
Cbfβ Is a Novel Modulator against Osteoarthritis by Maintaining Articular Cartilage Homeostasis through TGF-β Signaling
 , , , ,
, , , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Antibodies and Reagents
2.2. Mice and Experimental OA
2.3. Human Subjects
2.4. β-Galactosidase (β-gal) Staining
2.5. Assessment of OA Severity
2.6. Immunohistochemistry
2.7. Cell Culture
2.8. Western Blot Analyses
2.9. qRT-PCR Analyses
2.10. Co-Immunoprecipitation (Co-IP) Assay
2.11. Poly-Ubiquitination Assay
2.12. Statistical Analyses
3. Results
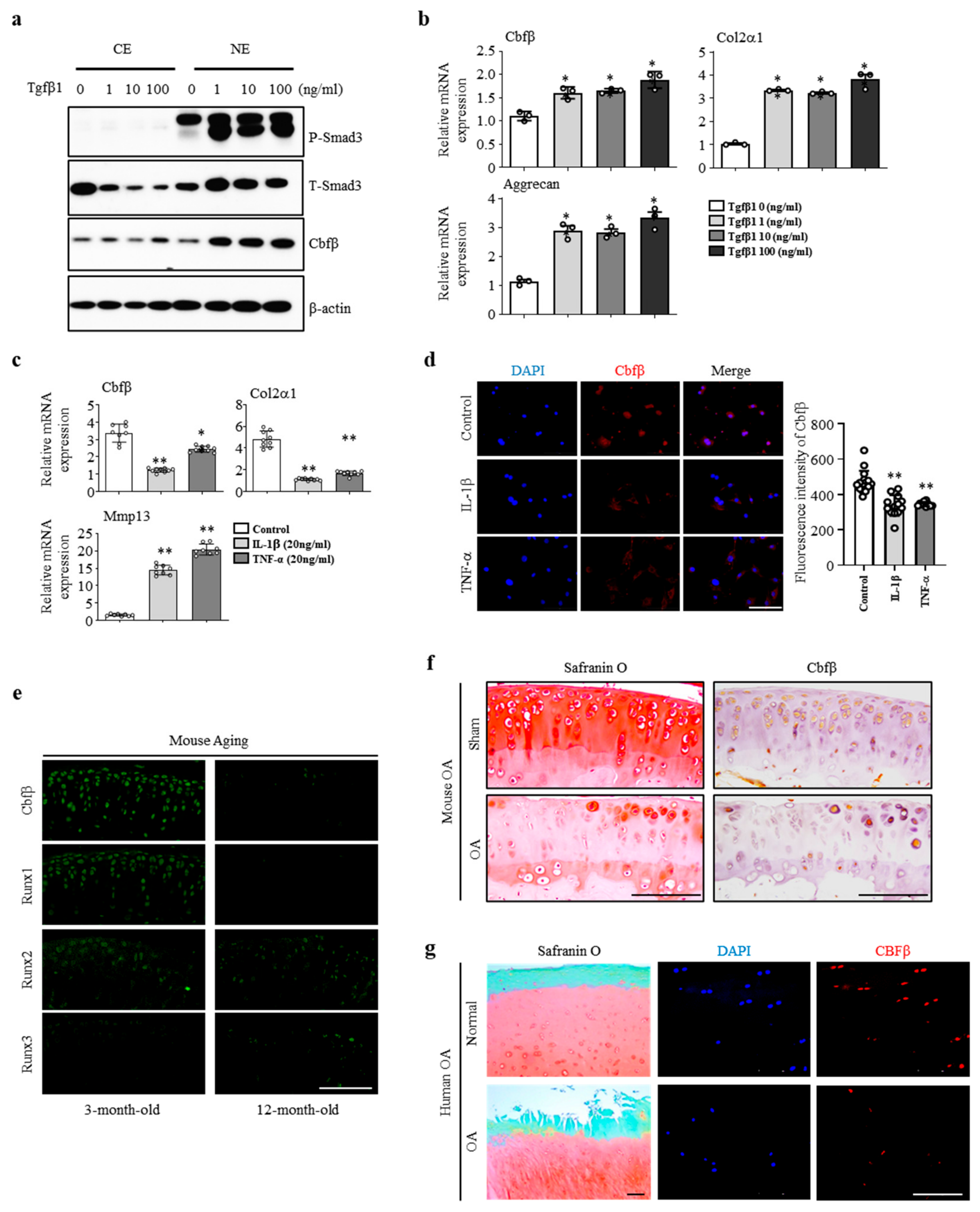
3.1. Cbfβ was Enhanced by Anabolism and Suppressed by Catabolism
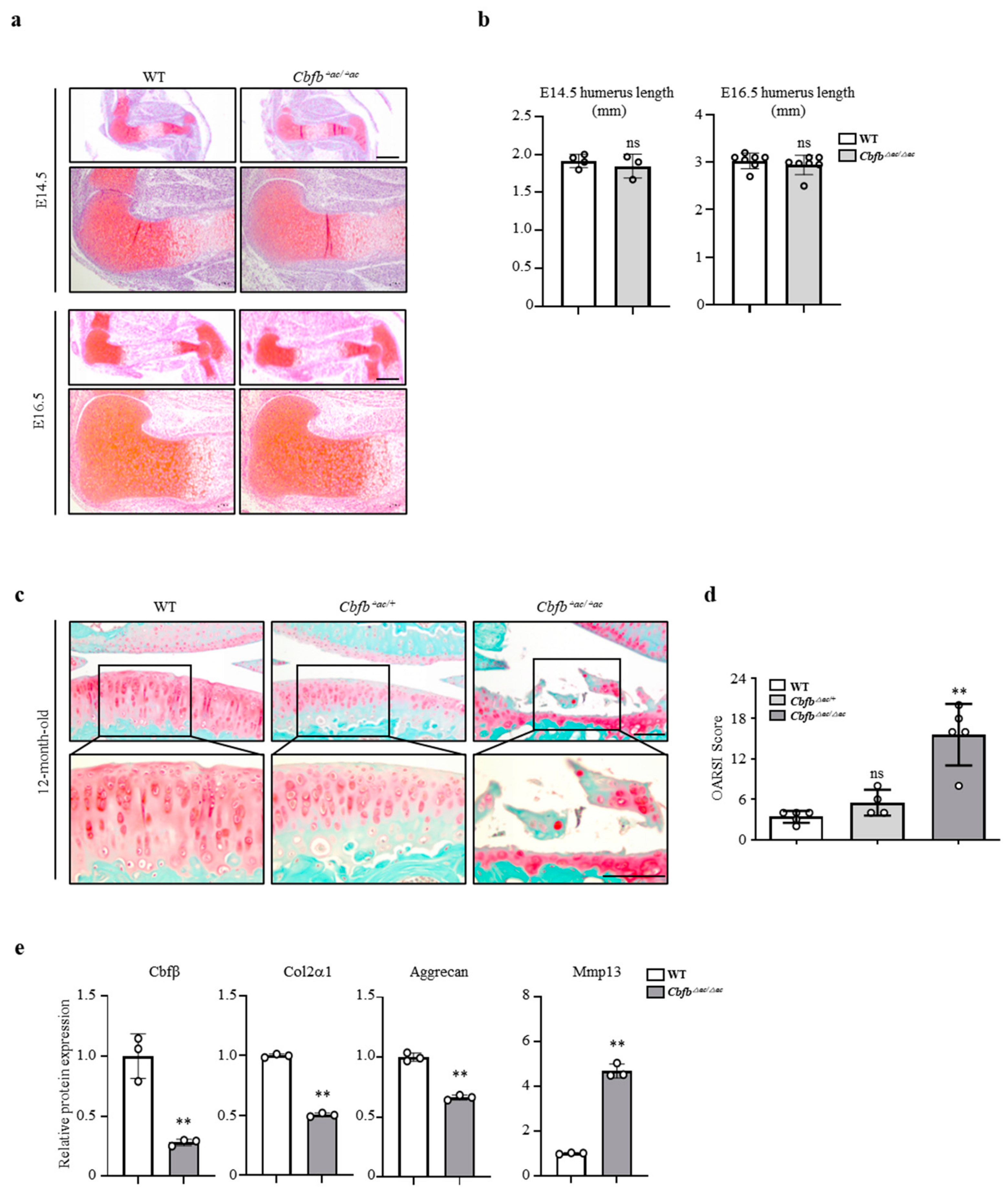
3.2. Cbfβ Loss Is Involved in Articular Cartilage Degeneration
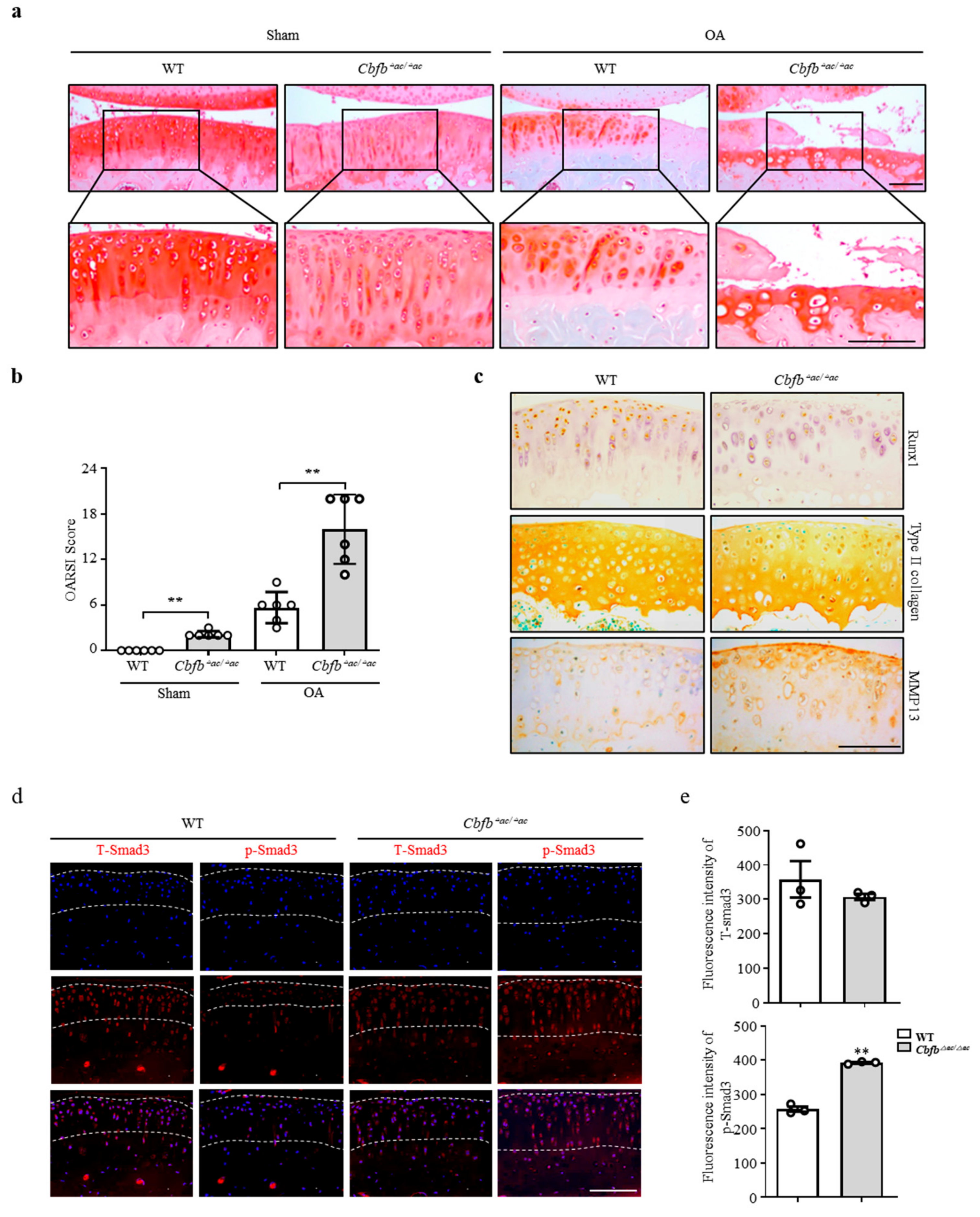
3.3. Genetic Deletion of Cbfb Accelerated OA Progression
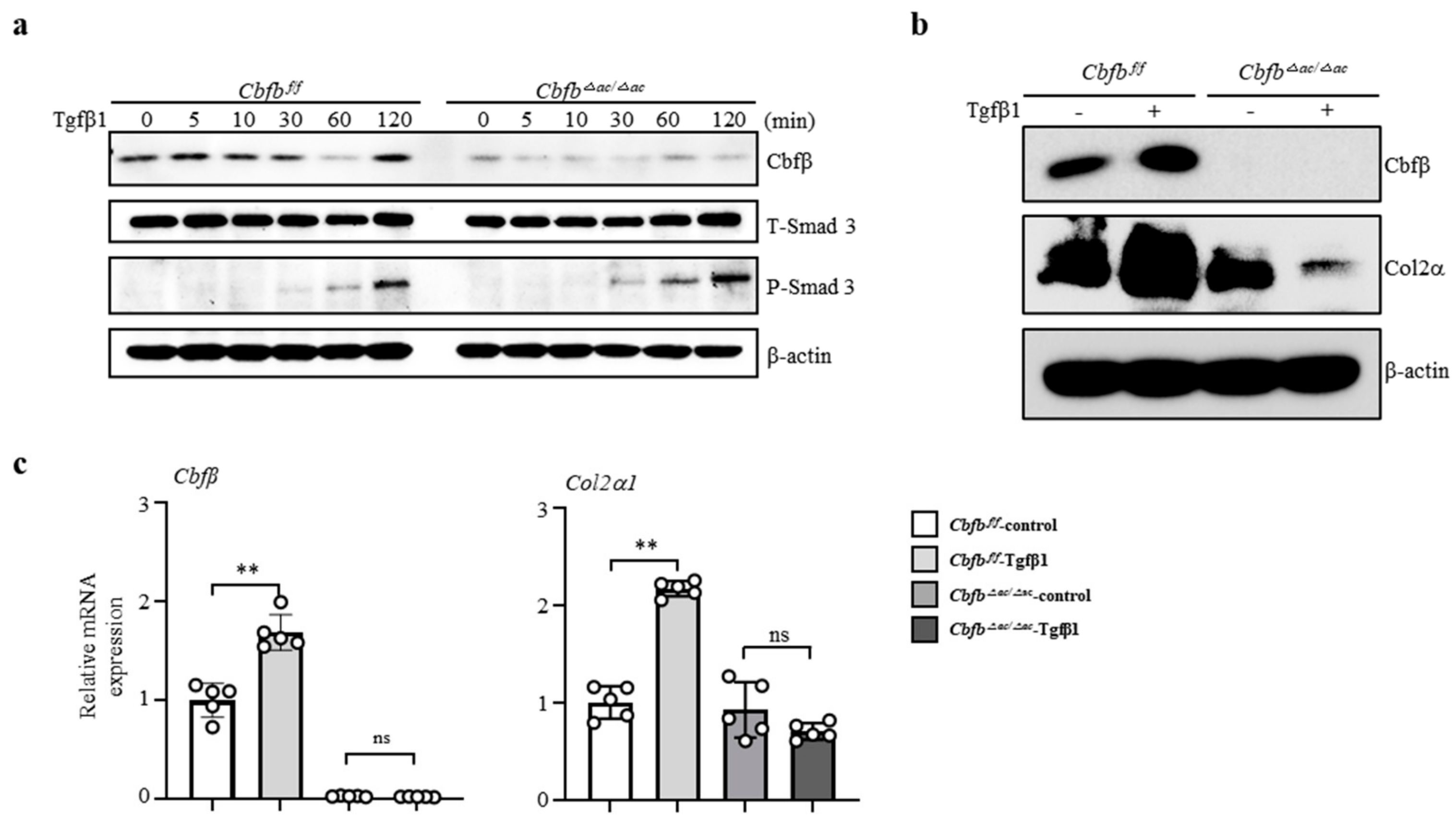
3.4. Cbfβ Modulates Articular Cartilage Integrity by Modulating TGF-β Signaling
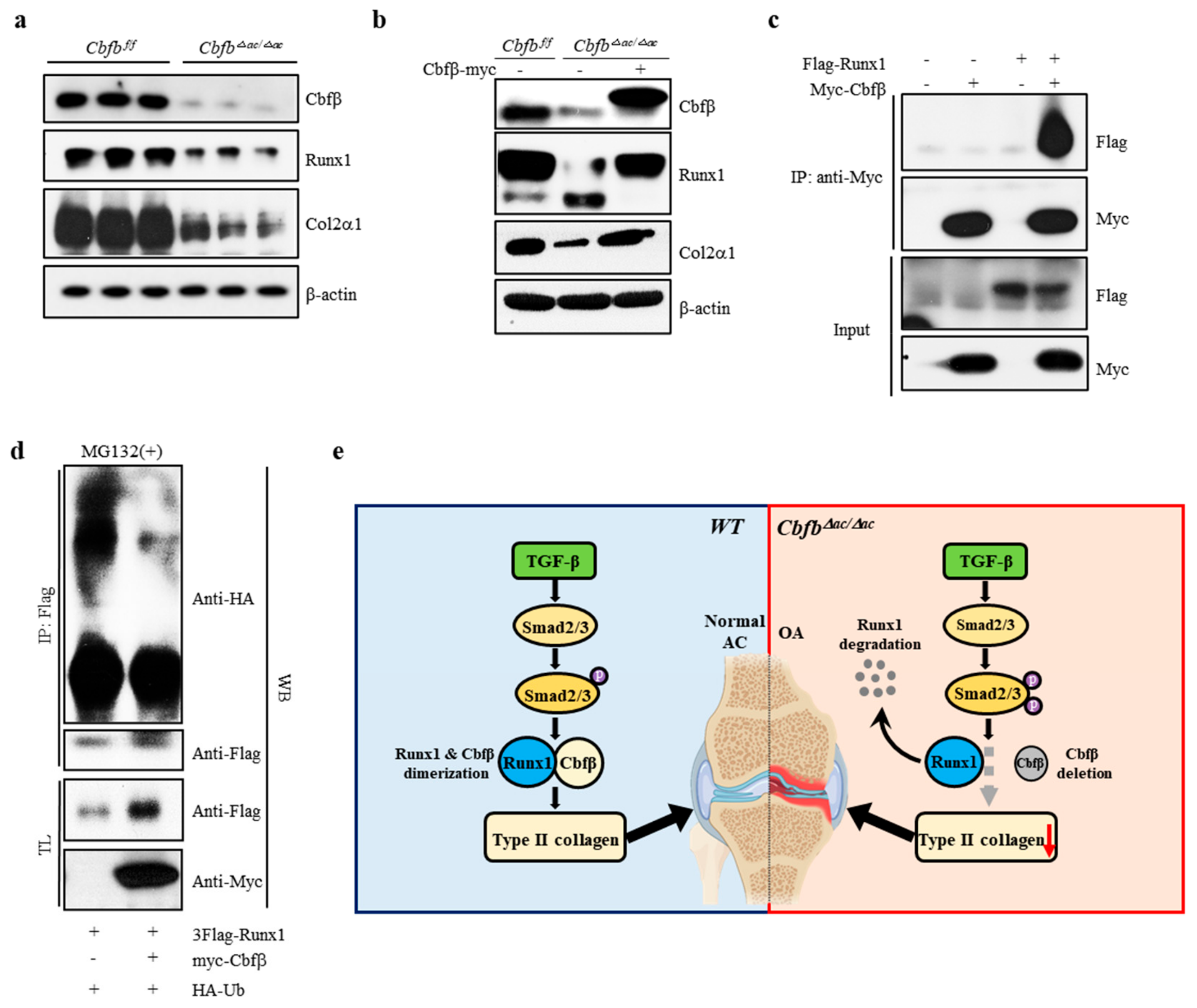
3.5. Cbfβ Stabilizes Runx1 in Articular Chondrocytes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Martel-Pelletier, J.; Barr, A.J.; Cicuttini, F.M.; Conaghan, P.G.; Cooper, C.; Goldring, M.B.; Jones, G.; Teichtahl, A.J.; Pelletier, J.P. Osteoarthritis. Nat. Rev. Dis. Prim. 2016, 2, 16072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, D.J.; Bierma-Zeinstra, S. Osteoarthritis. Lancet 2019, 393, 1745–1759. [Google Scholar] [CrossRef] [PubMed]
- Zhen, G.; Guo, Q.; Li, Y.; Wu, C.; Zhu, S.; Wang, R.; Guo, X.E.; Kim, B.C.; Huang, J.; Hu, Y.; et al. Mechanical stress determines the configuration of TGFβ activation in articular cartilage. Nat. Commun. 2021, 12, 1706. [Google Scholar] [CrossRef] [PubMed]
- Glyn-Jones, S.; Palmer, A.J.; Agricola, R.; Price, A.J.; Vincent, T.L.; Weinans, H.; Carr, A.J. Osteoarthritis. Lancet 2015, 386, 376–387. [Google Scholar] [CrossRef]
- Che, X.; Chi, L.; Park, C.Y.; Cho, G.H.; Park, N.; Kim, S.G.; Lee, B.H.; Choi, J.Y. A novel method to detect articular chondrocyte death during early stages of osteoarthritis using a non-invasive ApoPep-1 probe. Arthritis Res. Ther. 2015, 17, 309. [Google Scholar] [CrossRef] [Green Version]
- Hyttinen, M.M.; Toyras, J.; Lapvetelainen, T.; Lindblom, J.; Prockop, D.J.; Li, S.W.; Arita, M.; Jurvelin, J.S.; Helminen, H.J. Inactivation of one allele of the Type II collagen gene alters the collagen network in murine articular cartilage and makes cartilage softer. Ann. Rheum. Dis. 2001, 60, 262–268. [Google Scholar] [CrossRef] [Green Version]
- Goldring, S.R.; Goldring, M.B. Changes in the osteochondral unit during osteoarthritis: Structure, function and cartilage-bone crosstalk. Nat. Rev. Rheumatol. 2016, 12, 632–644. [Google Scholar] [CrossRef]
- Li, H.; Wang, D.; Yuan, Y.; Min, J. New insights on the MMP-13 regulatory network in the pathogenesis of early osteoarthritis. Arthritis Res. Ther. 2017, 19, 248. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Sampson, E.R.; Jin, H.; Li, J.; Ke, Q.H.; Im, H.J.; Chen, D. MMP13 is a critical target gene during the progression of osteoarthritis. Arthritis Res. Ther. 2013, 15, R5. [Google Scholar] [CrossRef] [Green Version]
- Thielen, N.G.M.; Neefjes, M.; Vitters, E.L.; van Beuningen, H.M.; Blom, A.B.; Koenders, M.I.; van Lent, P.L.E.M.; van de Loo, F.A.J.; Blaney Davidson, E.N.; van Caam, A.P.M.; et al. Identification of Transcription Factors Responsible for a Transforming Growth Factor-β-Driven Hypertrophy-like Phenotype in Human Osteoarthritic Chondrocytes. Cells 2022, 11, 1232. [Google Scholar] [CrossRef]
- van Beuningen, H.M.; van der Kraan, P.M.; Arntz, O.J.; van den Berg, W.B. Transforming growth factor-beta 1 stimulates articular chondrocyte proteoglycan synthesis and induces osteophyte formation in the murine knee joint. Lab. Investig. 1994, 71, 279–290. [Google Scholar]
- Scharstuhl, A.; Glansbeek, H.L.; van Beuningen, H.M.; Vitters, E.L.; van der Kraan, P.M.; van den Berg, W.B. Inhibition of endogenous TGF-beta during experimental osteoarthritis prevents osteophyte formation and impairs cartilage repair. J. Immunol. 2002, 169, 507–514. [Google Scholar] [CrossRef] [Green Version]
- Blaney Davidson, E.N.; Remst, D.F.; Vitters, E.L.; van Beuningen, H.M.; Blom, A.B.; Goumans, M.J.; van den Berg, W.B.; van der Kraan, P.M. Increase in ALK1/ALK5 ratio as a cause for elevated MMP-13 expression in osteoarthritis in humans and mice. J. Immunol. 2009, 182, 7937–7945. [Google Scholar] [CrossRef] [Green Version]
- Serra, R.; Johnson, M.; Filvaroff, E.H.; LaBorde, J.; Sheehan, D.M.; Derynck, R.; Moses, H.L. Expression of a truncated, kinase-defective TGF-beta type II receptor in mouse skeletal tissue promotes terminal chondrocyte differentiation and osteoarthritis. J. Cell Biol. 1997, 139, 541–552. [Google Scholar] [CrossRef]
- Yang, X.; Chen, L.; Xu, X.; Li, C.; Huang, C.; Deng, C.X. TGF-beta/Smad3 signals repress chondrocyte hypertrophic differentiation and are required for maintaining articular cartilage. J. Cell Biol. 2001, 153, 35–46. [Google Scholar] [CrossRef] [Green Version]
- Speck, N.A.; Terryl, S. A new transcription factor family associated with human leukemias. Crit. Rev. Eukaryot. Gene Expr. 1995, 5, 337–364. [Google Scholar] [CrossRef]
- Bae, S.C.; Ito, Y. Regulation mechanisms for the heterodimeric transcription factor, PEBP2/CBF. Histol. Histopathol. 1999, 14, 1213–1221. [Google Scholar]
- Wang, Y.; Belflower, R.M.; Dong, Y.F.; Schwarz, E.M.; O’Keefe, R.J.; Drissi, H. Runx1/AML1/Cbfa2 mediates onset of mesenchymal cell differentiation toward chondrogenesis. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2005, 20, 1624–1636. [Google Scholar] [CrossRef]
- LeBlanc, K.T.; Walcott, M.E.; Gaur, T.; O’Connell, S.L.; Basil, K.; Tadiri, C.P.; Mason-Savas, A.; Silva, J.A.; van Wijnen, A.J.; Stein, J.L.; et al. Runx1 Activities in Superficial Zone Chondrocytes, Osteoarthritic Chondrocyte Clones and Response to Mechanical Loading. J. Cell. Physiol. 2015, 230, 440–448. [Google Scholar] [CrossRef] [Green Version]
- Otto, F.; Thornell, A.P.; Crompton, T.; Denzel, A.; Gilmour, K.C.; Rosewell, I.R.; Stamp, G.W.H.; Beddington, R.S.P.; Mundlos, S.; Olsen, B.R.; et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell 1997, 89, 765–771. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.Y.; Pratap, J.; Javed, A.; Zaidi, S.K.; Xing, L.; Balint, E.; Dalamangas, S.; Boyce, B.; van Wijnen, A.J.; Lian, J.B.; et al. Subnuclear targeting of Runx/Cbfa/AML factors is essential for tissue-specific differentiation during embryonic development. Proc. Natl. Acad. Sci. USA 2001, 98, 8650–8655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komori, T.; Yagi, H.; Nomura, S.; Yamaguchi, A.; Sasaki, K.; Deguchi, K.; Shimizu, Y.; Bronson, R.T.; Gao, Y.-H.; Inada, M.; et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 1997, 89, 755–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, C.A.; Yamamoto, H.; Fujita, T.; Furuichi, T.; Ito, K.; Inoue, K.; Yamana, K.; Zanma, A.; Takada, K.; Ito, Y.; et al. Runx2 and Runx3 are essential for chondrocyte maturation, and Runx2 regulates limb growth through induction of Indian hedgehog. Genes Dev. 2004, 18, 952–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, G.; Shigesada, K.; Ito, K.; Wee, H.J.; Yokomizo, T.; Ito, Y. Dimerization with PEBP2beta protects RUNX1/AML1 from ubiquitin-proteasome-mediated degradation. EMBO J. 2001, 20, 723–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Stacy, T.; Miller, J.D.; Lewis, A.F.; Gu, T.L.; Huang, X.; Bushweller, J.H.; Bories, J.C.; Alt, F.W.; Ryan, G.; et al. The CBFbeta subunit is essential for CBFalpha2 (AML1) function in vivo. Cell 1996, 87, 697–708. [Google Scholar] [CrossRef] [Green Version]
- Tian, F.; Wu, M.; Deng, L.; Zhu, G.; Ma, J.; Gao, B.; Wang, L.; Li, Y.P.; Chen, W. Core binding factor beta (Cbfbeta) controls the balance of chondrocyte proliferation and differentiation by upregulating Indian hedgehog (Ihh) expression and inhibiting parathyroid hormone-related protein receptor (PPR) expression in postnatal cartilage and bone formation. J. Bone Miner. Res. 2014, 29, 1564–1574. [Google Scholar]
- Qin, X.; Jiang, Q.; Matsuo, Y.; Kawane, T.; Komori, H.; Moriishi, T.; Ito, K.; Kawai, Y.; Rokutanda, S.; Izumi, S.; et al. Cbfb regulates bone development by stabilizing Runx family proteins. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2015, 30, 706–714. [Google Scholar] [CrossRef]
- Wu, M.; Li, Y.P.; Zhu, G.; Lu, Y.; Wang, Y.; Jules, J.; McConnell, M.; Serra, R.; Shao, J.Z.; Chen, W. Chondrocyte-specific knockout of Cbfβ reveals the indispensable function of Cbfβ in chondrocyte maturation, growth plate development and trabecular bone formation in mice. Int. J. Biol. Sci. 2014, 10, 861–872. [Google Scholar] [CrossRef]
- Park, N.R.; Lim, K.E.; Han, M.S.; Che, X.; Park, C.Y.; Kim, J.E.; Taniuchi, I.; Bae, S.C.; Choi, J.Y. Core Binding Factor beta Plays a Critical Role During Chondrocyte Differentiation. J. Cell. Physiol. 2016, 231, 162–171. [Google Scholar] [CrossRef]
- Lim, K.E.; Park, N.R.; Che, X.; Han, M.S.; Jeong, J.H.; Kim, S.Y.; Park, C.Y.; Akiyama, H.; Kim, J.E.; Ryoo, H.M.; et al. Core binding factor β of osteoblasts maintains cortical bone mass via stabilization of Runx2 in mice. J. Bone Miner. Res. 2015, 30, 715–722. [Google Scholar] [CrossRef]
- Rountree, R.B.; Schoor, M.; Chen, H.; Marks, M.E.; Harley, V.; Mishina, Y.; Kingsley, D.M. BMP receptor signaling is required for postnatal maintenance of articular cartilage. PLoS Biol. 2004, 2, e355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naoe, Y.; Setoguchi, R.; Akiyama, K.; Muroi, S.; Kuroda, M.; Hatam, F.; Littman, D.R.; Taniuchi, I. Repression of interleukin-4 in T helper type 1 cells by Runx/Cbf beta binding to the Il4 silencer. J. Exp. Med. 2007, 204, 1749–1755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glasson, S.S.; Blanchet, T.J.; Morris, E.A. The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthr. Cartil. 2007, 15, 1061–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, J.H.; Jin, J.S.; Kim, H.N.; Kang, S.M.; Liu, J.C.; Lengner, C.J.; Otto, F.; Mundlos, S.; Stein, J.L.; van Wijnen, A.J.; et al. Expression of Runx2 transcription factor in non-skeletal tissues, sperm and brain. J. Cell. Physiol. 2008, 217, 511–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gosset, M.; Berenbaum, F.; Thirion, S.; Jacques, C. Primary culture and phenotyping of murine chondrocytes. Nat. Protoc. 2008, 3, 1253–1260. [Google Scholar] [CrossRef]
- Kim, H.J.; Lee, D.K.; Jin, X.; Che, X.; Ryu, S.H.; Choi, J.Y. Phospholipase D2 controls bone homeostasis by modulating M-CSF-dependent osteoclastic cell migration and microtubule stability. Exp. Mol. Med. 2022, 54, 1146–1155. [Google Scholar] [CrossRef]
- Che, X.; Park, N.; Jin, X.; Jung, Y.; Han, M.; Park, C.; Chun, J.; Kim, S.; Jin, J.; Kim, H.; et al. Hypoxia-inducible factor 2α is a novel inhibitor of chondrocyte maturation. J. Cell. Physiol. 2021, 236, 6963–6973. [Google Scholar] [CrossRef]
- Ito, Y.; Miyazono, K. RUNX transcription factors as key targets of TGF-beta superfamily signaling. Curr. Opin. Genet. Dev. 2003, 13, 43–47. [Google Scholar] [CrossRef]
- Zhang, Y.; Zuo, T.; McVicar, A.; Yang, H.; Li, Y.; Chen, W. Runx1 is a key regulator of articular cartilage homeostasis by orchestrating YAP, TGFβ, and Wnt signaling in articular cartilage formation and osteoarthritis. Bone Res. 2022, 10, 63. [Google Scholar] [CrossRef]
- Yano, F.; Ohba, S.; Murahashi, Y.; Tanaka, S.; Saito, T.; Chung, U.I. Runx1 contributes to articular cartilage maintenance by enhancement of cartilage matrix production and suppression of hypertrophic differentiation. Sci. Rep. 2019, 9, 7666. [Google Scholar] [CrossRef] [Green Version]
- Zhou, C.; Cui, Y.; Yang, Y.; Guo, D.; Zhang, D.; Fan, Y.; Li, X.; Zou, J.; Xie, J. Runx1 protects against the pathological progression of osteoarthritis. Bone Res. 2021, 9, 50. [Google Scholar] [CrossRef]
- Johnson, K.; Zhu, S.; Tremblay, M.S.; Payette, J.N.; Wang, J.; Bouchez, L.C.; Meeusen, S.; Althage, A.; Cho, C.Y.; Wu, X.; et al. A stem cell-based approach to cartilage repair. Science 2012, 336, 717–721. [Google Scholar] [CrossRef] [Green Version]
- Kawaguchi, H. Endochondral ossification signals in cartilage degradation during osteoarthritis progression in experimental mouse models. Mol. Cells 2008, 25, 1–6. [Google Scholar]
- Kamekura, S.; Kawasaki, Y.; Hoshi, K.; Shimoaka, T.; Chikuda, H.; Maruyama, Z.; Komori, T.; Sato, S.; Takeda, S.; Karsenty, G.; et al. Contribution of runt-related transcription factor 2 to the pathogenesis of osteoarthritis in mice after induction of knee joint instability. Arthritis Rheum. 2006, 54, 2462–2470. [Google Scholar] [CrossRef]
- Liao, L.; Zhang, S.; Gu, J.; Takarada, T.; Yoneda, Y.; Huang, J.; Zhao, L.; Oh, C.; Li, J.; Wang, B.; et al. Deletion of Runx2 in Articular Chondrocytes Decelerates the Progression of DMM-Induced Osteoarthritis in Adult Mice. Sci. Rep. 2017, 7, 2371. [Google Scholar] [CrossRef] [Green Version]
- Catheline, S.E.; Hoak, D.; Chang, M.; Ketz, J.P.; Hilton, M.J.; Zuscik, M.J.; Jonason, J.H. Chondrocyte-Specific RUNX2 Overexpression Accelerates Post-traumatic Osteoarthritis Progression in Adult Mice. J. Bone Miner. Res. 2019, 34, 1676–1689. [Google Scholar] [CrossRef]
- Dunbar, C.; High, K.; Joung, J.; Kohn, D.; Ozawa, K.; Sadelain, M. Gene therapy comes of age. Science 2018, 359, eaan4672. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reagent Type | Designation | Identifiers |
|---|---|---|
| Antibody | Anti-p-Smad3 (rabbit polyclonal) | Cell Signaling Technology #9520 |
| Antibody | Anti-smad3 (rabbit polyclonal) | Cell Signaling Technology #9513 |
| Antibody | Anti-Cbfβ (rabbit polyclonal) | Santa Cruz #20693 |
| Antibody | Anti-β-actin (mouse monoclonal) | Santa Cruz #47778 |
| Antibody | Anti-Col2α1 (rabbit polyclonal) | Abcam #53047 |
| Antibody | Anti-Runx1 (mouse monoclonal) | Santa Cruz #365644 |
| Antibody | Anti-Myc-Tag (rabbit polyclonal) | Abcam #9106 |
| Antibody | Anti-Myc-Tag (mouse monoclonal) | Invitrogen #P/N46-0603 |
| Antibody | Anti-Flag-Tag (mouse monoclonal) | Sigma #F1804 |
| Antibody | Anti-Mmp13 (mouse monoclonal) | Calbiochem #IM-78 |
| Protein ladder | Protein marker | Dokdo-1032 |
| Chemical compound, drug | Type IV collagenase | Gibco # 9001-12-1 |
| Chemical compound, drug | Lipofectamine 2000 | Invitrogen #11668019 |
| Chemical compound, drug | Hematoxylin | Sigma-Aldrich #H9627 |
| Chemical compound, drug | Fast green | Sigma-Aldrich #F7258 |
| Chemical compound, drug | Ferric chloride | Junsei #18510S0301 |
| Chemical compound, drug | Safranin O | Sigma-Aldrich #S-2255 |
| Chemical compound, drug | Mammalian cell lysis buffer | Thermo Fisher Scientific #78501 |
| Chemical compound, drug | Easy-Blue solution | iNtRON #17061 |
| Chemical compound, drug | DAB substrate-chromogen system | Dakocytomation #K3468 |
| Chemical compound, drug | Bovine Serum Albumin | Sigma-Aldrich #A9418 |
| Chemical compound, drug | 4% Paraformaldehyde | Biosolution #BP031 |
| Chemical compound, drug | Mayor’s Hematoxylin | Muto pure chemicals #3000-2 |
| Chemical compound, drug | Eosin | Muto pure chemicals #3200-2 |
| Chemical compound, drug | Ethylenediaminetetraacetic acid | Junsei #17388-0401 |
| Recombinant protein | IL-1β | R&D Systems #401-ML |
| Recombinant protein | TNF-α | R&D Systems #410-MT |
| Recombinant protein | Tgf-β1 | R&D Systems #7666-MB |
| No. | Age | Gender | ICRS Grade | Joint | Weight (kg) | Height (cm) | RA | Others |
|---|---|---|---|---|---|---|---|---|
| 1 | 65 | F | 4 | Knee | 66 | 158 | X | heart valve disease |
| 2 | 76 | F | 4 | Knee | 71 | 155 | X | HTN, DM, unstable angina, asthma |
| 3 | 64 | M | 3 | Knee | 67 | 168 | X | HTM |
| 4 | 83 | F | 4 | Knee | 55 | 151 | X | HTN |
| 5 | 76 | F | 3 | Knee | 55 | 155 | X | HTN, DM |
| 6 | 71 | F | 4 | Knee | 70 | 159 | X | HTM, DM, AF |
| Target Gene | Forward Sequence | Reverse Sequence |
|---|---|---|
| Gapdh | GCATCTCCCTCACAATTTCCA | GTGCAGCGAACTTTATTGATGG |
| Cbfb | TATGGGTTGCCTGGAGTT TG | AAGGCCTGTTGTGCTAATGC |
| Col2α1 | TTCCACTTCAGCTATGGCGA | GACGTTAGCGGTGTTGGGAG |
| Aggrecan | GAGAGAGGCGAATCGAACGA | CGTGAAGGGCAGCTGGTAAT |
| Mmp9 | AAACCAGACCCCAGACTCCTC | GAGGACACAGTCTGACCTGAA |
| Mmp13 | GCCAGAACTTCCCAACCATG | TCAGAGCCCAGAATTTTCTCC |
| Mmp14 | GGATGGACACAGAGAACTTCGTG | CGAGAGGTAGTTCTGGGTTGAG |
| Mmp15 | CTGAGCAGCTATGGCACAGACA | TGCTGTGTCTCCTCGTTGAAGC |
| IL-6 | TTGCCTTCTTGGGACTGATG | CTGAAGGACTCTGGCTTTGT |
| IL-17 | CTCAAAGCTCAGCGTGTCCAAACA | TATCAGGGTCTTCATTGCGGTGGA |
| IL-18 | CAGGCCTGACATCTTCTGCAA | TTTGATGTAAGTTAGTGAGAGTGA |
| IL-22 | GGTGACGACCAGAACATCCA | GACGTTAGCTTCTCACTTTCCTT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Che, X.; Jin, X.; Park, N.R.; Kim, H.-J.; Kyung, H.-S.; Kim, H.-J.; Lian, J.B.; Stein, J.L.; Stein, G.S.; Choi, J.-Y. Cbfβ Is a Novel Modulator against Osteoarthritis by Maintaining Articular Cartilage Homeostasis through TGF-β Signaling. Cells 2023, 12, 1064. https://doi.org/10.3390/cells12071064
Che X, Jin X, Park NR, Kim H-J, Kyung H-S, Kim H-J, Lian JB, Stein JL, Stein GS, Choi J-Y. Cbfβ Is a Novel Modulator against Osteoarthritis by Maintaining Articular Cartilage Homeostasis through TGF-β Signaling. Cells. 2023; 12(7):1064. https://doi.org/10.3390/cells12071064
Chicago/Turabian StyleChe, Xiangguo, Xian Jin, Na Rae Park, Hee-June Kim, Hee-Soo Kyung, Hyun-Ju Kim, Jane B. Lian, Janet L. Stein, Gary S. Stein, and Je-Yong Choi. 2023. "Cbfβ Is a Novel Modulator against Osteoarthritis by Maintaining Articular Cartilage Homeostasis through TGF-β Signaling" Cells 12, no. 7: 1064. https://doi.org/10.3390/cells12071064