. Cancer Biol. 2020, 4, 177–196. [Google Scholar] [CrossRef] [Green Version]
- Takada, R.; Satomi, Y.; Kurata, T.; Ueno, N.; Norioka, S.; Kondoh, H.; Takao, T.; Takada, S. Monounsaturated Fatty Acid Modification of Wnt Protein: Its Role in Wnt Secretion. Dev. Cell 2006, 11, 791–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herr, P.; Basler, K. Porcupine-Mediated Lipidation Is Required for Wnt Recognition by Wls. Dev. Biol. 2012, 361, 392–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niehrs, C. The Complex World of WNT Receptor Signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 767–779. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt Signaling in Cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef]
- Huels, D.J.; Ridgway, R.A.; Radulescu, S.; Leushacke, M.; Campbell, A.D.; Biswas, S.; Leedham, S.; Serra, S.; Chetty, R.; Moreaux, G.; et al. E-Cadherin Can Limit the Transforming Properties of Activating β-Catenin Mutations. EMBO J. 2015, 34, 2321–2333. [Google Scholar] [CrossRef]
- Li, V.S.W.; Ng, S.S.; Boersema, P.J.; Low, T.Y.; Karthaus, W.R.; Gerlach, J.P.; Mohammed, S.; Heck, A.J.R.; Maurice, M.M.; Mahmoudi, T.; et al. Wnt Signaling through Inhibition of β-Catenin Degradation in an Intact Axin1 Complex. Cell 2012, 149, 1245–1256. [Google Scholar] [CrossRef] [Green Version]
- Nusse, R.; Clevers, H. Wnt/b-Catenin Signaling, Disease, and Emerging Therapeutic Modalities Roel. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Y.Y.; Fu, C.; Ishikawa, S.; Stella, A.; Kojima, M.; Shitoh, K.; Schreiber, E.M.; Day, B.W.; Liu, B. APC Is Essential for Targeting Phosphorylated β-Catenin to the SCFβ-TrCP Ubiquitin Ligase. Mol. Cell 2008, 32, 652–661. [Google Scholar] [CrossRef] [PubMed]
- Cliffe, A.; Hamada, F.; Bienz, M. A Role of Dishevelled in Relocating Axin to the Plasma Membrane during Wingless Signaling. Curr. Biol. 2003, 13, 960–966. [Google Scholar] [CrossRef]
- van Tienen, L.M.; Mieszczanek, J.; Fiedler, M.; Rutherford, T.J.; Bienz, M. Constitutive Scaffolding of Multiple Wnt Enhanceosome Components by Legless. Elife 2017, 6, e20882. [Google Scholar] [CrossRef]
- Valenta, T.; Hausmann, G.; Basler, K. The Many Faces and Functions of B-Catenin. EMBO J. 2012, 31, 2714–2736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, T.; Sparks, A.; Rago, C.; Hermeking, H.; Zawel, L.; da Costa, L.; Morin, P.; Vogelstein, B.; Kinzler, K. Identification of C-MYC as a Target of the APC Pathway. Science 1998, 281, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Tetsu, O.; McCormick, F. Beta-Catenin Regulates Expression of Cyclin D1 in Colon Carcinoma Cells. Nature 1999, 398, 422–426. [Google Scholar] [CrossRef]
- de Lau, W.; Peng, W.C.; Gros, P.; Clevers, H. The R-Spondin/Lgr5/Rnf43 Module: Regulator of Wnt Signal Strength. Genes. Dev. 2014, 28, 305–316. [Google Scholar] [CrossRef] [Green Version]
- Yan, K.S.; Janda, C.Y.; Chang, J.; Zheng, G.X.Y.; Larkin, K.A.; Luca, V.C.; Chia, L.A.; Mah, A.T.; Han, A.; Terry, J.M.; et al. Non-Equivalence of Wnt and R-Spondin Ligands during Lgr5 + Intestinal Stem-Cell Self-Renewal. Nature 2017, 545, 238–242. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Cui, J.; Yu, W.; Wu, L.; Carmon, K.S.; Liu, Q.J. Differential Activities and Mechanisms of the Four R-Spondins in Potentiating Wnt/-Catenin Signaling. J. Biol. Chem. 2018, 293, 9759–9769. [Google Scholar] [CrossRef] [Green Version]
- Kleeman, S.O.; Leedham, S.J. Not All Wnt Activation Is Equal: Ligand-Dependent versus Ligand-Independent Wnt Activation in Colorectal Cancer. Cancers 2020, 12, 3355. [Google Scholar] [CrossRef] [PubMed]
- Cruciat, C.M.; Niehrs, C. Secreted and Transmembrane Wnt Inhibitors and Activators. Cold Spring Harb. Perspect. Biol. 2013, 5, a015081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakugawa, S.; Langton, P.F.; Zebisch, M.; Howell, S.A.; Chang, T.-H.; Liu, Y.; Feizi, T.; Bineva, G.; O’Reilly, N.; Snijders, A.P.; et al. Notum Deacylates Wnt Proteins to Suppress Signalling Activity. Nature 2015, 519, 187–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flanagan, D.J.; Pentinmikko, N.; Luopajärvi, K.; Willis, N.J.; Gilroy, K.; Raven, A.P.; Mcgarry, L.; Englund, J.I.; Webb, A.T.; Scharaw, S.; et al. NOTUM from Apc-Mutant Cells Biases Clonal Competition to Initiate Cancer. Nature 2021, 594, 430–435. [Google Scholar] [CrossRef]
- Getz, G.; Gabriel, S.B.; Cibulskis, K.; Lander, E.; Sivachenko, A.; Sougnez, C.; Lawrence, M.; Kandoth, C.; Dooling, D.; Fulton, R.; et al. Integrated Genomic Characterization of Endometrial Carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Jeong, S. Mutation Hotspots in the β-Catenin Gene: Lessons from the Human Cancer Genome Databases. Mol. Cells 2019, 42, 8–16. [Google Scholar]
- van der Zee, M.; Jia, Y.; Wang, Y.; Heijmans-Antonissen, C.; Ewing, P.C.; Franken, P.; Demayo, F.J.; Lydon, J.P.; Burger, C.W.; Fodde, R.; et al. Alterations in Wnt-β-Catenin and Pten Signalling Play Distinct Roles in Endometrial Cancer Initiation and Progression. J. Pathol. 2013, 230, 48–58. [Google Scholar] [CrossRef]
- Aguilera, O.; Fraga, M.F.; Ballestar, E.; Paz, M.F.; Herranz, M.; Espada, J.; García, J.M.; Mũoz, A.; Esteller, M.; González-Sancho, J.M. Epigenetic Inactivation of the Wnt Antagonist DICKKOPF-1 (DKK-1) Gene in Human Colorectal Cancer. Oncogene 2006, 25, 4116–4121. [Google Scholar] [CrossRef] [Green Version]
- Nojima, M.; Suzuki, H.; Toyota, M.; Watanabe, Y.; Maruyama, R.; Sasaki, S.; Sasaki, Y.; Mita, H.; Nishikawa, N.; Yamaguchi, K.; et al. Frequent Epigenetic Inactivation of SFRP Genes and Constitutive Activation of Wnt Signaling in Gastric Cancer. Oncogene 2007, 26, 4699–4713. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Arman Aksoy, B.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the CBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The CBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Näthke, I.S. The Adenomatous Polyposis Coli Protein: The Achilles Heel of the Gut Epithelium. Annu. Rev. Cell Dev. Biol. 2004, 20, 337–366. [Google Scholar] [CrossRef] [PubMed]
- Christie, M.; Jorissen, R.N.; Mouradov, D.; Sakthianandeswaren, A.; Li, S.; Day, F.; Tsui, C.; Lipton, L.; Desai, J.; Jones, I.T.; et al. Different APC Genotypes in Proximal and Distal Sporadic Colorectal Cancers Suggest Distinct WNT/β-Catenin Signalling Thresholds for Tumourigenesis. Oncogene 2013, 32, 4675–4682. [Google Scholar] [CrossRef] [PubMed]
- Albuquerque, C.; Breukel, C.; van der Luijt, R.; Fidalgo, P.; Lage, P.; Fodde, R.; Smits, R.; Slors, F.J.M.; Leita, C.N. The ‘Just-Right’ Signaling Model: APC Somatic Mutations Are Selected Based on a Specific Level of Activation of the β-Catenin Signaling Cascade. Hum. Mol. Genet. 2002, 11, 1549–1560. [Google Scholar] [CrossRef] [Green Version]
- Leedham, S.J.; Rodenas-cuadrado, P.; Howarth, K.; Lewis, A.; Mallappa, S.; Segditsas, S.; Davis, H.; Jeffery, R.; Rodriguez-justo, M.; Keshav, S.; et al. A Basal Gradient of Wnt and Stem-Cell Number Influences Regional Tumour Distribution in Human and Mouse Intestinal Tracts. Gut 2011, 62, 83–94. [Google Scholar] [CrossRef]
- Moser, A.M.Y.R.; Pitot, H.C.; Dove, W.F.; Clo, D. A Dominant Mutation That Predisposes Intestinal Neoplasia in the Mouse. Science 1990, 247, 4–6. [Google Scholar] [CrossRef]
- Pollard, P.; Deheragoda, M.; Segditsas, S.; Lewis, A.; Rowan, A.; Howarth, K.; Willis, L.; Nye, E.; McCart, A.; Mandir, N.; et al. The Apc 1322T Mouse Develops Severe Polyposis Associated with Submaximal Nuclear Beta-Catenin Expression. Gastroenterology 2009, 136, 2204–2213.e13. [Google Scholar] [CrossRef]
- Trautmann, M.; Rehkämper, J.; Gevensleben, H.; Becker, J.; Wardelmann, E.; Hartmann, W.; Grünewald, I.; Huss, S. Novel Pathogenic Alterations in Pediatric and Adult Desmoid-Type Fibromatosis—A Systematic Analysis of 204 Cases. Sci. Rep. 2020, 10, 3368. [Google Scholar] [CrossRef] [Green Version]
- Fatima, I.; Barman, S.; Rai, R.; Thiel, K.W.; Chandra, V. Targeting Wnt Signaling in Endometrial Cancer. Cancers 2021, 13, 2351. [Google Scholar] [CrossRef]
- Rebouissou, S.; Franconi, A.; Calderaro, J.; Letouz, E.; Imbeaud, S.; Pilati, C.; Nault, J.-C.; Couchy, G.; Laurent, A.; Balabaud, C.; et al. Genotype-Phenotype Correlation of CTNNB1 Mutations Reveals Different ß-Catenin Activity Associated with Liver Tumor Progression. Hepatology 2016, 64, 2047–2061. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.; Kurnit, K.C.; Djordjevic, B.; Singh, C.; Munsell, M.F.; Wang, W.L.; Lazar, A.J.; Zhang, W.; Broaddus, R. Nuclear β-Catenin Localization and Mutation of the CTNNB1 Gene: A Context-Dependent Association. Mod. Pathol. 2018, 31, 1553–1559. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liu, P.; Lavrijsen, M.; Li, S.; Zhang, R.; Li, S.; van de Geer, W.S.; van de Werken, H.J.G.; Peppelenbosch, M.P.; Smits, R. Evaluation of AXIN1 and AXIN2 as Targets of Tankyrase Inhibition in Hepatocellular Carcinoma Cell Lines. Sci. Rep. 2021, 11, 7470. [Google Scholar] [CrossRef] [PubMed]
- Feng, G.J.; Cotta, W.; Wei, X.Q.; Poetz, O.; Evans, R.; Jardé, T.; Reed, K.; Meniel, V.; Williams, G.T.; Clarke, A.R.; et al. Conditional Disruption of Axin1 Leads to Development of Liver Tumors in Mice. Gastroenterology 2012, 143, 1650–1659. [Google Scholar] [CrossRef] [PubMed]
- Salahshor, S.; Woodgett, J.R. The Links between Axin and Carcinogenesis. J. Clin. Pathol. 2005, 58, 225–236. [Google Scholar] [CrossRef] [Green Version]
- Giannakis, M.; Hodis, E.; Jasmine Mu, X.; Yamauchi, M.; Rosenbluh, J.; Cibulskis, K.; Saksena, G.; Lawrence, M.S.; Qian, Z.R.; Nishihara, R.; et al. RNF43 Is Frequently Mutated in Colorectal and Endometrial Cancers. Nat. Genet. 2014, 46, 1264–1266. [Google Scholar] [CrossRef]
- Wang, K.; Yuen, S.T.; Xu, J.; Lee, S.P.; Yan, H.H.N.; Shi, S.T.; Siu, H.C.; Deng, S.; Chu, K.M.; Law, S.; et al. Whole-Genome Sequencing and Comprehensive Molecular Profiling Identify New Driver Mutations in Gastric Cancer. Nat. Genet. 2014, 46, 573–582. [Google Scholar] [CrossRef]
- van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective Derivation of a Living Organoid Biobank of Colorectal Cancer Patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Lavrijsen, M.; Bakker, A.; Magierowski, M.; Magierowska, K.; Liu, P.; Wang, W.; Peppelenbosch, M.P.; Smits, R. Commonly Observed RNF43 Mutations Retain Functionality in Attenuating Wnt/β-Catenin Signaling and Unlikely Confer Wnt-Dependency onto Colorectal Cancers. Oncogene 2020, 39, 3458–3472. [Google Scholar] [CrossRef]
- Seshagiri, S.; Stawiski, E.W.; Durinck, S.; Modrusan, Z.; Storm, E.E.; Conboy, C.B.; Chaudhuri, S.; Guan, Y.; Janakiraman, V.; Jaiswal, B.S.; et al. Recurrent R-Spondin Fusions in Colon Cancer. Nature 2012, 488, 660–664. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, T.; Ogawa, R.; Yoshida, H.; Taniguchi, H.; Kojima, M.; Saito, Y.; Sekine, S. EIF3E–RSPO2 and PIEZO1–RSPO2 Fusions in Colorectal Traditional Serrated Adenoma. Histopathology 2019, 75, 266–273. [Google Scholar] [CrossRef]
- Kleeman, S.O.; Koelzer, V.H.; Jones, H.J.S.; Vazquez, E.G.; Davis, H.; East, J.E.; Arnold, R.; Koppens, M.A.J.; Blake, A.; Domingo, E.; et al. Exploiting Differential Wnt Target Gene Expression to Generate a Molecular Biomarker for Colorectal Cancer Stratification. Gut 2020, 69, 1092–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, T.; Schatoff, E.M.; Murphy, C.; Zafra, M.P.; Wilkinson, J.E.; Elemento, O.; Dow, L.E. R-Spondin Chromosome Rearrangements Drive Wnt-Dependent Tumour Initiation and Maintenance in the Intestine. Nat. Commun. 2017, 8, 15945. [Google Scholar] [CrossRef] [PubMed]
- Longerich, T.; Endris, V.; Neumann, O.; Rempel, E.; Kirchner, M.; Abadi, Z.; Uhrig, S.; Kriegsmann, M.; Weiss, K.H.; Breuhahn, K.; et al. RSPO2 Gene Rearrangement: A Powerful Driver of β-Catenin Activation in Liver Tumours. Gut 2019, 68, 1287–1296. [Google Scholar] [CrossRef]
- Li, C.; Cao, J.; Zhang, N.; Tu, M.; Xu, F.; Wei, S.; Chen, X.; Xu, Y. Identification of RSPO2 Fusion Mutations and Target Therapy Using a Porcupine Inhibitor. Sci. Rep. 2018, 8, 14244. [Google Scholar] [CrossRef] [Green Version]
- Robinson, D.; van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ter Steege, E.J.; Bakker, E.R.M. The Role of R-Spondin Proteins in Cancer Biology. Oncogene 2021, 40, 6469–6478. [Google Scholar] [CrossRef]
- Martin-Orozco, E.; Sanchez-Fernandez, A.; Ortiz-Parra, I.; Ayala-San Nicolas, M. WNT Signaling in Tumors: The Way to Evade Drugs and Immunity. Front. Immunol. 2019, 10, 2854. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.S.; Park, J.I. Wnt Signaling in Cancer: Therapeutic Targeting of Wnt Signaling beyond β-Catenin and the Destruction Complex. Exp. Mol. Med. 2020, 52, 183–191. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Pan, S.; Hsieh, M.H.; Ng, N.; Sun, F.; Wang, T.; Kasibhatla, S.; Vanasse, G.; Harris, J.L. Targeting Wnt-Driven Cancer through the Inhibition of Porcupine by LGK974. Proc. Natl. Acad. Sci. USA 2013, 110, 20224–20229. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Hao, H.X.; Growney, J.D.; Woolfenden, S.; Bottiglio, C.; Ng, N.; Lu, B.; Hsieh, M.H.; Bagdasarian, L.; Meyer, R.; et al. Inactivating Mutations of RNF43 Confer Wnt Dependency in Pancreatic Ductal Adenocarcinoma. Proc. Natl. Acad. Sci. USA 2013, 110, 12649–12654. [Google Scholar] [CrossRef] [Green Version]
- Madan, B.; Ke, Z.; Harmston, N.; Ho, S.Y.; Frois, A.O.; Alam, J.; Jeyaraj, D.A.; Pendharkar, V.; Ghosh, K.; Virshup, I.H.; et al. Wnt Addiction of Genetically Defined Cancers Reversed by PORCN Inhibition. Oncogene 2016, 35, 2197–2207. [Google Scholar] [CrossRef] [Green Version]
- Koo, B.K.; van Es, J.H.; van den Born, M.; Clevers, H. Porcupine Inhibitor Suppresses Paracrine Wnt-Driven Growth of Rnf43;Znrf3-Mutant Neoplasia. Proc. Natl. Acad. Sci. USA 2015, 112, 7548–7550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proffitt, K.D.; Madan, B.; Ke, Z.; Pendharkar, V.; Ding, L.; Lee, M.A.; Hannoush, R.N.; Virshup, D.M. Pharmacological Inhibition of the Wnt Acyltransferase PORCN Prevents Growth of WNT-Driven Mammary Cancer. Cancer Res. 2013, 73, 502–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huels, D.J.; Bruens, L.; Hodder, M.C.; Cammareri, P.; Campbell, A.D.; Ridgway, R.A.; Gay, D.M.; Solar-Abboud, M.; Faller, W.J.; Nixon, C.; et al. Wnt Ligands Influence Tumour Initiation by Controlling the Number of Intestinal Stem Cells. Nat. Commun. 2018, 9, 1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funck-Brentano, T.; Nilsson, K.H.; Brommage, R.; Henning, P.; Lerner, U.H.; Koskela, A.; Tuukkanen, J.; Cohen-Solal, M.; Movérare-Skrtic, S.; Ohlsson, C. Porcupine Inhibitors Impair Trabecular and Cortical Bone Mass and Strength in Mice. J. Endocrinol. 2018, 238, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Madan, B.; McDonald, M.J.; Foxa, G.E.; Diegel, C.R.; Williams, B.O.; Virshup, D.M. Bone Loss from Wnt Inhibition Mitigated by Concurrent Alendronate Therapy. Bone Res. 2018, 6, 17. [Google Scholar] [CrossRef] [Green Version]
- Le, P.N.; McDermott, J.D.; Jimeno, A. Targeting the Wnt Pathway in Human Cancers: Therapeutic Targeting with a Focus on OMP-54F28. Pharmacol. Ther. 2015, 146, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Gurney, A.; Axelrod, F.; Bond, C.J.; Cain, J.; Chartier, C.; Donigan, L.; Fischer, M.; Chaudhari, A.; Ji, M.; Kapoun, A.M.; et al. Wnt Pathway Inhibition via the Targeting of Frizzled Receptors Results in Decreased Growth and Tumorigenicity of Human Tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 11717–11722. [Google Scholar] [CrossRef] [Green Version]
- Flanagan, D.J.; Barker, N.; di Costanzo, N.S.; Mason, E.A.; Gurney, A.; Meniel, V.S.; Koushyar, S.; Austin, C.R.; Ernst, M.; Pearson, H.B.; et al. Frizzled-7 Is Required for Wnt Signaling in Gastric Tumors with and without APC Mutations. Cancer Res. 2019, 79, 970–981. [Google Scholar] [CrossRef] [Green Version]
- Storm, E.E.; Durinck, S.; de Sousa E Melo, F.; Tremayne, J.; Kljavin, N.; Tan, C.; Ye, X.; Chiu, C.; Pham, T.; Hongo, J.A.; et al. Targeting PTPRK-RSPO3 Colon Tumours Promotes Differentiation and Loss of Stem-Cell Function. Nature 2016, 529, 97–100. [Google Scholar] [CrossRef]
- Huang, S.-M.A.; Mishina, Y.M.; Liu, S.; Cheung, A.; Stegmeier, F.; Michaud, G.A.; Charlat, O.; Wiellette, E.; Zhang, Y.; Wiessner, S.; et al. Tankyrase Inhibition Stabilizes Axin and Antagonizes Wnt Signalling. Nature 2009, 461, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Waaler, J.; Machon, O.; Tumova, L.; Dinh, H.; Korinek, V.; Wilson, S.R.; Paulsen, J.E.; Pedersen, N.M.; Eide, T.J.; Machonova, O.; et al. A Novel Tankyrase Inhibitor Decreases Canonical Wnt Signaling in Colon Carcinoma Cells and Reduces Tumor Growth in Conditional APC Mutant Mice. Cancer Res. 2012, 72, 2822–2833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, T.; Chan, E.; Callow, M.; Waaler, J.; Boggs, J.; Blake, R.A.; Magnuson, S.; Sambrone, A.; Schutten, M.; Firestein, R.; et al. A Novel Tankyrase Small-Molecule Inhibitor Suppresses APC Mutation—Driven Colorectal Tumor Growth. Cancer Res. 2013, 73, 3132–3145. [Google Scholar] [CrossRef] [Green Version]
- Shen, C.; Li, B.; Astudillo, L.; Deutscher, M.P.; Cobb, M.H.; Capobianco, A.J.; Lee, E.; Robbins, D.J. The CK1α Activator Pyrvinium Enhances the Catalytic Efficiency (Kcat/ Km) of CK1α. Biochemistry 2019, 58, 5102–5106. [Google Scholar] [CrossRef] [PubMed]
- Thorne, C.A.; Hanson, A.J.; Schneider, J.; Tahinci, E.; Orton, D.; Cselenyi, C.S.; Jernigan, K.K.; Meyers, K.C.; Hang, B.I.; Waterson, A.G.; et al. Small-Molecule Inhibition of Wnt Signaling through Activation of Casein Kinase 1α. Nat. Chem. Biol. 2010, 6, 829–836. [Google Scholar] [CrossRef] [Green Version]
- Wiegering, A.; Uthe, F.W.; Hüttenrauch, M.; Mühling, B.; Linnebacher, M.; Krummenast, F.; Germer, C.T.; Thalheimer, A.; Otto, C. The Impact of Pyrvinium Pamoate on Colon Cancer Cell Viability. Int. J. Color. Dis. 2014, 29, 1189–1198. [Google Scholar] [CrossRef]
- Li, B.; Flaveny, C.A.; Giambelli, C.; Liang Fei, D.; Han, L.; Hang, B.I.; Bai, F.; Pei, X.H.; Nose, V.; Burlingame, O.; et al. Repurposing the FDA-Approved Pinworm Drug Pyrvinium as a Novel Chemotherapeutic Agent for Intestinal Polyposis. PLoS ONE 2014, 9, e0101969. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Ma, L.; Paik, H.; Sirota, M.; Wei, W.; Chua, M.S.; So, S.; Butte, A.J. Reversal of Cancer Gene Expression Correlates with Drug Efficacy and Reveals Therapeutic Targets. Nat. Commun. 2017, 8, 16022. [Google Scholar] [CrossRef] [Green Version]
- Emami, K.H.; Nguyen, C.; Ma, H.; Kim, D.H.; Jeong, K.W.; Eguchi, M.; Moon, R.T.; Teo, J.-L.; Oh, W.; Kim, Y.; et al. A Small Molecule Inhibitor Of-CateninCREB-Binding Protein Transcription. Proc. Natl. Acad. Sci. USA 2004, 101, 12682–12687. [Google Scholar] [CrossRef] [Green Version]
- Gabata, R.; Harada, K.; Mizutani, Y.; Ouchi, H.; Yoshimura, K.; Sato, Y.; Kitao, A.; Kimura, K.; Kouji, H.; Miyashita, T.; et al. Anti-Tumor Activity of the Small Molecule Inhibitor PRI-724 against β-Catenin-Activated Hepatocellular Carcinoma. Anticancer. Res. 2020, 40, 5211–5219. [Google Scholar] [CrossRef]
- Mieszczanek, J.; van Tienen, L.M.; Ibrahim, A.E.K.; Winton, D.J.; Bienz, M. Bcl9 and Pygo Synergise Downstream of Apc to Effect Intestinal Neoplasia in FAP Mouse Models. Nat. Commun. 2019, 10, 724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gay, D.M.; Ridgway, R.A.; Müeller, M.; Hodder, M.C.; Hedley, A.; Clark, W.; Leach, J.D.; Jackstadt, R.; Nixon, C.; Huels, D.J.; et al. Loss of BCL9/9l Suppresses Wnt Driven Tumourigenesis in Models That Recapitulate Human Cancer. Nat. Commun. 2019, 10, 723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, K.; Fallahi, M.; Dai, X. Chromatin Effector Pygo2 Regulates Mammary Tumor Initiation and Heterogeneity in MMTV-Wnt1 Mice. Oncogene 2014, 33, 632–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takada, K.; Zhu, D.; Bird, G.H.; Sukhdeo, K.; Zhao, J.-J.; Mani, M.; Lemieux, M.; Carrasco, D.E.; Ryan, J.; Horst, D.; et al. Targeted Disruption of the BCL9/β-Catenin Complex Inhibits Oncogenic Wnt Signaling. Sci. Transl. Med. 2012, 4, 148ra117. [Google Scholar] [CrossRef] [Green Version]
- de la Roche, M.; Ibrahim, A.E.; Mieszczanek, J.; Bienz, M. LEF1 and B9L Shield β-Catenin from Inactivation by Axin, Desensitizing Colorectal Cancer Cells to Tankyrase Inhibitors. Cancer Res. 2014, 74, 1495–1505. [Google Scholar] [CrossRef] [Green Version]
- Tanton, H.; Sewastianik, T.; Seo, H.-S.; Remillard, D.; St Pierre, R.; Bala, P.; Aitymbayev, D.; Dennis, P.; Adler, K.; Geffken, E.; et al. A Novel β-Catenin/BCL9 Complex Inhibitor Blocks Oncogenic Wnt Signaling and Disrupts Cholesterol Homeostasis in Colorectal Cancer. Sci. Adv. 2022, 8, eabm3108. [Google Scholar] [CrossRef]
- Bhat, M.; Robichaud, N.; Hulea, L.; Sonenberg, N.; Pelletier, J.; Topisirovic, I. Targeting the Translation Machinery in Cancer. Nat. Rev. Drug Discov. 2015, 14, 261–278. [Google Scholar] [CrossRef]
- Bruno, P.M.; Liu, Y.; Park, G.Y.; Murai, J.; Koch, C.E.; Eisen, T.J.; Pritchard, J.R.; Pommier, Y.; Lippard, S.J.; Hemann, M.T. A Subset of Platinum-Containing Chemotherapeutic Agents Kills Cells by Inducing Ribosome Biogenesis Stress. Nat. Med. 2017, 23, 461–471. [Google Scholar] [CrossRef] [Green Version]
- Jackson, R.J.; Hellen, C.U.T.; Pestova, T.V. The Mechanism of Eukaryotic Translation Initiation and Principles of Its Regulation. Nat. Rev. Mol. Cell Biol. 2010, 11, 113–127. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; de Souza Alves, V.; von der Haar, T.; O’Keefe, L.; Lenchine, R.V.; Jensen, K.B.; Liu, R.; Coldwell, M.J.; Wang, X.; Proud, C.G. Regulation of the Elongation Phase of Protein Synthesis Enhances Translation Accuracy and Modulates Lifespan. Curr. Biol. 2019, 29, 737–749.e5. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.C.L.; Kanellos, G.; Vlahov, N.; Alexandrou, C.; Willis, A.E.; Knight, J.R.P.; Sansom, O.J. Translation Initiation in Cancer at a Glance. J. Cell Sci. 2021, 134, jcs248476. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.R.P.; Garland, G.; Pöyry, T.; Mead, E.; Vlahov, N.; Sfakianos, A.; Grosso, S.; De-Lima-Hedayioglu, F.; Mallucci, G.R.; von der Haar, T.; et al. Control of Translation Elongation in Health and Disease. Dis. Model. Mech. 2020, 13, dmm043208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruggero, D. Translational Control in Cancer Etiology. Cold Spring Harb. Perspect. Biol. 2013, 5, a012336. [Google Scholar] [CrossRef]
- Gay, D.M.; Lund, A.H.; Jansson, M.D. Translational Control through Ribosome Heterogeneity and Functional Specialization. Trends Biochem. Sci. 2021, 47, 66–81. [Google Scholar] [CrossRef]
- Sansom, O.J.; Meniel, V.S.; Muncan, V.; Phesse, T.J.; Wilkins, J.A.; Reed, K.R.; Vass, J.K.; Athineos, D.; Clevers, H.; Clarke, A.R. Myc Deletion Rescues Apc Deficiency in the Small Intestine. Nature 2007, 446, 676–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minnee, E.; Faller, W.J. Translation Initiation and Its Relevance in Colorectal Cancer. FEBS J. 2021, 288, 6635–6651. [Google Scholar] [CrossRef] [PubMed]
- van Riggelen, J.; Yetil, A.; Felsher, D.W. MYC as a Regulator of Ribosome Biogenesis and Protein Synthesis. Nat. Rev. Cancer 2010, 10, 301–309. [Google Scholar] [CrossRef]
- Rosenwald, I.B.; Rhoads, D.B.; Callanan, L.D.; Isselbacher, K.J.; Schmidt, E.V. Increased Expression of Eukaryotic Translation Initiation Factors EIF-4E and EIF-2a in Response to Growth Induction by c-Myc (Delayed Early Genes/Gene Expression/Protein Synthesis). Proc. Natl. Acad. Sci. USA 1993, 90, 6175–6178. [Google Scholar] [CrossRef] [Green Version]
- Grandori, C.; Gomez-Roman, N.; Felton-Edkins, Z.A.; Ngouenet, C.; Galloway, D.A.; Eisenman, R.N.; White, R.J. C-Myc Binds to Human Ribosomal DNA and Stimulates Transcription of RRNA Genes by RNA Polymerase I. Nat. Cell Biol. 2005, 7, 311–318. [Google Scholar] [CrossRef]
- Harold, C.M.; Buhagiar, A.F.; Cheng, Y.; Baserga, S.J. Ribosomal Rna Transcription Regulation in Breast Cancer. Genes 2021, 12, 502. [Google Scholar] [CrossRef]
- Ruggero, D.; Pandolfi, P.P. Does the Ribosome Translate Cancer? Nat. Rev. Cancer 2003, 3, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The Consensus Molecular Subtypes of Colorectal Cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Laurentius Smit, W.; Spaan, C.N.; Johannes De Boer, R.; Ramesh, P.; Martins Garcia, T.; Meijer, J.; Ludovicus, J.; Vermeulen, M.; Lezzerini, M.; Macinnes, A.W.; et al. Driver Mutations of the Adenoma-Carcinoma Sequence Govern the Intestinal Epithelial Global Translational Capacity. Proc. Natl. Acad. Sci. USA 2020, 117, 25560–25570. [Google Scholar] [CrossRef]
- Faller, W.J.; Jackson, T.J.; Knight, J.R.P.; Ridgway, R.A.; Jamieson, T.; Karim, S.A.; Jones, C.; Radulescu, S.; Huels, D.J.; Myant, K.B.; et al. MTORC1-Mediated Translational Elongation Limits Intestinal Tumour Initiation and Growth. Nature 2015, 517, 497–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiegering, A.; Uthe, F.W.; Jamieson, T.; Ruoss, Y.; Hüttenrauch, M.; Küspert, M.; Pfann, C.; Nixon, C.; Herold, S.; Walz, S.; et al. Targeting Translation Initiation Bypasses Signaling Crosstalk Mechanisms That Maintain High MYC Levels in Colorectal Cancer. Cancer Discov. 2015, 5, 768–781. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, S.; Gay, D.; Uthe, F.W.; Denk, S.; Paauwe, M.; Matthes, N.; Diefenbacher, M.E.; Bryson, S.; Warrander, F.C.; Erhard, F.; et al. A MYC–GCN2–EIF2α Negative Feedback Loop Limits Protein Synthesis to Prevent MYC-Dependent Apoptosis in Colorectal Cancer. Nat. Cell Biol. 2019, 21, 1413–1424. [Google Scholar] [CrossRef]
- Knight, J.R.P.; Alexandrou, C.; Skalka, G.L.; Vlahov, N.; Pennel, K.; Officer, L.; Teodosio, A.; Kanellos, G.; Gay, D.M.; May-Wilson, S.; et al. MNK Inhibition Sensitizes KRAS-Mutant Colorectal Cancer to MTORC1 Inhibition by Reducing EIF4E Phosphorylation and c-MYC Expression. Cancer Discov. 2020, 11, 1228–1247. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.R.; Vlahov, N.; Gay, D.M.; Ridgway, R.A.; Liam Faller, W.; Proud, C.; Mallucci, G.R.; von der Haar, T.; Mark Smales, C.; Willis, A.E.; et al. Rpl24 Bst Mutation Suppresses Colorectal Cancer by Promoting EEF2 Phosphorylation via EEF2K. Elife 2021, 10, e69729. [Google Scholar] [CrossRef] [PubMed]
- el Khoury, W.; Nasr, Z. Deregulation of Ribosomal Proteins in Human Cancers. Biosci. Rep. 2021, 41, BSR20211577. [Google Scholar] [CrossRef]
- Morral, C.; Stanisavljevic, J.; Hernando-Momblona, X.; Mereu, E.; Álvarez-Varela, A.; Cortina, C.; Stork, D.; Slebe, F.; Turon, G.; Whissell, G.; et al. Zonation of Ribosomal DNA Transcription Defines a Stem Cell Hierarchy in Colorectal Cancer. Cell Stem Cell 2020, 26, 845–861.e12. [Google Scholar] [CrossRef]
- Madan, B.; Harmston, N.; Nallan, G.; Montoya, A.; Faull, P.; Petretto, E.; Virshup, D.M. Temporal Dynamics of Wnt-Dependent Transcriptome Reveal an Oncogenic Wnt/MYC/Ribosome Axis. J. Clin. Investig. 2018, 128, 5620–5633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weeks, S.E.; Kammerud, S.C.; Metge, B.J.; AlSheikh, H.A.; Schneider, D.A.; Chen, D.; Wei, S.; Mobley, J.A.; Ojesina, A.I.; Shevde, L.A.; et al. Inhibiting β-Catenin Disables Nucleolar Functions in Triple-Negative Breast Cancer. Cell Death Dis. 2021, 12, 242. [Google Scholar] [CrossRef] [PubMed]
- Genuth, N.R.; Barna, M. Heterogeneity and Specialized Functions of Translation Machinery: From Genes to Organisms. Nat. Rev. Genet. 2018, 19, 431–452. [Google Scholar] [CrossRef]
- Ferretti, M.B.; Karbstein, K. Does Functional Specialization of Ribosomes Really Exist? RNA 2019, 25, 521–538. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Fujii, K.; Kovary, K.M.; Genuth, N.R.; Röst, H.L.; Teruel, M.N.; Barna, M. Heterogeneous Ribosomes Preferentially Translate Distinct Subpools of MRNAs Genome-Wide. Mol. Cell 2017, 67, 71–83.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopes, T.; Norris, K.; Agapiou, M.; McCarthy, C.G.P.; Lewis, P.A.; O’Connell, M.J.; Fontana, J.; Aspden, J.L. Ribosome Heterogeneity in Drosophila Melanogaster Gonads through Paralog-Switching. Nucleic Acids Res. 2022, 50, 2240–2257. [Google Scholar] [CrossRef]
- Genuth, N.R.; Shi, Z.; Kunimoto, K.; Hung, V.; Xu, A.F.; Kerr, C.H.; Tiu, G.C.; Oses-Prieto, J.A.; Salomon-Shulman, R.E.A.; Axelrod, J.D.; et al. A Stem Cell Roadmap of Ribosome Heterogeneity Reveals a Function for RPL10A in Mesoderm Production. Nat. Commun. 2022, 13, 5491. [Google Scholar] [CrossRef]
- Bretones, G.; Alvarez, M.G.; Arango, J.R.; Rodríguez, D.; Nadeu, F.; Prado, M.A.; Valdés, R.; Valdés-Mas, V.; Puente, D.A.; Paulo, J.A.; et al. Altered Patterns of Global Protein Synthesis and Translational Fidelity in RPS15-Mutated Chronic Lymphocytic Leukemia. Blood 2018, 132, 2375–2388. [Google Scholar]
- Kampen, K.R.; Sulima, S.O.; Vereecke, S.; de Keersmaecker, K. Hallmarks of Ribosomopathies. Nucleic Acids Res. 2020, 48, 1013–1028. [Google Scholar] [CrossRef]
- Elhamamsy, A.R.; Metge, B.J.; Alsheikh, H.A.; Shevde, L.A.; Samant, R.S. Ribosome Biogenesis: A Central Player in Cancer Metastasis and Therapeutic Resistance. Cancer Res. 2022, 82, 2344–2353. [Google Scholar] [CrossRef]
- Parks, M.M.; Kurylo, C.M.; Dass, R.A.; Bojmar, L.; Lyden, D.; Vincent, C.T.; Blanchard, S.C. Variant Ribosomal RNA Alleles Are Conserved and Exhibit Tissue-Specific Expression. Sci. Adv. 2018, 4, eaao0665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothschild, D.; Susanto, T.; Spence, J.P.; Genuth, N.R.; Sinnott-Armstrong, N.; Pritchard, J.K.; Barna, M. A Comprehensive RRNA Variation Atlas in Health and Disease. bioRxiv 2023. [Google Scholar] [CrossRef]
- Krogh, N.; Asmar, F.; Côme, C.; Munch-Petersen, H.F.; Grønbæk, K.; Nielsen, H. Profiling of Ribose Methylations in Ribosomal RNA from Diffuse Large B-Cell Lymphoma Patients for Evaluation of Ribosomes as Drug Targets. NAR Cancer 2020, 2, zcaa035. [Google Scholar] [CrossRef]
- Marcel, V.; Kielbassa, J.; Marchand, V.; Natchiar, K.S.; Paraqindes, H.; Nguyen Van Long, F.; Ayadi, L.; Bourguignon-Igel, V.; lo Monaco, P.; Monchiet, D.; et al. Ribosomal RNA 2′O-Methylation as a Novel Layer of Inter-Tumour Heterogeneity in Breast Cancer. NAR Cancer 2020, 2, zcaa036. [Google Scholar] [CrossRef]
- Jansson, M.D.; Häfner, S.J.; Altinel, K.; Tehler, D.; Krogh, N.; Jakobsen, E.; Andersen, J.V.; Andersen, K.L.; Schoof, E.M.; Ménard, P.; et al. Regulation of Translation by Site-Specific Ribosomal RNA Methylation. Nat. Struct. Mol. Biol. 2021, 28, 889–899. [Google Scholar] [CrossRef]
- Esguerra, J.; Warringer, J.; Blomberg, A. Functional Importance of Individual RRNA 2′-O-Ribose Methylations Revealed by High-Resolution Phenotyping. RNA 2008, 14, 649–656. [Google Scholar] [CrossRef] [Green Version]
- Babaian, A.; Rothe, K.; Girodat, D.; Minia, I.; Djondovic, S.; Milek, M.; Spencer Miko, S.E.; Wieden, H.J.; Landthaler, M.; Morin, G.B.; et al. Loss of M1acp3Ψ Ribosomal RNA Modification Is a Major Feature of Cancer. Cell Rep. 2020, 31, 107611. [Google Scholar] [CrossRef]
- McMahon, M.; Contreras, A.; Holm, M.; Uechi, T.; Forester, C.M.; Pang, X.; Jackson, C.; Calvert, M.E.; Chen, B.; Quigley, D.A.; et al. A Single H/ACA Small Nucleolar RNA Mediates Tumor Suppression Downstream of Oncogenic RAS. Elife 2019, 8, e48847. [Google Scholar] [CrossRef]
- Ford, D. Ribosomal Heterogeneity—A New Inroad for Pharmacological Innovation. Biochem. Pharmacol. 2020, 175, 113874. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Groenewald, W.; Lund, A.H.; Gay, D.M. The Role of WNT Pathway Mutations in Cancer Development and an Overview of Therapeutic Options. Cells 2023, 12, 990. https://doi.org/10.3390/cells12070990
Groenewald W, Lund AH, Gay DM. The Role of WNT Pathway Mutations in Cancer Development and an Overview of Therapeutic Options. Cells. 2023; 12(7):990. https://doi.org/10.3390/cells12070990
Chicago/Turabian StyleGroenewald, Wibke, Anders H. Lund, and David Michael Gay. 2023. "The Role of WNT Pathway Mutations in Cancer Development and an Overview of Therapeutic Options" Cells 12, no. 7: 990. https://doi.org/10.3390/cells12070990