
Crosstalk between Autophagy and RLR Signaling

1
Department of Biochemistry & Molecular Biology, Graduate Institute of Biomedical Sciences, College of Medicine, Chang Gung University, Taoyuan 33302, Taiwan
2
Liver Research Center, Chang Gung Memorial Hospital, Taoyuan 33305, Taiwan
Cells 2023, 12(6), 956; https://doi.org/10.3390/cells12060956
Submission received: 14 February 2023
/
Revised: 17 March 2023
/
Accepted: 20 March 2023
/
Published: 21 March 2023
(This article belongs to the Special Issue Autophagy in Cell Survival and Growth)
Abstract
:Autophagy plays a homeostatic role in regulating cellular metabolism by degrading unwanted intracellular materials and acts as a host defense mechanism by eliminating infecting pathogens, such as viruses. Upon viral infection, host cells often activate retinoic acid-inducible gene I (RIG-I)-like receptor (RLR) signaling to induce the transcription of type I interferons, thus establishing the first line of the innate antiviral response. In recent years, numerous studies have shown that virus-mediated autophagy activation may benefit viral replication through different actions on host cellular processes, including the modulation of RLR-mediated innate immunity. Here, an overview of the functional molecules and regulatory mechanism of the RLR antiviral immune response as well as autophagy is presented. Moreover, a summary of the current knowledge on the biological role of autophagy in regulating RLR antiviral signaling is provided. The molecular mechanisms underlying the crosstalk between autophagy and RLR innate immunity are also discussed.
1. Introduction
Autophagy is a fundamentally catabolic process that degrades unnecessary intracellular materials to support the recycling of nutrients, thus promoting the regeneration of energy sources and balancing metabolic homeostasis. In addition to basal autophagy, a variety of stimuli, including deprivation of nutrients, aggregation and/or unfolding of proteins, damage to organelles, and infection by pathogens, can specifically activate autophagy to resolve cellular stresses. Deregulation of autophagy is involved in the pathogenesis of human diseases, such as cancer, neurodegenerative diseases, infectious diseases, and metabolic disorders. Several viral infections may induce autophagy to reconstitute the membranous compartments for virus replication and to repress the innate antiviral response, which benefits virus replication. In contrast, virus-triggered autophagy may be used to restrict viral growth by eliminating the infecting virus, acting as a host defense mechanism against viral infection. Upon virus infection, pattern recognition receptors (PRRs), such as retinoic acid-inducible I (RIG-I)-like receptors (RLRs), recognize pathogen-associated molecular patterns (PAMPs) and subsequently trigger a downstream signal cascade to induce the production of interferons (IFNs) and cytokines, thus stimulating innate antiviral immunity. However, to achieve successful replication, viruses have evoked strategies to repress the RLR-triggered antiviral response and may also interfere with autophagic degradation. In recent years, numerous studies have provided insights into the molecular interactions between virus-activated autophagy and RLR innate immune signaling. In this review, we first summarize the current understanding of the molecular regulation of RLR signaling and autophagy. Then, we provide an overview of the physiological importance and molecular mechanism by which autophagy regulates the antiviral response of RLRs.
2. RLR Antiviral Signaling
2.1. Functional Molecules Involved in RLR Signaling
During viral infection, the binding of virus genome-containing PAMPs by PRRs triggers a signaling cascade that transcriptionally activates the gene expression of IFNs and cytokines in infected cells, ultimately promoting the synthesis and secretion of IFNs and cytokines [1,2,3,4]. PRRs are sensing molecules localized in the cytosol and membranous compartments within cells and are responsible for recognizing viral-derived PAMPs and stress cell-produced danger-associated molecular patterns (DAMPs) [5,6,7,8,9]. To date, three classes of PRRs have been shown to activate the innate immune response, including the membrane-bound Toll-like receptors (TLRs) responsible for sensing viral PAMPs in endosomes [7,8,9,10,11,12,13], cytosolic RLRs responsible for sensing viral RNA [6,7,8,9,14,15], and cyclic GMP-AMP (cGAMP) synthases (cGASs) responsible for sensing DNA in the cytosol [7,8,9,16,17]. In addition to these PRRs, the cytosolic NBD leucine-rich repeat-containing receptor (NLR) family, such as absent in melanoma 2 (AIM2)—an initiator of the assembly of inflammasome complex in immune cells—contributes to PAMP and DAMP sensing and leads to proteolytic activation of caspases, thereby activating immune responses and inflammation [18,19,20].
Three RLRs that possess DExD/H box helicase activity, RIG-I [21], melanoma differentiation association gene 5 (MDA5) [22], and laboratory of genetics and physiology 2 (LGP2) [23], were identified in the 2000s. All three RLRs contain an ATP-dependent DExD/H box-containing RNA helicase domain for the recognition of double-stranded RNA (dsRNA) in the central region and a C-terminal repressor domain (CTD) for binding to RNA- and Zinc ligands (Figure 1A) [22,24,25,26,27,28,29]. A pincer-link domain connects the C-terminus of the helicase domain (HELICc) and the CTD (Figure 1A) [22,24,25]. Additionally, RIG-I and MDA5 contain two caspase activation and recruitment domains (CARDs) at the N-terminus, which transduce signaling to downstream effectors to induce the IFN response [22,24,26,27,30]. Moreover, these tandem CARDs of RIG-I interact with the helical insertion segment of the helicase domain in cells under steady-state conditions, forming an autorepressor conformation which interferes with the binding of ligand RNA to RIG-I [26,27,28,29,31]. Upon viral infection, binding of the RNA ligand to the CTD of RIG-I induces an ATP-dependent conformational change in RIG-I and relieves the N-terminal CARDs, which associates with the downstream signaling molecule [26,27,28,29,31,32]. In contrast, LGP2 lacks these N-terminal CARDs, but it retains the core helicase domain that can bind to dsRNA and has been shown to regulate the biological functions of RIG-I and MDA-5 (Figure 1A) [25,33,34].
In addition to RIG-I and MDA5, mitochondrial antiviral signaling (MAVS, also known as IFN-beta promoter stimulator-1 [IPS-1], virus-induced signaling adaptor [VISA], and CARD adaptor inducing IFN-β [Cardif]) also contains a CARD at its N-terminal regions (Figure 1A), which can interact with the CARDs of RIG-I and MDA5 and a transmembrane domain at the C-terminus for localization on the outer membrane of mitochondria (MOM) (Figure 1B) [35,36,37,38]. The association between the CARDs of RIG-I/MDA5 and MAVS activates downstream signaling. Moreover, oligomerization of MAVS through N-terminal CARDs assembles into protease- and detergent-resistant prion-like structures on the MOM and promotes the recruitment of the ubiquitin E3 ligases tumor necrosis factor (TNF) receptor-associated factor 2 (TRAF2), TRAF5, and TRAF6 [39,40]. Subsequently, these ubiquitin E3 ligases produce the polyubiquitin chain that recruits the nuclear factor kappa-light-chain-enhancer of activated B (NF-kB) essential modulator (NEMO), which in turn activates TANK binding kinase 1 (TBK1) and inhibitor of nuclear factor kappa B (Ik-B) kinase (IKK) to phosphorylate interferon response factor 3 (IRF3) and Ik-B, respectively (Figure 1B) [39,40]. The homodimers of phosphorylated IRF3 and NF-kB then translocate into the nucleus and transactivate the gene expression of IFN and proinflammatory cytokines, respectively (Figure 1B) [39,40]. Then, type I IFNs, such as IFN-α and IFN-β, are expressed and secreted in virus-infected cells and bind to the IFN-α receptor (IFNAR), which consists of IFNAR1 and IFNAR2 subunits [1,2,3,41].
The binding of IFNs to IFNAR activates the IFNAR-associated receptor tyrosine kinases tyrosine kinase 2 (TYK2) and Janus kinase 1 (JAK1) to phosphorylate signal transducer and activator of transcription 1 (STAT1) and STAT2. Subsequently, the tyrosine phosphorylation of STAT1 and STAT2 induces the formation of homodimers and triggers translocation into the nucleus. Before entering the nucleus, this STAT1-STAT2 homodimer associates with interferon response factor 9 (IRF9), forming a heterotrimeric protein complex, IFN-stimulated gene factor 3 (ISGF3). ISGF3 is able to bind to IFN-stimulated response elements (containing the consensus sequence TTTCNNTTTC) and thus transcriptionally activate the gene expression of IFN-stimulated genes (ISGs). Several types of ISGs have been identified, and their biological functions, including antiviral activity, are less understood, although some of them have been shown to repress virus entry and replication [1,2,42,43].
2.2. Recognition of Viral RNA by RLRs
RLRs are ubiquitously expressed in most tissues and are mostly dominantly expressed in the cytosol [6,14,15]. RIG-I has been shown to recognize dsRNA or single-stranded RNA (ssRNA) with triphosphate or diphosphate moieties at the 5′-end and is approximately 10–300 bp in length [44,45,46,47]. Additionally, the poly-uridine (U)/U-core (UC) (poly-U/UC) located within the 3′-untranslated region of the hepatitis C virus (HCV) RNA genome (3′-UTR) [48,49,50] and the adenine-uridine (AU)-rich sequence in the viral RNA of measles virus [51,52] serve as additional signatures for determining the specificity of viral PAMP binding to RIG-I. Moreover, the cleavage of viral RNA by RNase L promotes the release of small PAMP RNA containing a 5′-triphosphate (5′-ppp) moiety to induce the RIG-I-mediated antiviral IFN response [53,54]. In contrast to RIG-I, MDA5 binds to longer dsRNA sequences (approximately 1–2 kilobase [Kb] pairs), which presumably are replication intermediates of viral RNA that form a high-order RNA structure [55,56,57,58]. Genetic knockout studies in mice showed that RIG-I and MDA5 play distinct role(s) in the recognition of different RNA viruses [59,60]. To date, RIG-I has been extensively shown to recognize several human pathogenic viruses such as influenza A virus (IAV) [46,61,62,63,64], HCV [48,49,50,54], dengue virus (DENV) [65,66,67], Zika virus (ZIKV) [65,68], and West Nile virus (WNV) [69,70,71], and activate antiviral immunity in infected human cells. Similarly, several viruses that can lead to human diseases, including picornavirus [55,72], HCV [73,74], ZIKV [68], WNV [71], and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [75,76], also trigger antiviral IFN responses through MDA5.
2.3. Post-Translational Modification(s) of RLR and Regulation of RLR Signaling
Several post-translational modifications (PTMs) have been found to extensively modulate RLR-triggered antiviral immunity, such as phosphorylation, acetylation, sumoylation, deamination, ISGylation, and ubiquitination [6,7,8,9,14,15]. Conformational changes, including oligomerization and the formation of highly ordered and/or filamentous structures of RIG-I, MDA5, and MAVS, have been shown to regulate the activation of the RLR antiviral IFN response [6,7,8,9,14,15]. Protein kinase C (PKC)-α and PKC-β-induced protein phosphorylation at serine (Ser) residue 8 and threonine (Thr) residue 170 within the CARD was shown to repress the signal transduction of RIG-I antiviral immunity by interrupting the binding of RIG-I to tripartite motif (TRIM) 25 (TRIM25), a ubiquitin ligase required to positively regulate RIG-I downstream IFN signaling (Figure 2) [77,78,79]. Additionally, phosphorylation at Thr770, Ser854, and Ser855 within the CTD of RIG-I by casein kinase II (CK-II) was found to be necessary for maintaining the autorepressor conformation of RIG-I (Figure 2) [80], thereby preventing the improper activation of RIG-I antiviral immunity in the absence of viral infection. In addition, the induction of RIG-I signaling promotes the activation of death-associated protein kinase 1 (DAPK1) to phosphorylate Thr667 within the helicase domain and interfere with RIG-I recognition of 5′-triphosphate-dsRNA, acting as feedback inhibition of the RIG-I antiviral response (Figure 2) [81]. Similarly, MDA5 is also negatively regulated by the phosphorylation of Ser88 on the CARD and Ser828 on the CTD of MDA5 (Figure 2) [82,83]. In contrast, dephosphorylation of RIG-I at Ser8 and Thr170 and MDA5 at Ser88 and Ser828 by protein phosphatase 1 (PP1)-α and PP1-γ was demonstrated to trigger RIG-I and MDA5-mediated antiviral immunity (Figure 2) [83,84].
Acetylation of RIG-I at lysine (Lys) residue 858 (Lys858) and Lys909 within the CTD domain was shown to interrupt the oligomer formation of RIG-I, which is necessary to activate RIG-I, thus repressing antiviral immunity (Figure 2) [85,86]. In contrast, histone deacetylase 6 (HDAC6)-mediated deacetylation of RIG-I reversely triggers the sensing of viral RNAs by RIG-I and activates downstream antiviral signaling (Figure 2) [85,86]. The sumoylation of RIG-I at Lys96 and Lys888 and MDA5 at Lys43 and Lys865 was demonstrated to inhibit the degradation of RIG-I and MDA5 in the cell resting state and promote the PP1-mediated dephosphorylation of RIG-I and MDA5 during viral infection (Figure 2) [87]. Conversely, sentrin/SUMO-specific protease 2 (SENP2)-mediated desumoylation of RIG-I and MDA5 led to Lys48 polyubiquitination and proteasomal degradation at the late stage of viral infection (Figure 2) [87]. In addition, the conjugation of IFN-stimulated gene 15 (ISG15) to RIG-I by ISGylation and deamination of RIG-I represent additional regulatory mechanisms for activating RIG-I antiviral immunity [88,89]. On the other hand, adenosine deaminase acting on RNA 1 (ADAR1), an ISG with RNA editing, was shown to restrict the recognition of viral RNAs by RLRs and repress the downstream antiviral immune response [90,91,92].
Ubiquitination has been widely demonstrated to play multiple roles in regulating RLR signaling. Linkage of the Lys-63-linked polyubiquitin chain onto Lys172 within the N-terminal CARDs of RIG-I by TRIM25 was first reported to serve as a prerequisite signal for binding to MAVS (Figure 2), thus controlling the induction of the downstream antiviral IFN response [93]. In addition to TRIM25, another ubiquitin E3 ligase, RING finger protein 135 (RNF135, also named Riplet and REUL), induces Lys-63-linked polyubiquitination of RIG-I CARDs (Lys154, Lys164, and Lys172) and CTD (Lys788, Lys849, and Lys851) to activate the antiviral IFN response (Figure 2) [94,95,96,97]. Moreover, mex-3 RNA binding family member C (MEX3C, also known as RNF194) also triggers the linkage of Lys-63-linked polyubiquitin to Lys45, Lys99, and Lys169 within the CARDs of RIG-I to promote the immune response against viral infection (Figure 2) [98]. In contrast to the positive role of Lys-63-linked polyubiquitination, the linkage of the Lys-48-linked polyubiquitin chain to RIG-I promotes the degradation of RIG-I, thereby negatively regulating the activation of the antiviral IFN response [99,100,101]. To date, at least three ubiquitin ligases, including RNF122, RNF125, and casitas B-lineage lymphoma proto-oncogene (Cbl), have been indicated to promote Lys-48-linked polyubiquitination at the Lys115 and Lys146 residues of RIG-I CARDs and Lys813 on the CTD of RIG-I, thus destabilizing RIG-I (Figure 2) [99,100,101]. In contrast, deubiquitination (DUB) enzymes, such as ubiquitin-specific protease (USP) 4 (USP4) and USP15, enhance RLR antiviral immunity by removing the Lys48-linked polyubiquitination of RIG-I (Figure 2) [102,103]. Other DUB enzymes, USP3 and CYLD lysine 63 deubiquitinase (CYLD), bind to CARDs of RIG-I and cleave Lys-63-linked polyubiquitin to repress the RIG-I-mediated IFN response (Figure 2) [104,105]. Analogously, TRIM65-mediated Lys-63-linked polyubiquitin at Lys743 within the helicase domain of MDA5 was shown to pivotally activate MDA5 antiviral signaling [106], whereas RNF125 represses MDA5 antiviral function by promoting Lys-48-linked polyubiquitination (Figure 2) [99]. USP17 has been identified as a DUB enzyme involved in the positive regulation of the IFN response by deubiquitinating MDA5 as well as RIG-I [107]. Very recently, ovarian tumor protease (OTU) deubiquitinating enzyme 3 (OTUD3), a deubiquitination enzyme, was reported to bind to MDA5 and RIG-I and ultimately remove their Lys-63-linked polyubiquitination to diminish the signal transduction of RLRs, thus inhibiting the antiviral immune response against RNA viruses (Figure 2) [108]. Apart from its function of complexing with Lys on RLRs, the unanchored Lys-63-linked polyubiquitin chain also serves as a ligand of RLR CARDs and potentiates RLR antiviral signaling [109,110].
In the past decade, mounting lines of evidence have demonstrated that the assembly of filamentous and highly ordered structures of RLRs, including RIG-I and MDA5, critically activates RLR antiviral signaling by enhancing their binding affinity to dsRNA and MAVS [110,111,112,113,114,115,116,117,118]. In the presence of an unanchored Lys-63-linked polyubiquitin chain and ATP hydrolysis, the CARDs of RIG-I assemble on RNA and form a tightly bound tetramer, which forms single-competent aggregates with the CARDs of MAVS [110,111,112,118]. In contrast to RIG-I, the head-to-tail assembly of MDA5 forms a filament along the long dsRNA sequence, allowing MDA5 to discriminate between self- and nonself RNA via assembly and disassembly dynamics [113,114,115,116]. It is also noted that ATP hydrolysis is required to disassemble MDA5 filaments rather than to oligomerize MDA5 [113,114,115,116]. In addition to RLRs, viral infection induces the aggregation of MAVS on the MOM and forms prion-like fibers that are resistant to detergent and proteases [39,40]. MAVS aggregates on the MOM and activates RLR antiviral signaling not only by associating with the RIG-I tetramer to form single-competent aggregates [110,111,112,118] but also by recruiting ubiquitin E3 ligases to produce polyubiquitin chains [39,40]. In addition to mitochondria, MAVS localized on peroxisomes and mitochondria-associated endoplasmic reticulum (ER) membranes (MAMs) also serves as a platform for innate antiviral immunity [119,120,121]. In particular, viral infection induces the assembly of a RIG-I translocon consisting of RIG-I, TRIM25, and 14-3-3ε on MAMs to activate the RLR antiviral response by promoting RIG-I polyubiquitination and interaction with MAVS [121].
2.4. Gene Polymorphism(s) of RLR and Modulation of RLR Antiviral Signaling
Single nucleotide polymorphisms (SNPs) in RLR have been identified and shown to regulate IFN response and affect viral susceptibility. The mutation of serine at residue 183 of RIG-I to isoleucine (Ser183Ile) has been shown to inhibit the polyinosinic acid-polycytidylic acid (poly ([I:C]), 5′-triphosphate-RNA, Sendai virus (SeV), and IAV-induced activation of IFN-β promoter, presumably increasing the formation of RIG-I self association [122,123]. In addition, the mutation of arginine at cysteine (Arg7Cys) also reportedly decreases the activation of the IFN-β promoter in IAV-infected cells [124]. In contrast, a nonsense mutation of glutamic acid at residue 627 (E627) leads to deletion of the C-terminal region and dsRNA binding activity of MDA5, which represses the poly (I:C)-triggered activation of the IFN-β promoter [123]. In addition, an inherited missense mutation in MDA5 (mutation of lysine at residue 365 to glutamic acid, K365E) also reportedly represses the poly (I:C)-induced activation of the IFN-β promoter and the IFN-driven ISRE activation of the ISRE promoter [125]. Additionally, K365E mutant MDA5 increases the replication of human rhinovirus (HRV) in patient-derived nasal epithelial cells [125]. Type 1 diabetes (T1D)-associated variants in MDA5, including mutations of alanine at residue 946 to threonine (A946T), isoleucine at residue 923 to valine (I923V), and glutamic acid at residue 627 to the stop codon (E627X), have also been demonstrated to decrease the rotavirus (RV)-induced upregulation of IFN-β mRNA and increase the replication of RV in infected cells [126]. These studies collectively indicate that SNPs and germline mutations of RLR may disturb innate antiviral immunity and enhance susceptibility to virus infection.
3. Autophagy
3.1. Types of Autophagy and Autophagy-Related Molecules
Autophagy is a self-digestion process that recycles nutrients and regenerates energy through lysosomal degradation of unwanted intracellular components, such as damaged organelles and aggregated/unfolded proteins [127,128,129]. Proper and precise control of autophagy primarily maintains cellular homeostasis, whereas dysregulation of autophagy has been widely shown to be associated with human diseases [129,130,131]. Three types of autophagy have been identified in eukaryotes, including microautophagy, macroautophagy, and chaperone-mediated autophagy (CMA); all three kinds of autophagy involve lysosomal degradation (Figure 3) [129,132,133,134]. Microautophagy involves the protrusion and/or invagination of the lysosomal membrane to sequester the components of the cytosol, including organelles, and deliver them to the lysosomal lumen for degradation (Figure 3A) [135,136,137,138]. CMA is also a lysosome-dependent degradation process that selectively eliminates substrates that possess the pentapeptide “Lys-Phe-Glu-Arg-Gln” (KFERQ) motif (Figure 3B) [139,140,141]. After recognition by heat shock cognate 70 kDa protein (HSC70), which specifically binds to lysosomal membrane protein 2A (LAMP2A) on the lysosomal membrane, CMA cargoes are then transported into the lumen of lysosomes and degraded by lysosomal proteases (Figure 3B) [139,140,141]. Macroautophagy, the most common form of autophagy, is a stepwise vacuole biogenesis process involving the formation of double-membranous autophagosomes that sequester cytoplasmic portions and ultimately deliver them to lysosomes for decomposition (Figure 3C) [129,134,142,143]. The completion of autophagy relies on the coordinated regulation of functional autophagy-related genes (ATGs) and the successful rearrangement of intracellular membranes, which is initiated by the nucleation of the isolation membrane (IM)/phagophore [129,142,143,144,145]. When cells are starved of nutrients, suppression of the serine/threonine protein kinase mammalian target of rapamycin (mTOR) leads to translocation of the unc-51-like kinase (ULK) complex containing ULK1/2, ATG13, ATG101, and RB1-inducible coiled-coil 1 (RB1CC1, otherwise known as FIP200) from the cytoplasm to a unique compartment derived from the ER membrane [146,147,148,149]. Then, the ULK complex activates the class III phosphatidylinositol-3-OH kinase (PI(3)K) complex (containing Vps34/PI3KC3, ATG14, Beclin 1, and Vps15) on the ER-reconstituted microdomain, leading to the biosynthesis of phosphatidylinositol-3-phosphate (PtdIns(3)P) [145,150,151,152]. The newly generated PtdIns(3)P subsequently induces the relocalization of double-FYVE-containing protein 1 (DFCP1) and WD-repeat domain PtdIns(3)P-interacting (WIPI, also known as ATG18) to induce the emergence of an ER-reconstituted and omegasome-like IM/phagophore [145,150,151,153,154]. Furthermore, the ER-associated vacuole membrane protein 1 (VMP1) promotes crosstalk between the ER and IM/phagophore and the recruitment of the class III PI(3)K complex by direct binding to Beclin-1 [155,156,157]. Moreover, the multi-spanning membrane protein ATG9a triggers membrane movement and lipid mobilization from other organelles, including the trans-Golgi network (TGN), plasma membrane, and lipid droplet to ER subdomain, thus supporting the formation of IM/phagophore [158,159,160,161,162]. In addition to the ER [163,164], various types of intracellular membranous compartments, including mitochondria [165], Golgi apparatus [166], recycling endosome [167,168], MAMs [169], and plasma membrane [170], also provide the required membranes for the emergence of IM/phagophore.
When IM/phagophores are nucleated on the preautophagosomal (PAS) structure, two ubiquitin-like (UBL) conjugation systems subsequently facilitate the expansion and enclosure of IM/phagophores into double membranous autophagosomes (Figure 3C) [149,171,172,173,174,175]. The enzymatic reaction composed of ubiquitination activating enzyme 1 (E1), ATG7, and ubiquitin-conjugating enzyme 2 (E2), ATG10, induces the covalent conjugation of the UBL molecule ATG12 to ATG5, forming the ATG12-ATG5 conjugate. Then, the ATG12-ATG5 conjugate further interacts with ATG16L to form an ATG12-ATG5-ATG16L heterotrimeric complex [171,172,176,177,178]. Moreover, the conjugation of phosphatidylethanolamine (PE) to ATG8/LC3 family proteins (so-called lipidation of ATG8/LC3) requires another UBL conjugation machinery involving ATG7 (E1) and ATG3 (E2) [179,180,181]. The C-termini of ATG8/LC3 proteins are first cleaved by ATG4 family proteases, producing ATG8/LC3-I, which is then conjugated to PE to generate ATG8/LC3-II, namely, lipidated ATG8/LC3 (also known as ATG8/LC3-PE) [179,180,181]. Then, ATG8/LC3-II promotes the expansion of the PAS membrane and membrane fusion to facilitate the maturation of autophagosomes [174,182,183], whereas ATG12-ATG5-ATG16L (presumably functions as an E3 ligase) facilitates the conjugation of PE to ATG8/LC3-I [184,185]. Despite these UBL conjugation systems, UV radiation resistance-associated gene (UVRAG) protein interacts with the class III (PI(3)K) complex and participates in the trafficking of endosomes and autophagosome formation [151,186,187]. Mature autophagosomes subsequently fuse with lysosomes (Figure 3C), forming autolysosomes in which acidic lysosomal proteases degrade the sequestrated components to recycle nutrients and regenerate energy. Several classes of proteins have been shown to function in autophagosome–lysosome fusion [175,188,189,190,191,192]. The Rab-interacting lysosomal protein (RILP) and FYVE and coiled-coil domain-containing 1 (FYCO1), two regulators involved in microtubule dynamics, have been shown to interact with the autophagosome-bound small GTPase Ras-related protein 7 (Rab7), thereby promoting the movement of autophagosomes along with microtubules [193,194,195,196,197]. In addition, actin remodeling by HDAC6 also facilitates the maturation of autolysosomes [198]. In addition to autophagosome transport through the cytoskeleton, pleckstrin homology domain-containing protein family member 1 (PLEKHM1) and homotypic fusion and protein sorting (HOPS) complex associated with syntaxin 17 (STX17) promote the fusion of autophagosomes with lysosomes [199,200,201]. Moreover, the protein complex composed of ATG14L, vesicle-associated membrane protein 8 (VAMP8), synaptosome-associated protein 29 (SNAP29), and STX17 stimulates the tethering of membranes involved in autophagosome–lysosome fusion [202,203]. Furthermore, ATG8/LC3 family proteins play pivotal roles in the fusion of autophagosomes with lysosomes by recruiting PLEKHM1 [204]. When the autophagy process is completed, the reactivation of mTOR by nutrient recycling subsequently terminates autophagy by coordinately suppressing the initiation of autophagy and activating autophagic lysosome reformation (ALR) [205]. Several molecules, including phospholipid PtdIns(4,5)P(2), the functional molecules involved in endocytosis, clathrin, and motor movement, kinesin, the lysosomal efflux permease, and the ubiquitin E3 ligase Cullin 3-Kelch-like protein 20 (KLHL20), have been shown to participate in ALR activation at the termination stage of autophagy [206,207,208,209,210,211,212,213,214,215,216].
3.2. Selective Autophagy
Although autophagy is known as a non-specific degradation process, accumulating lines of evidence indicate that autophagy may selectively sequester intracellular components and deliver them to lysosomes for degradation, a process called “selective autophagy” (Figure 3C) [217,218,219,220,221]. Through recognition of protein ubiquitination and/or association with adaptor proteins and the specific interaction between the autophagosomal ATG8/LC3 family proteins, the cargo receptors of selective autophagy can specifically target proteins and organelles for autophagic degradation (Figure 3C) [220,221,222,223,224,225,226,227,228]. Several types of cargo receptors, such as the neighbor of BRCA1 (NBR1), calcium-binding and coiled-coil domain-containing protein 2 (Calcoco2, also named NDP52), p62/sequestosome 1 (p62/SQSTM1), optineurin (OPTN), BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3)-like (BNIP3L/Nix), and Tax1-binding protein 1 (Tax1 bp1), have been identified to function in selective autophagy [220,221,224,225,227,228]. Most of these cargo receptors contain the ubiquitin-associated domain (UBA) that is responsible for the recognition of ubiquitinated proteins and the LC3-interacting regions (LIRs), which are required for ATG8/LC3 family protein binding [220,221,224,225,227,228]. In addition to LIRs located within cargo receptors, two unique interacting domains for binding to ATG8/LC3 family proteins, including ATG8-interacting motifs (AIMs) and GABARAP-interacting motifs (GIMs) within ATGs and adaptor proteins, have been shown to participate in the sequestration of cargoes within selective autophagy and the local biogenesis of autophagosomes [229,230,231,232]. Specific elimination of organelles, including mitochondria, peroxisomes, lysosomes, ER, ribosomes, lipid droplets (LDs), and nuclei, by selective autophagy (referred to as “organellophagy”) not only maintains the integrity of organelles but also promotes the regeneration of them (Figure 3C) [217,219,233].
Mitophagy is a specific kind of selective autophagy that uniquely targets mitochondria for degradation [234,235]. The fission and depolarization of mitochondria induced by mitochondrial damage inhibit the proper proteolysis of PTEN-induced putative kinase 1 (PINK1) by presenilin-associated rhomboid-like protein (PARL) within mitochondria, leading to stabilization of PINK1 on the MOM [236,237]. MOM-bound PINK1 induces the recruitment of Parkin, a ubiquitin E3 ligase, and phosphorylates serine 65 residues within Parkin and ubiquitin [238,239,240,241,242,243]. Parkin in turn promotes the ubiquitination of mitochondrial proteins [238,239,240,241,244], thereby inducing the recruitment of selective autophagy cargo receptors, such as p62/SQSTM1, NDP52/Calcoco2, and OPTN, for the removal of damaged mitochondria in a PINK1/Parkin-dependent manner (Figure 3C) [235,245]. The TBK1-induced phosphorylation of Ser177, Ser473, and Ser513 within OPTN and Ser403 within p62/SQSTM1 facilitates the recruitment of cargo receptors for mitophagy [246,247,248]. In addition to these cargo receptors, Toll-interacting protein (Tollip) and the mitochondrial adenine nucleotide translocator (ANT) complex serve as other mitophagy receptors for PINK1/Parkin-dependent mitophagy (Figure 3C) [249,250,251,252,253,254]. To facilitate the sequestration of ubiquitinated mitochondria by autophagy, DFCP1 and WIPI/ATG18 family proteins, two regulators of initiating autophagy, are coincidently recruited to facilitate the emergence of IM/phagophore proximal to damaged mitochondria [245]. Moreover, the spatiotemporal translocations of FIP200 and ATG13 by TBK1 phosphorylation and OPTN [255] and the omegasome-proximal PtdIns(4,5)P2 and F-actin organization [256] also promote the constitution of an omegasome-like structure and the disassembly of mitoaggregates to facilitate the removal of damaged mitochondria during mitophagy. However, deubiquitination of MOM proteins by DUB enzymes USP30 and USP35 suppresses mitophagy [257,258]. In contrast, various mitochondrial proteins, including BNIP3, FUN14 domain-containing 1 (FUNDC1), BNIP3L, yeast ATG32, prohibitin 2 (PHB2), and nitrophenylphosphatase domain and nonneuronal SNAP25-like protein homolog (NIPSNAP) family proteins, activate mitophagy by interacting with ATG8/LC3 family proteins (Figure 3C) [259,260,261,262,263,264,265,266].
To remove the damaged ER by organellophagy (referred to as ER-phagy) [267,268,269], ATG39, ATG11, and ATG40 in yeast [270] and the family with sequence similarity 134, member B (FAM134B), and reticulon family proteins in mammals [271,272,273] serve as cargo receptors for targeting the stressed ER for degradation (Figure 3C). In addition, testis-expressed sequence 264 protein (TEX264) [274,275], Atlastins 3 (ATL3) [276,277], cell-cycle progression gene 1 (CCPG1) [278], translocon, SEC62 [279], and progesterone receptor membrane component 1 (PGRMC1) [280] act as cargo receptors of ER-phagy (Figure 3C). Moreover, the elimination of harmful peroxisomes by organellophagy, so-called pexophagy, relies on several identified cargo receptors, including yeast ATG36 [281,282,283], mammalian NBR1 [284], and mammalian p62/SQSTM1 (Figure 3C) [285]. Additionally, two kinases, yeast Hrr25 and mammalian ataxia-telangiectasia-mutated (ATM), induce the phosphorylation of ATG36 and p62/SQSTM1, respectively, to positively regulate pexophagy [281,285,286]. In addition to phosphorylation, polyubiquitination of several peroxisomal (PEX) membrane proteins, such as PEX5 and the 70-kDa PEX membrane protein (PMP70) by the ubiquitin E3 ligase PEX2, also promotes NBR1-dependent pexophagy in mammals [287]. Very recently, the induced translocation of a ubiquitin E3 ligase, STIP1 Homology and U-Box Containing Protein 1 (Stub1), was shown to promote the turnover of oxidized peroxisomes through ubiquitination-dependent pexophagy (Figure 3C) [288]. Organellophagy is also involved in the turnover of other organelles and proteins, including lysosomes (lysophagy) [289,290,291], nuclei (nucleophagy) [270,292], ribosomes (ribophagy) [293,294,295], protein aggregates (aggrephagy) [198,296,297,298,299], LDs (lipophagy) [300,301,302,303], and ferritin (ferritinophagy) [304,305,306,307], through specific cognate cargo receptors (Figure 3C).
In addition to clearing damaged organelles by organellophagy, selective autophagy may eliminate invading pathogens, including bacteria and viruses, referred to as “xenophagy” [308,309,310,311,312]. Several types of cargo receptors, such as p62/SQSTM1, NDP52/Calcoco2, OPTN, and NBR1, participate in the process of xenophagy, in which Salmonella [313], group A Streptococcus [314], Mycobacterium tuberculosis [315], and Sindbis virus (SINV) [316] are eliminated. Phosphorylation of p62/SQSTM1 and OPTN [312,313,317] and ubiquitination of pathogen-associated proteins [318,319,320] play critical roles in the regulation of xenophagy (Figure 3C).
3.3. Virophagy
Several viral infections have been extensively shown to activate host autophagy to eliminate the infecting viruses and thus restrict viral growth in the infected cells, which is called “virophagy” [321,322,323]. The targeting of invading viruses for degradation through virophagy represents one type of cell defense against numerous human pathogenic viruses, including herpes simplex virus-1 (HSV-1) [324,325,326], SINV [327], human cytomegalovirus (HCMV) [328], Chikungunya virus (CHIKV) [329], and coxsackievirus B3 (CVB3) [330]. The RNA-dependent protein kinase (PKR) and eukaryotic translation initiation factor 2 subunit 1 (eIF2alpha)-dependent activation of virophagy degrades the HSV-1 proteins, thus restricting HSV-1 infection [324]. Conversely, ICP34.5 can antagonize HSV-1-induced virophagy by binding to Beclin-1 to inhibit autophagy initiation [331], thereby promoting HSV-1 replication and neurovirulence. Moreover, Ames et al. recently showed that OPTN promoted the degradation of HSV-1 VP16 and gB proteins through autophagy, and deficiency of OPTN led to impairment of host immunity, an increase in susceptibility to encephalitis, and loss of cognition [332]. SINV infection induces UBL-dependent virophagy to degrade viral capsid proteins through SMAD ubiquitin regulatory factor 1 (SMURF1)-mediated capsid ubiquitination and the recognition of ubiquitinated capsids by p62/SQSTM1 [316,327]. In a similar fashion, virophagy also promotes the degradation of the CHIKV capsid protein and CVB3 VP1 protein [329,330], indicating that virophagy may act as an innate antiviral immune response to negatively repress viral replication and protect infected cells from virus-induced cell death.
Since autophagy involves the rearrangement of intracellular membranes, several viruses exploit activated autophagy in infected cells to form multi-vesicular compartments for viral RNA replication, such as poliovirus and HRV [333,334]. Analogously, other viruses that cause a major burden in the human population, including HCV [335,336,337], hepatitis B virus (HBV) [338,339], DENV [340,341], and enteroviruses [342,343], were also shown to utilize autophagy in order to organize membranous structures for virus replication. Notably, SARS-CoV-2 infection has recently been reported to induce the accumulation of autophagosomes, in which viral RNAs replicate, presumably through the repression of autophagosome–lysosome fusion by the viral genome-encoded protein open reading frame 3a (ORF3a) [344,345,346]. Similarly, CVB3 infection has been shown to block autophagosome fusion through the viral protease-mediated proteolysis of SNARE complex, ultimately promoting accumulated autophagosome for the replication of the virus [347]. Therefore, viral-induced autophagy may play a distinct role in positively promoting virus growth by generating membranous vesicles for the replication of viral RNA. In addition, viral-induced autophagy functions in the nonlytic and intercellular spread of nonenveloped viruses, such as poliovirus, through an unconventional secretion pathway [348]. Moreover, autophagy reportedly participates in the viral envelopment of HBV [349], the egress of HCV [350], the maturation of infectious particles of DENV [351], and the prevention of cell death induced by DENV and CHIKV [352,353]. Together, these studies indicate that viral-induced autophagy may affect many aspects of the virus life cycle to benefit viral growth.
4. Regulation of RLR Signaling by Autophagy
In recent years, autophagy has emerged as playing a functional role(s) in negatively regulating RLR-mediated antiviral signaling. Several viruses activate autophagy and thus target the degradation of functional molecules and organelles involved in the regulation of the RLR IFN response. In this section, we therefore summarize the current knowledge on how autophagy represses RLR innate immunity according to the molecular targets of RLR signaling degraded by autophagy. The molecular mechanism and physiological significance of autophagy in RLR antiviral immunity is also discussed.
4.1. The Interaction between the ATG12-ATG5 Conjugate and CARDs of RIG-I and MDA5
The integration of autophagy into the RLR antiviral immune response was first uncovered by findings showing the enhancement of the vesicular stomatitis virus (VSV)-induced RLR-mediated IFN response in the infected ATG5-deficient mouse embryonic fibroblasts (MEFs) (Table 1) [354]. The increased RLR antiviral IFN immunity in VSV-infected cells lacking autophagy was characterized by increases in IFN-β mRNA levels and IRF3 phosphorylation, which were accompanied by a decrease in the viral infectivity of infected cells. Apart from VSV-infected cells, gene knockout of ATG5 also leads to hyperactivation of the dsRNA (poly[I:C])-induced type IFN I response, as shown by the increased mRNA levels of IFN-4α, IFN-β, interleukin-6 (IL-6), and C-X-C motif chemokine 10 (CXCL-10, also named IP-10). Reciprocally, overexpression of wild-type (WT) ATG5, rather than ATG5 K103R (Lys130 mutated to arginine [Arg]), which is unable to conjugate with ATG12 in cells, repressed the dsRNA-induced increase in promoter activities of IFN-4α, NF-kB, and IFN-β. In addition, the ATG12-ATG5 conjugate was shown to physically interact with RLRs in human embryonic kidney (HEK293) cells, including RIG-I and MAVS, through CARD binding. These studies collectively suggest that the ATG12-ATG5 conjugate, an important regulator of autophagosome maturation, may specifically bind to the CARDs of RIG-I and MAVS, leading to repression of RLR signaling and inhibition of the antiviral response (Table 1) (Figure 4) [354].
4.2. Removal of Mitochondria by Autophagy
Mitochondria and the associated ER membrane represent platforms for the assembly of RLR-associated molecules and signaling transduction of RLR antiviral immunity [119,120,121]. In recent years, it has been interesting and remains questionable whether alteration of mitochondrial dynamics and mitochondrial turnover regulate RLR antiviral signaling. Tal et al. firstly showed that interference with autophagy led to an increase in type I IFN response, which was associated with the accumulation of mitochondria [355]. In this study, the authors reported that gene silencing of ATG5 in MEF increased the dsRNA-induced production and secretion of IFN-α and IL-6, thus enhancing VSV infection (Table 1) [355]. In addition, ATG5-knockdown cells were shown to contain accumulated mitochondria, as demonstrated by the enhanced fluorescence intensity of MitoTracker (MitoTracker Green and MitoTracker Red)-labeled mitochondria in a flow cytometry assay and the upregulated mitochondrial DNA expression in a Southern blotting analysis. Along with these results, the expression of MAVS was indicated to be increased in ATG5 knockout MEFs, as shown by Western blotting and immunofluorescence (IFA) staining coupled flow cytometry assays. Additionally, overexpression of MAVS increased IFN-α mRNA levels in ATG5-deficient cells. Moreover, MitoSOX fluorescence probe labeling and flow cytometry experimental results showed that interruption of the autophagic process in MEF cells by ATG5 knockout led to an increase in the level of mitochondrial reactive oxygen species (ROS). In addition, treatment of ATG5-deficient MEFs with the antioxidant N-acetyl-L-cysteine (NAC) not only decreased the ROS level in mitochondria but also alleviated dsRNA-induced upregulation of IFN-α expression. In contrast, induction of mitochondrial ROS accumulation by an inhibitor of the mitochondrial electron transfer complex 1, rotenone, in autophagy-competent cells increased the dsRNA-activated IFN-α mRNA levels, which was further potentiated in rotenone-treated ATG5-deficient cells. In conclusion, these findings imply that autophagy plays a homeostatic role in the regulation of RLR antiviral immunity by eliminating mitochondria containing ROS, which may trigger overexpression of MAVS and hyperactivation of RLR signaling (Table 1) (Figure 4) [355].
4.3. Repression of Flaviviral PAMP-Triggered RLR Innate Immunity by Autophagy
In addition to VSV and dsRNA-induced IFN antiviral responses, viral-activated autophagy was also demonstrated to repress the HCV PAMP-induced IFN antiviral response (Table 1) [356,357]. The RIG-I N-terminal fragment (RIG-I N) has been previously shown to sufficiently activate the type I IFN response [22] and gene knockdown of ATG5 increased the RIG-I N-induced activation of IFN-β and ISRE promoters in human hepatoma Huh7 cells. Additionally, the HCV (JFH1 strain, genotype 2a) PAMP (3′-UTR and PU/UC within HCV RNA genome)-triggered IFN antiviral response was potentiated in cells with ATG5 gene silencing, as shown by the enhancement of IFN-β promoter activity and an increase in IFN-β mRNA level. The upregulated transactivation of IFN-β led to the upregulated expression of IFN-induced protein with tetratricopeptide repeats 1 (IFIT1, also named ISG56) and triggered paracrine antiviral immunity against HCV replication. In addition to HCV PAMPs, ATG5 knockdown also enhanced the DENV PAMP (3′-UTR)-induced activation of the IFN-β promoter and upregulated the expression of ISG56/IFIT1. Moreover, nutrient-starvation-induced autophagy triggered by treatment with Earle’s balanced salt solution (EBSS), Hank’s balanced salt solution (HBSS), or the mTOR inhibitor rapamycin was shown to repress HCV PAMP-activated IFN-β promoter activity. In a similar fashion, activation of autophagy through the unfolded protein response (UPR) by dithiothreitol (DTT) and tunicamycin treatment also inhibited the activation of the IFN-β promoter in HCV PAMP-treated cells. Furthermore, interference with autophagosome–lysosome fusion by the chemicals chloroquine (CQ) and bafilomycin (BAF-A1) and gene silencing of Rab7 and LAMP2 significantly inhibited IFN-β promoter activation. Overall, these results provide the first line of evidence showing that HCV activates autophagy to repress the RLR-mediated IFN response and suggest that pharmacological modulation of autophagy may alter innate antiviral immunity (Table 1) [356,357]. Soon after this study, another study showed that gene knockdown of Beclin-1 in HCV (H77 strain, genotype 1a)-infected immortalized human hepatocytes (IHHs) inhibited viral-activated autophagy and virus infectivity, which coincidently increased the mRNA levels of IFN-α, IFN-β, 2’-5’-oligoadenylate synthetase 1 (OAS1), and IFN-α-inducible protein 27 (IFI27) [358]. Additionally, inhibition of autophagy by silencing ATG7 gene expression analogously upregulated the mRNA expression of IFN-α, OAS1, and IFI27 and simultaneously reduced the infectious titer of HCV. Moreover, interference with HCV-induced autophagy promoted the apoptosis of infected IHHs. Again, these studies indicate that HCV may use autophagy to repress the RLR innate immune response (Table 1) [358]; however, whether and how RLR signaling molecules are regulated by autophagy in HCV-infected cells remain largely unclear.
4.4. Autophagic Degradation of TRAF6 by p62/SQSTM1
HCV (JFH1) infection was shown to induce autophagy through activating unfolded protein response (UPR) in an in vitro cell culture model [356,376]. Gene silencing of the molecules involved in autophagy and UPR inhibits the replication of HCV viral RNA [356,376], suggesting that HCV-activated autophagy benefits viral growth. In addition, HCV (JFH1) infection leads to mitochondrial fission and promotes the PINK1/Parkin-dependent mitophagy [377,378], thus attenuating cell apoptosis in the infected cells and promoting the establishment of viral persistence. These studies collectively indicate that HCV infection induces general autophagy and triggers selective autophagy to promote mitochondrial turnover.
Chan et al. first discovered that HCV (JFH1) infection led to the degradation of TRAF6, a ubiquitin E3 ligase necessary for producing polyubiquitin chains that recruit NEMO and activate RLR antiviral signaling (Table 1) [359]. Treatment with an autolysosome inhibitor, BAF-A1, restored the expression of TRAF6 in HCV-infected cells. Further studies revealed that HCV infection led to the colocalization of TRAF6 within viral-induced autophagic vacuoles and its interaction with p62/SQSTM1. Overexpression of TRAF6 inhibited the replication of HCV viral RNA, whereas depletion of TRAF6 gene expression increased the levels of HCV viral RNA and the viral infectivity of infected cells. Furthermore, TRAF6 knockdown was shown to reduce NF-kB promoter activity and decrease the mRNA and protein levels of IL-6 and tumor necrosis factor-α (TNF-α) in HCV-infected cells. These findings suggest that autophagy may negatively regulate the RLR-mediated type I IFN response by promoting the degradation of TRAF6 (Table 1) (Figure 4) [359].
4.5. Autophagic Degradation of RIG-I
RIG-I is a degradative substrate of autophagy induced by RNA viruses, including VSV and H1N1 IAV (Table 1) [360]. Du et al. reported that leucine-rich repeat containing protein 25 (LRRC25) significantly repressed RIG-I N-induced activation of the ISRE promoter in HEK293 cells (Table 1) [360]. The challenge of human leukemia monocytes, THP-1 cells with VSV and poly (I:C) resulted in the upregulation of LRRC25. Overexpression of LRRC25 repressed the poly (I:C)- and SeV-induced activation of the IFN-β and ISRE promoters, accompanied by enhanced VSV replication in HEK293 cells. In contrast, gene knockout of LRRC25 in THP-1 cells further increased VSV-triggered IRF3 phosphorylation and IFN-β production and simultaneously decreased the replication of VSV. Analogously, gene silencing of LRRC25 in human peripheral blood mononuclear cells (PBMCs) also led to the hyperactivation of H1N1 IAV-induced IRF3 phosphorylation and upregulation of IFN-β, IFIT1, and IFIT2 mRNA expression. In addition, VSV infection in THP-1 and PBMC cells and treatment with poly (I:C) in HEK293 cells promoted the interaction between LRRC25 and the CARDs of RIG-I and MDA5. Moreover, overexpression of LRRC25 in HEK293 cells promoted the degradation of RIG-I, which was abrogated by treatment with CQ and NH4Cl, inhibitors of autolysosome maturation and 3-MA, and gene knockout of ATG5 and Beclin-1. Moreover, the authors demonstrated that LRRC25 can mediate the interaction between RIG-I and p62/SQSTM1 in poly (I:C)-treated and VSV-infected cells by simultaneously binding to these two proteins. Gene knockout of p62/SQSTM1 reversed the LRRC25-induced degradation of RIG-I. It was noted that protein ubiquitination of RIG-I was not required for p62/SQSTM1-mediated targeting. Furthermore, gene knockout of ISG15 remarkably blocked the interaction between LRRC25 and RIG-I and reduced LRRC25-triggered RIG-I degradation, implying that ISG15 serves as a critical signal for the degradation of RIG-I by LRRC25. These studies collectively indicate that LRRC25 inhibits type I IFN response by promoting the degradation of RIG-I via ISG15 and p62/SQSTM1 (Table 1) [360]. In addition to LRRC25, the mitochondria-associated ubiquitin E3 ligase, membrane-associated ring-CH-type finger 5 (MARCH5), was also shown to promote the Lys-48-linked ubiquitination of RIG-I at the Lys193 and Lys203 residues within the CARDs of RIG-I, leading to the degradation of RIG-I and inhibition of RLR antiviral immunity (Table 1) (Figure 4) [361].
4.6. Degradation of RIG-I and MDA5 by Autophagy and PINK1/Parkin-Dependent Mitophagy
4.6.1. Mitophagic Degradation of RIG-I and MDA5 by PINK/Parkin-Dependent Pathway
Induction of mitochondrial depolarization and mitophagy was recently reported to antagonize the type I IFN response (Table 1) [362]. Bu et al. reported that treatment of HEK293 cells with the mitochondrial uncoupler carbonyl cyanide 3-chlorophenylhydrazone (CCCP) activated mitophagy and simultaneously reduced SeV-triggered activation of the IFN-β, ISRE, and NF-kB promoters. Similarly, CCCP treatment also inhibited the increase in IFN-β, ISG56/IFIT1, and ISG15 mRNA levels in SeV-infected cells in a dose- and time-dependent manner. Notably, CCCP treatment led to a dramatic decrease in the protein levels of RIG-I, MDA5, and MAVS and the phosphorylation of TBK1 and IRF3 in cells infected with SeV. Ectopic expression of Parkin repressed the SeV-induced type I IFN response and promoted viral replication in infected cells, whereas gene silencing of Parkin potentiated RLR antiviral immunity in VSV-infected cells and repressed the proliferation of VSV. In addition, ectopic expression of PINK1 along with Parkin further repressed the IFN antiviral response in SeV-infected cells. The PINK1/Parkin-induced repression of the type I IFN response and degradation of RIG-I and MDA5 were reversed by BAF-A1 treatment. Moreover, CCCP also promoted the interaction between Parkin and RIG-I and MDA5 on mitochondria, thus promoting Lys-48-linked ubiquitination of RIG-I and MDA5. These studies suggest that PINK1/Parkin-dependent mitophagy may repress RLR IFN immunity by promoting the ubiquitination and degradation of RIG-I and MDA5 (Table 1) (Figure 4) [362]. Very recently, Glon et al. reported that in addition to CCCP, oligomycin/antimycin A (O/A), AMBRA1-ActA, and Epstein–Barr virus (EBV) BHRF1 protein also inhibited IFN antiviral response through inducing mitophagy [363], arguing again that mitophagy plays a negative role in regulating RLR IFN response (Table 1) (Figure 4).
4.6.2. Autophagic Degradation of RIG-I and MDA5 by CCDC50
Apart from the known cargo receptors for selective autophagy, coiled-coil domain-containing protein 50 (CCDC50) was recently reported to function as a novel cargo receptor of selective autophagy for targeting RIG-I and MDA5 for degradation (Table 1) [364]. Gene silencing of CCDC50 in mouse BMDMs and BMDCs amplified the SeV-induced increase in IFN-β mRNA levels and production of IFN-β and increased the phosphorylation of TBK1 in SeV-infected cells. SeV infection led to increases in the mRNA and protein levels of CCDC50. In a similar fashion, gene knockout of CCDC50 in mice potentiated the SeV- and VSV-induced type 1 IFN response, restricted the viral replication of VSV, and protected VSV-infected mice from death. Overexpression of CCDC50 in HEK293 cells diminished the SeV-triggered activation of the IFN-β, ISRE, and NF-kB promoters, whereas gene knockout of CCDC50 increased the IFN-β mRNA and protein levels and repressed the production of infectious titers in VSV-infected cells. In addition, overexpression of CCDC50 led to the degradation of RIG-I and MDA5, while CCDC50 knockout prolonged the protein half-lives of RIG-I and MDA5. Moreover, SeV infection enhanced the interaction of CCDC50 with RIG-I and MDA5, and ectopic expression of CCDC50 in SeV-infected cells promoted the degradation of these two proteins, which was attenuated by 3-MA, CQ, and NH4Cl treatment. Additionally, SeV infection resulted in the interaction of CCDC50 with LC3B and p62/SQSTM1 and enhanced the colocalization between CCDC50 and autophagic vacuoles containing GFP-LC3B and p62/SQSTM1 puncta. Moreover, CCDC50 specifically bound to Lys-63-linked ubiquitinated RIG-I and MDA5 and interacted with LC3B, thereby targeting RIG-I and MDA5 for autophagy for degradation. These findings collectively suggest that CCDC50 may decrease the antiviral IFN response of RLR by targeting RIG-I and MDA5 for autophagic degradation (Table 1) (Figure 4) [364].
4.7. Autophagic Degradation of MAVS
4.7.1. Autophagic Degradation of MAVS by NDP52/Calcoco2
In addition to RIG-I and MDA5, MAVS have been extensively shown to be degraded by autophagy in repressing RLR antiviral response. Jin et al. first reported that selective autophagy may negatively regulate RLR-triggered IFN antiviral immunity through Tetherin (Table 1) [365]. In this study, the authors demonstrated that ectopic expression of Tetherin promoted the viral replication of VSV and SeV in infected A549 cells (human adenocarcinoma alveolar basal epithelial cells), accompanied by repression of IFN-β activation. Overexpression of Tetherin also reduced the RIG-I N-triggered activation of IFN-β and ISRE promoters in large T antigen-immortalized HEK293 (HEK293T) cells, whereas silencing of Tetherin in HEK293T and A549 cells potentiated the poly (I:C) and 5′-triphosphate-RNA-induced activation of IFN-β and ISRE promoters and elevated the mRNA levels of IFN-β, ISG56/IFIT1, and ISG54 (also named IFIT2). Similarly, gene knockdown of Tetherin led to elevated IFN-β and ISG56/IFIT1 mRNA levels, enhanced phosphorylation of TBK1 and IRF3 in SeV- and H1N1 IAV-infected cells, and coincidentally attenuated the replication of these two viruses. Tetherin abrogated RIG-I N, MDA5 N (the N-terminal fragment of MDA5 has been previously shown to sufficiently activate the type I IFN response [22]), and MAVS-triggered inductions on IFN-β and ISRE promoters. Further studies showed the specific interaction between Tetherin and MAVS, which was enhanced by H1N1 IAV infection. Additionally, poly (I:C) treatment induced the colocalization of Tetherin with MAVS in mitochondria. Ectopic expression of Tetherin resulted in MAVS degradation, which was reversed by treating cells with 3-methyladenine (3-MA), an inhibitor of the class III (PI(3)K) complex, autolysosome inhibitors, CQ, BAF-A1, and ammonia chloride (NH4Cl), and gene silencing of ATG5 and Beclin-1. In addition, Tetherin promoted the physical interaction between MAVS and NDP52/Calcoco2. Downregulation of NDP52/Calcoco2 gene expression alleviated the Tetherin-induced degradation of MAVS, attenuated the Tetherin-mediated repression of SeV-triggered IFN-β, ISG56/IFIT1, and ISG54/IFIT2 mRNA levels, and repressed the Tetherin-triggered enhancement of virus replication in SeV-infected cells. Moreover, Tetherin promoted the Lys-27-linked ubiquitination of MAVS at the Lys7 residue through the ubiquitin E3 ligase membrane-associated ring finger (C3HC4) 8 (MARCH8). Interference with the ubiquitination of MAVS by gene knockout of MARCH8 and a mutation of the ubiquitination site of MAVS (K7R) protected MAVS from degradation by SeV infection and Tetherin, and amplified SeV-triggered RLR antiviral IFN immunity, including elevated phosphorylation of TBK1 and IRF3 and upregulated mRNA levels of IFN-β, ISG56/IFIT1, and ISG54/IFIT2. Collectively, these studies conclude that Tetherin functions as a negative regulator of the RLR downstream IFN response through MARCH8-mediated ubiquitination of MAVS and autophagic degradation of MAVS via interaction with NDP52/Calcoco2 (Table 1) (Figure 4) [365].
In addition to SeV and VSV, NDP52/Calcoco2 also targets MAVS for degradation to inhibit RLR antiviral signaling in CVB3-infected cells [330]. Mukherjee et al. first reported that CVB3 may attenuate the type I IFN response through 3Cpro protease-mediated cleavage of MAVS and Toll receptor domain-containing adaptor inducing interferon-beta (TRIF) [379]. In addition to MAVS, CVB3 3Cpro targets MDA5 for proteolysis and leads to inhibition of the activation of IFN antiviral immunity [380]. These studies suggest that CVB3 represses the type I IFN immune response through the viral protease-mediated proteolysis of MAVS, TRIF, and MDA5. Interestingly, CVB3 3Cpro was reported to cleave SNAP29 and PLEKHM1, two molecules that critically function in autophagosome–lysosome fusion, thus inhibiting autophagic flux in the infected cells and inducing the accumulation of autophagosome for virus replication [347]. Mohamud et al. showed that p62/SQSTM1 and NDP52/Calcoco2 differentially regulated virus proliferation in CVB3-infected HeLa cells (Table 1) [330]. Gene silencing of NDP52/Calcoco2 dramatically reduced CVB3 replication in the infected cells, whereas p62/SQSTM1 knockdown in CVB3-infected cells elevated viral replication. In addition, overexpression of p62/SQSTM1 reduced CVB3 replication in the infected cells, while ectopic expression of NDP52/Calcoco2 enhanced it in CVB3-infected cells. In addition, both NDP52/Calcoco2 and p62/SQSTM1 were demonstrated to interact with the CVB3 VP1 protein. Further analysis revealed that CVB3 infection induced Lys-48- and Lys-63-linked protein ubiquitination of VP1. In addition, gene knockdown of NDP52/Calcoco2, rather than p62/SQSTM1, in CVB3-infected cells led to an increase in MAVS protein levels and the phosphorylation of TBK1. CVB3 infection induced the cleavage of NDP52/Calcoco2 after Q139 treatment by viral protease 3C, and the free C-terminal fragment of NDP52/Calcoco2 was able to promote the degradation of MAVS and the viral replication of CVB3 and repress the activation of poly(I:C)-induced IFN-β production in a similar fashion as the full-length form. These findings suggest that CVB3 infection may mediate the degradation of MAVS through NDP52/Calcoco2, thereby attenuating antiviral signaling downstream of RLR (Table 1) (Figure 4) [330].
4.7.2. Autophagic Degradation of MAVS by the RNF34-NDP52/Calcoco2 Axis
He et al. found that gene silencing of RNF34 in VSV-infected THP-1 cells remarkably potentiated the production of IFN-β and IL-6, accompanied by a decrease in the infectious titer (Table 1) [366]. Overexpression of WT RNF34 but not the E3 ligase-knockout mutant (H342A) of RNF34 significantly reduced the VSV-triggered activation of the IFN-β and NF-kB promoters. In addition, RNF34 overexpression repressed RIG-I N, MAVS, and poly (I:C)-triggered IFN-β activation. Reciprocally, knockdown of RNF34 gene expression elevated the phosphorylation of TBK1 and IRF3 and increased the expression of ISG54/IFIT2 and ISG56/IFIT1 in VSV-infected cells. RNF34 was shown to directly bind to MAVS, as indicated by yeast two-hybrid and glutathione S-transferase (GST) pull-down assays. VSV infection led to colocalization and interaction of RNF34 with MAVS. Additionally, RNF34 promoted Lys-27- and Lys-63-linked ubiquitination of MAVS at Lys311. Moreover, VSV infection led to the degradation of MAVS, which was reversed by NH4Cl treatment and gene silencing of RNF34. Furthermore, VSV infection also induced the interaction of MAVS with WT NDP52/Calcoco2, but not with the UBA mutant (D439R/C443K) of NDP52/Calcoco2. In addition, ectopic expression of WT NDP52/Calcoco2, but not the UBA mutant of NDP52/Calcoco2, repressed MAVS-induced activation of the IFN-β promoter. In contrast, gene knockdown of NDP52/Calcoco2 inhibited the VSV- and RNF34-induced degradation of MAVS. Furthermore, gene knockdown of RNF34 attenuated the VSV-induced degradation of the mitochondrial proteins TOMM20 and HSP60 and coincidently enhanced IRF3 phosphorylation. Conversely, ectopic expression of RNF34 facilitated the degradation of TOMM20 and HSP60, accompanied by a dramatic decrease in MAVS protein levels. Moreover, the accumulation of deformed mitochondria was found in the RNF34-silenced VSV-infected cells. Additionally, RNF34 was shown to induce mitophagy, as indicated by the increased acidic signal of an mt-Keima reporter, which is generally used for analyzing mitophagic processes [381,382]. These studies collectively suggest that RNF34 promotes the degradation of MAVS through NDP52/Calcoco2 and mitophagy (Table 1) (Figure 4) [366].
4.7.3. Degradation of MAVS by IAV PB1-F2/TUFM Axis-Induced Mitophagy
Varga et al. first showed that IAV (H1N1) PB1-F2 protein repressed the RIG-I N and MAVS-induced type I IFN response, and IAV PB1-F2 colocalized with MAVS, which suggests that IAV PB1-F2 may interact with MAVS and interfere with MAVS downstream antiviral signaling (Table 1) [368]. Recently, Wang et al. reported that the IAV (H1N1 and H5N1 strains) PB1-F2 protein specifically activated autophagy through the formation of autolysosomes (Table 1) [367], as demonstrated by an increased level of LC3-II and enhanced RFP+/GFP- fluorescence intensity of an mCherry-GFP-LC3 autophagy reporter, which is typically used to interpret autophagic flux [383]. Further analysis revealed the colocalization of IAV PB1-F2-induced autophagic vacuoles with mitochondria. The IAV PB1-F2 proteins also induced the degradation of mitochondrially encoded cytochrome C oxidase II (MTCO20) and translocase of the outer mitochondrial membrane complex subunit 20 (TOMM20). Moreover, the IAV PB1-F2 protein also increased the RFP+/GFP+ signal of the pmRFP-GFP-Mito mitophagy reporter. These results collectively suggest that the IAV PB1-F2 protein can activate mitophagy [367].
In addition, the authors demonstrated that the IAV PB1-F2 protein interacted with the Tu translation elongation factor, mitochondrial (TUFM) [367]. TUFM is a mitochondrial protein which has been shown to bind to NLR family member X1 (NLRX1) and the ATG12-ATG5-ATG16 trimeric complex (Table 1) [369]. TUFM was also demonstrated to activate autophagy and inhibit RLR downstream type I IFN response (Table 1) [369]. Downregulation of endogenous TUFM expression by small interference RNA (siRNA) gene knockdown and CRISPR/Cas9-sgRNA gene knockout inhibited IAV PB1-F2-induced mitophagy, as indicated by the restored protein levels of MTCO2 and TOMM20, and disturbed colocalization of autophagic vacuoles with mitochondria [367]. Moreover, overexpression of IAV PB1-F2 repressed MAVS-induced activation of the IFN-β promoter, whereas knockdown of TUFM reversed this inhibitory effect. The IAV PB1-F2-induced repression of the MAVS-activated IFN-β promoter was reversed by gene silencing of ATG5. Additionally, ectopic expression of IAV PB1-F2 resulted in the degradation of MAVS, which was restored by TUFM knockdown. Gene knockout of TUFM significantly increased the production of IFN in IAV-infected cells, accompanied by the repression of infected cell viral infectivity. Conversely, overexpression of TUFM benefited the viral growth of IAV-infected cells. Finally, the authors demonstrated that IAV PB1-F2 contained an LIR for binding to LC3B and served as a mitophagy receptor. Together, these results indicate that IAV infection may activate PB1-F2/TUFM axis-induced mitophagy to promote the degradation of MAVS, thus repressing RLR IFN immunity (Table 1) (Figure 4).
4.7.4. Autophagic Degradation of MAVS by NBR1 and RNF5
Analogously, ectopic expression of the H7N9 PB1 protein was also shown to inhibit SeV infection and poly(I:C)-induced activation of the IFN-β promoter, ISRE, and NF-κB promoters in HEK293 cells (Table 1) [370]. Accordingly, the overexpression of PB1 potentiated the SeV- and poly(I:C)-triggered upregulation of IFN-β, IFN-stimulated gene 15 (ISG15), ISG56/IFIT1, regulated upon activation normal T-cell expressed and secreted factor (RANTES), and oligoadenylate synthetase-like protein (OASL) mRNA levels. PB1 also diminished the phosphorylation of IRF3, TBK1, and Ik-Bα and reduced RIG-I N, MDA5 N, and MAVS-triggered activation of the IFN-β promoter, suggesting that PB1 significantly inhibited RLR antiviral signaling. Coimmunoprecipitation (co-IP), in vitro GST pull-down, and IFA assays revealed that PB1 directly interacted with MAVS. In addition, PB1 promoted the degradation of MAVS, which was restored by treatment with CQ and BAF-A1, two autolysosome inhibitors, and gene knockout of ATG7. Reciprocally, activation of autophagy by EBSS further enhanced PB1-triggered MAVS degradation. In addition, PB1 promoted the autophagic degradation of MAVS through the induction of RNF5-mediated Lys27-linked ubiquitination of MAVS and the physical interaction with NBR1. Moreover, overexpression of NBR1 reduced RIG-I N-induced IFN-β promoter activation and enhanced the infectivity of H7N9 IAV-infected cells. In contrast, gene knockout of NBR1 increased the SeV-triggered activation of the IFN-β promoter and decreased the H7N9 IAV infectious titer of infected cells. Finally, ectopic expression of RNF5 increased the virus titer of H7N9 IAV-infected cells, whereas RNF5 knockout repressed the viral infectivity of H7N9 IAV-infected cells. These findings indicate that IAV PB1 induces K27-linked ubiquitination of MAVS by RNF5 and promotes the autophagic degradation of MAVS through NBR1, thereby repressing the antiviral response of RLR (Table 1) (Figure 4) [370].
4.7.5. Autophagic Degradation of MAVS by p62/SQSTM1 and HFE
Liu et al. showed that H7N9 IAV infection in mice decreased the mRNA and protein levels of the hereditary hemochromatosis gene (HFE). The HFE protein has been shown to regulate iron metabolism by binding to the transferrin receptor (TFRC/TFR1) (Table 1) [371]. Gene knockout of HFE in H7N9 IAV-infected mice inhibited viral replication and protected the infected mice from H7N9 IAV-induced death. Notably, depletion of MAVS in H7N9 IAV-infected HFE knockout (HFE−/−) mice restored the infectious titer of infected cells. Further studies demonstrated that HFE gene knockout further potentiated the H7N9 IAV-induced mRNA and protein levels of IFN-β in infected mice, as well as the phosphorylation of TBK1 and STAT1 in H7N9 IAV-infected mouse bone marrow-derived macrophages (BMDMs), suggesting that H7N9 IAV infection induced HFE expression to repress the antiviral IFN response in RLR. In a similar fashion, depletion of HFE gene expression amplified the poly (I:C)-stimulated IFN-β mRNA and MAVS protein levels and VSV-induced TBK1, IRF3, and STAT1 phosphorylation. Ectopic expression of HFE led to a dramatic degradation of mitochondria- and peroxisome-associated MAVS in HEK293 cells. In contrast, HFE gene knockout resulted in the stabilization of MAVS in H7N9 IAV-infected mouse BMDMs, mouse bone marrow-derived dendritic cells (BMDCs), and MEFs. Moreover, HFE was shown to interact with and promote autophagic degradation of MAVS, which was reversed by treatment with CQ and gene silencing of ATG5 and ATG7. Furthermore, p62/SQSTM1 targeted MAVS for degradation by interacting with HFE, and gene knockout of p62/SQSTM1 diminished the HFE-induced degradation of MAVS and inhibited HFE-repressed MAVS-triggered activation of IFN-β and ISRE promoters. Overall, this study shows that HFE promotes autophagic degradation of MAVS by interacting with p62/SQSTM1 in IAV-infected cells, thus repressing downstream RLR antiviral immunity (Table 1) (Figure 4) [371].
4.8. Degradation of RIG-I and MAVS by CSFV-Activated Autophagy
Classical swine fever virus (CSFV), an enveloped and positive-stranded RNA virus, was shown to activate autophagy in infected swine kidney cells, PK-15 cells, and 3D4/2 porcine macrophage cells, as indicated by the upregulated level of LC3B-II and the increased number of GFP-LC3-labeled punctate (Table 1) [372]. Interference with the autophagic process by gene silencing of Beclin-1 or LC3B triggered cell apoptosis in CSFV-infected cells and increased the mRNA levels of IFN-α, IFN-β, and ISGs, TNF superfamily member 10 (TNFSF10), and tumor necrosis factor receptor superfamily member 6 (TNFRSF6, also known as Fas/CD95). In addition, deficiency of autophagy by gene knockdown of Beclin-1 or LC3B in CSFV-infected cells resulted in accumulation of ROS, upregulation of mitochondrial DNA, and increased protein levels of RIG-I and MAVS, which were reversed by NAC, an inhibitor of ROS release. In contrast, the induction of ROS by rotenone in CSFV-infected cells increased the level of RIG-I. Moreover, treatment with NAC reduced cell apoptosis and increased viral replication in CSFV-infected autophagy-deficient cells. In contrast, rotenone repressed the production of CSFV in autophagy-repressed cells. Thereafter, Xie et al. showed that CSFV activated autophagy through repression of mTOR and the calcium/calmodulin-dependent protein kinase 2 (CAMKK2/CaMKKβ)-protein kinase AMP-activated catalytic subunit alpha (PRKAA/AMPK) axis [373]. Moreover, the authors demonstrated that CSFV infection repressed the production of type I IFN through the interaction between Beclin-1 and MAVS. These findings indicate that CSFV may activate autophagy to remove ROS, thus preventing the infected cell from undergoing apoptosis and repressing the activation of the RLR antiviral immune response (Table 1) (Figure 4).
4.9. Degradation of IRF3 Protein Stability by TRIM21 and NDP52/Calcoco2
Wu et al. reported that SeV infection of A549 cells, THP-1 cells, and PBMCs led to the degradation of IRF3, which was restored by 3-MA treatment and gene knockout of ATG5 and Beclin-1 (Table 1) [374]. Additionally, SeV infection promoted the binding of IRF3 to NDP52/Calcoco2 and enhanced their colocalization. Gene knockout of NDP52/Calcoco2 resulted in the stabilization and dimerization of IRF3 in SeV-infected cells. In addition, SeV infection triggered the Lys-27-linked ubiquitination of IRF3 at K313 by TRIM21 ubiquitin E3 ligase. The ubiquitination of IRF3 by TRIM21 was shown to serve as a signal for NDP52/Calcoco2-mediated autophagic degradation in cells upon SeV infection. In contrast, the DUB enzyme proteasome 26S subunit, non-ATPase 14 (PSMD14), reversed the degradation of IRF3. Gene silencing and gene knockout of PSMD14 destabilized IRF3 in SeV-infected cells. Further analysis revealed that gene silencing of PSMD14 led to an increase in IRF3 protein ubiquitination in SeV-infected cells, and PSMD14 directly removed the conjugation of the polyubiquitin chain from K313 of IRF3, suggesting that PSMD14 is required for maintaining the stability of IRF3. Moreover, the reduction in VSV-induced ISRE promoter activation in PSMD14-knockout HEK293 cells, the decreased TBK1 and IRF3 phosphorylation by gene knockdown of PSMD14 in VSV-infected cells, the repression of VSV-triggered IFN-β production in PSMD14-silenced A549 cells, and the reduction in VSV-activated secretion of IFN-β in PSMD14-knockdown A549 cells together implied that PSMD14 expression is required for maintaining the basal level of IRF3 to induce the RLR IFN response. Gene knockout of NDP52/Calcoco2 and interference with the protein ubiquitination of IRF3 by a mutation on K313 (K313) diminished the PSMD14-enhanced SeV-infection-triggered IFN response. Hence, these findings suggest that autophagy regulates IRF3 stability and the antiviral IFN response of RLR (Table 1) (Figure 4) [374].
4.10. Autophagic Degradation of TBK1 through NEDD4 and NDP52/Calcoco2
In addition to IRF3, TBK1 was shown to be regulated by selective autophagy. Xie et al. recently reported that a ubiquitin E3 ligase, neural precursor cell-expressed developmentally downregulated gene 4 (NEDD4), was capable of attenuating the type I antiviral IFN response (Table 1) [375]. Ectopic expression of NEDD4 repressed SeV-, VSV-, and poly (I:C)-triggered activation of IFN-β and ISRE promoters in HEK293T cells. In particular, NEDD4 overexpression in SeV-infected cells diminished the upregulation of IFN-β, ISG56/IFIT1, and ISG54/IFIT2 mRNA levels and simultaneously enhanced viral replication in the infected cells. Additionally, ectopic expression of NEDD4 repressed the RIG-I N, MDA5, MAVS, and TBK1-induced activation of the IFN-β and ISRE promoters. In addition, NEDD4 was shown to bind to TBK1 physically, and SeV infection promoted the interaction and colocalization between these two proteins. Moreover, NEDD4 induced the Lys-27-linked ubiquitination of TBK1 at the K344 residue and promoted the recognition of TBK1 by NDP52/Calccoco2. Loss of NEDD4-triggered ubiquitination of TBK1 and gene knockout of NDP52/Calccoco2 resulted in resistance of TBK1 to degradation by NEDD4 in SeV-infected cells and relieved the repressive effect of NEDD4 on SeV-induced IFN-β, ISG56/IFIT1, and ISG54/IFIT2 mRNA levels. Together, these studies indicate a repressive role of the NEDD4/NDP52 axis in the regulation of RLR antiviral immunity by targeting TBK1 for degradation (Table 1) (Figure 4) [375].
5. Conclusions and Perspectives
In the past decade, multiple lines of evidence have implied that autophagy plays functional roles in the regulation of RLR antiviral signaling. Both general and selective autophagy may negatively control the activation of RLR innate immunity by targeting RLR signaling-related molecules for degradation, including RIG-I, MDA5, MAVS, IRF3, and TBK1. Additionally, several fundamental cellular events are involved in the autophagic degradation of these RLR signaling components: (1) the ubiquitin E3 ligases mediate the conjugations of different types of polyubiquitin chain linkages, such as Lys-48, Lys-63, and Lys-27-linked ubiquitination, to the degradative substrates; (2) the targeting of ubiquitinated molecules to degradation within the autophagic process by specific cargo receptors; and (3) the attenuation of degradation by DUB-mediated removal of protein ubiquitination. Although the molecular mechanisms underlying the regulation of RLR innate immunity by autophagy have been proposed, several fundamental questions remain. For instance, whether and how oragnellophagy-mediated turnover of damaged organelles, such as mitochondria and peroxisomes, mainly and/or sufficiently represses the antiviral signaling of RLR is still unclear. In addition, it remains questionable whether these identified molecular mechanisms control RLR innate immunity against virus infection by autophagy in a physiologically relevant context, and these mechanisms should be tested in viral infection experimental models using small animals with competent immune systems. Moreover, the spatial and temporal autophagic regulation of the RLR-mediated type I IFN response remains unresolved. Furthermore, numerous SNPs have been extensively identified in the genes encoding autophagic regulators, and most of these have been shown to be relevant to the development and pathogenesis of human diseases, such as cancer and Crohn’s disease [128,384]; however, the underlying molecular mechanism remains unclear. Most importantly, whether the SNPs and germline mutations of autophagy-related molecules involved in the repression of RLR antiviral signaling regulate the viral susceptibility and pathogenicity of infecting viruses remains largely unknown. Therefore, further investigations are urgently needed to comprehensively understand the physiological importance of the crosstalk between autophagy and RLR signaling in the balance of host cell-virus interactions and the pathogenesis of human diseases.
Funding
This study was supported by research grants from the National Science and Technology Council (MOST 109-2320-B-182-010-MY3), Taipei, National Health Research Institute (NHRI-EX103-10322SC, NHRI-EX104-10322SC, NHRI-EX105-10322SC and NHRI-EX106-10322SC), Miaoli, and Chang Gung Memorial Hospital (CMRPD1K0371, CMRPD1L0301, and CMRPD1M0671), Taoyuan, Taiwan.
Acknowledgments
The author apologizes to colleagues whose studies could not be cited due to space limitations. The author thanks the research grants supported by the National Science and Technology Council, National Health Research Institute, and Chang Gung Memorial Hospital. The author is grateful to Chih-Wei Chang for the help on preparing the figures and the resources from the microscopy center of Chang Gung University.
Conflicts of Interest
The authors declare no conflict of interest.
References
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Mesev, E.V.; LeDesma, R.A.; Ploss, A. Decoding type I and III interferon signalling during viral infection. Nat. Microbiol. 2019, 4, 914–924. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katze, M.G.; He, Y.; Gale, M., Jr. Viruses and interferon: A fight for supremacy. Nat. Rev. Immunol. 2002, 2, 675–687. [Google Scholar] [CrossRef]
- Barbalat, R.; Ewald, S.E.; Mouchess, M.L.; Barton, G.M. Nucleic acid recognition by the innate immune system. Annu. Rev. Immunol. 2011, 29, 185–214. [Google Scholar] [CrossRef] [PubMed]
- Chow, K.T.; Gale, M., Jr.; Loo, Y.M. RIG-I and Other RNA Sensors in Antiviral Immunity. Annu. Rev. Immunol. 2018, 36, 667–694. [Google Scholar] [CrossRef]
- Tan, X.; Sun, L.; Chen, J.; Chen, Z.J. Detection of Microbial Infections Through Innate Immune Sensing of Nucleic Acids. Annu. Rev. Microbiol. 2018, 72, 447–478. [Google Scholar] [CrossRef]
- Wu, J.; Chen, Z.J. Innate immune sensing and signaling of cytosolic nucleic acids. Annu. Rev. Immunol. 2014, 32, 461–488. [Google Scholar] [CrossRef]
- Li, D.; Wu, M. Pattern recognition receptors in health and diseases. Signal Transduct. Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef]
- Lind, N.A.; Rael, V.E.; Pestal, K.; Liu, B.; Barton, G.M. Regulation of the nucleic acid-sensing Toll-like receptors. Nat. Rev. Immunol. 2021, 22, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Sato, S. Toll-like receptors and their signaling mechanisms. Scand. J. Infect. Dis. 2003, 35, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.A.; Kagan, J.C. Toll-like Receptors and the Control of Immunity. Cell 2020, 180, 1044–1066. [Google Scholar] [CrossRef]
- Rehwinkel, J.; Gack, M.U. RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef]
- Onomoto, K.; Onoguchi, K.; Yoneyama, M. Regulation of RIG-I-like receptor-mediated signaling: Interaction between host and viral factors. Cell. Mol. Immunol. 2021, 18, 539–555. [Google Scholar] [CrossRef]
- Decout, A.; Katz, J.D.; Venkatraman, S.; Ablasser, A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 2021, 21, 548–569. [Google Scholar] [CrossRef]
- Hopfner, K.P.; Hornung, V. Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat. Rev. Mol. Cell Biol. 2020, 21, 501–521. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.K.; Wen, H.; Ting, J.P. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu. Rev. Immunol. 2011, 29, 707–735. [Google Scholar] [CrossRef] [Green Version]
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome activation and regulation: Toward a better understanding of complex mechanisms. Cell Discov. 2020, 6, 36. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Kikuchi, M.; Matsumoto, K.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Foy, E.; Loo, Y.M.; Gale, M., Jr.; Akira, S.; et al. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol. 2005, 175, 2851–2858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murali, A.; Li, X.; Ranjith-Kumar, C.T.; Bhardwaj, K.; Holzenburg, A.; Li, P.; Kao, C.C. Structure and function of LGP2, a DEX(D/H) helicase that regulates the innate immunity response. J. Biol. Chem. 2008, 283, 15825–15833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, S.; Eisenacher, K.; Kirchhofer, A.; Brzozka, K.; Lammens, A.; Lammens, K.; Fujita, T.; Conzelmann, K.K.; Krug, A.; Hopfner, K.P. The C-terminal regulatory domain is the RNA 5′-triphosphate sensor of RIG-I. Mol. Cell 2008, 29, 169–179. [Google Scholar] [CrossRef] [Green Version]
- Saito, T.; Hirai, R.; Loo, Y.M.; Owen, D.; Johnson, C.L.; Sinha, S.C.; Akira, S.; Fujita, T.; Gale, M., Jr. Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc. Natl. Acad. Sci. USA 2007, 104, 582–587. [Google Scholar] [CrossRef] [Green Version]
- Jiang, F.; Ramanathan, A.; Miller, M.T.; Tang, G.Q.; Gale, M., Jr.; Patel, S.S.; Marcotrigiano, J. Structural basis of RNA recognition and activation by innate immune receptor RIG-I. Nature 2011, 479, 423–427. [Google Scholar] [CrossRef] [Green Version]
- Kowalinski, E.; Lunardi, T.; McCarthy, A.A.; Louber, J.; Brunel, J.; Grigorov, B.; Gerlier, D.; Cusack, S. Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell 2011, 147, 423–435. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ludwig, J.; Schuberth, C.; Goldeck, M.; Schlee, M.; Li, H.; Juranek, S.; Sheng, G.; Micura, R.; Tuschl, T.; et al. Structural and functional insights into 5′-ppp RNA pattern recognition by the innate immune receptor RIG-I. Nat. Struct. Mol. Biol. 2010, 17, 781–787. [Google Scholar] [CrossRef] [Green Version]
- Yoneyama, M.; Fujita, T. Structural mechanism of RNA recognition by the RIG-I-like receptors. Immunity 2008, 29, 178–181. [Google Scholar] [CrossRef] [Green Version]
- Luo, D.; Kohlway, A.; Vela, A.; Pyle, A.M. Visualizing the determinants of viral RNA recognition by innate immune sensor RIG-I. Structure 2012, 20, 1983–1988. [Google Scholar] [CrossRef] [Green Version]
- Luo, D.; Ding, S.C.; Vela, A.; Kohlway, A.; Lindenbach, B.D.; Pyle, A.M. Structural insights into RNA recognition by RIG-I. Cell 2011, 147, 409–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Civril, F.; Bennett, M.; Moldt, M.; Deimling, T.; Witte, G.; Schiesser, S.; Carell, T.; Hopfner, K.P. The RIG-I ATPase domain structure reveals insights into ATP-dependent antiviral signalling. EMBO Rep. 2011, 12, 1127–1134. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Kato, H.; Kumagai, Y.; Yoneyama, M.; Sato, S.; Matsushita, K.; Tsujimura, T.; Fujita, T.; Akira, S.; Takeuchi, O. LGP2 is a positive regulator of RIG-I- and MDA5-mediated antiviral responses. Proc. Natl. Acad. Sci. USA 2010, 107, 1512–1517. [Google Scholar] [CrossRef] [Green Version]
- Venkataraman, T.; Valdes, M.; Elsby, R.; Kakuta, S.; Caceres, G.; Saijo, S.; Iwakura, Y.; Barber, G.N. Loss of DExD/H box RNA helicase LGP2 manifests disparate antiviral responses. J. Immunol. 2007, 178, 6444–6455. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.J.; Takeuchi, O.; Akira, S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef]
- Meylan, E.; Curran, J.; Hofmann, K.; Moradpour, D.; Binder, M.; Bartenschlager, R.; Tschopp, J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005, 437, 1167–1172. [Google Scholar] [CrossRef]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.G.; Wang, Y.Y.; Han, K.J.; Li, L.Y.; Zhai, Z.; Shu, H.B. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell 2005, 19, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Hou, F.; Sun, L.; Zheng, H.; Skaug, B.; Jiang, Q.X.; Chen, Z.J. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 2011, 146, 448–461. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Chen, J.; Cai, X.; Wu, J.; Chen, X.; Wu, Y.T.; Sun, L.; Chen, Z.J. MAVS recruits multiple ubiquitin E3 ligases to activate antiviral signaling cascades. eLife 2013, 2, e00785. [Google Scholar] [CrossRef]
- Sadler, A.J.; Williams, B.R. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 2008, 8, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W. Interferon-Stimulated Genes: What Do They All Do? Annu. Rev. Virol. 2019, 6, 567–584. [Google Scholar] [CrossRef] [PubMed]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hornung, V.; Ellegast, J.; Kim, S.; Brzozka, K.; Jung, A.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.K.; Schlee, M.; et al. 5′-Triphosphate RNA is the ligand for RIG-I. Science 2006, 314, 994–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Naslund, T.I.; Liljestrom, P.; Weber, F.; Reis e Sousa, C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 2006, 314, 997–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlee, M.; Roth, A.; Hornung, V.; Hagmann, C.A.; Wimmenauer, V.; Barchet, W.; Coch, C.; Janke, M.; Mihailovic, A.; Wardle, G.; et al. Recognition of 5′ triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity 2009, 31, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Goubau, D.; Schlee, M.; Deddouche, S.; Pruijssers, A.J.; Zillinger, T.; Goldeck, M.; Schuberth, C.; Van der Veen, A.G.; Fujimura, T.; Rehwinkel, J.; et al. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5′-diphosphates. Nature 2014, 514, 372–375. [Google Scholar] [CrossRef] [Green Version]
- Saito, T.; Owen, D.M.; Jiang, F.; Marcotrigiano, J.; Gale, M., Jr. Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature 2008, 454, 523–527. [Google Scholar] [CrossRef] [Green Version]
- Schnell, G.; Loo, Y.M.; Marcotrigiano, J.; Gale, M., Jr. Uridine composition of the poly-U/UC tract of HCV RNA defines non-self recognition by RIG-I. PLoS Pathog. 2012, 8, e1002839. [Google Scholar] [CrossRef] [Green Version]
- Kell, A.; Stoddard, M.; Li, H.; Marcotrigiano, J.; Shaw, G.M.; Gale, M., Jr. Pathogen-Associated Molecular Pattern Recognition of Hepatitis C Virus Transmitted/Founder Variants by RIG-I Is Dependent on U-Core Length. J. Virol. 2015, 89, 11056–11068. [Google Scholar] [CrossRef] [Green Version]
- Runge, S.; Sparrer, K.M.; Lassig, C.; Hembach, K.; Baum, A.; Garcia-Sastre, A.; Soding, J.; Conzelmann, K.K.; Hopfner, K.P. In vivo ligands of MDA5 and RIG-I in measles virus-infected cells. PLoS Pathog. 2014, 10, e1004081. [Google Scholar] [CrossRef] [Green Version]
- Sanchez David, R.Y.; Combredet, C.; Sismeiro, O.; Dillies, M.A.; Jagla, B.; Coppee, J.Y.; Mura, M.; Guerbois Galla, M.; Despres, P.; Tangy, F.; et al. Comparative analysis of viral RNA signatures on different RIG-I-like receptors. eLife 2016, 5, e11275. [Google Scholar] [CrossRef] [PubMed]
- Malathi, K.; Dong, B.; Gale, M., Jr.; Silverman, R.H. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 2007, 448, 816–819. [Google Scholar] [CrossRef] [Green Version]
- Malathi, K.; Saito, T.; Crochet, N.; Barton, D.J.; Gale, M., Jr.; Silverman, R.H. RNase L releases a small RNA from HCV RNA that refolds into a potent PAMP. RNA 2010, 16, 2108–2119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Q.; Hato, S.V.; Langereis, M.A.; Zoll, J.; Virgen-Slane, R.; Peisley, A.; Hur, S.; Semler, B.L.; van Rij, R.P.; van Kuppeveld, F.J. MDA5 detects the double-stranded RNA replicative form in picornavirus-infected cells. Cell Rep. 2012, 2, 1187–1196. [Google Scholar] [CrossRef] [Green Version]
- Kato, H.; Takeuchi, O.; Mikamo-Satoh, E.; Hirai, R.; Kawai, T.; Matsushita, K.; Hiiragi, A.; Dermody, T.S.; Fujita, T.; Akira, S. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 2008, 205, 1601–1610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Rehwinkel, J.; Kato, H.; Takeuchi, O.; Akira, S.; Way, M.; Schiavo, G.; Reis e Sousa, C. Activation of MDA5 requires higher-order RNA structures generated during virus infection. J. Virol. 2009, 83, 10761–10769. [Google Scholar] [CrossRef] [Green Version]
- Triantafilou, K.; Vakakis, E.; Kar, S.; Richer, E.; Evans, G.L.; Triantafilou, M. Visualisation of direct interaction of MDA5 and the dsRNA replicative intermediate form of positive strand RNA viruses. J. Cell Sci. 2012, 125, 4761–4769. [Google Scholar] [CrossRef] [Green Version]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef]
- Loo, Y.M.; Fornek, J.; Crochet, N.; Bajwa, G.; Perwitasari, O.; Martinez-Sobrido, L.; Akira, S.; Gill, M.A.; Garcia-Sastre, A.; Katze, M.G.; et al. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J. Virol. 2008, 82, 335–345. [Google Scholar] [CrossRef] [Green Version]
- Rehwinkel, J.; Tan, C.P.; Goubau, D.; Schulz, O.; Pichlmair, A.; Bier, K.; Robb, N.; Vreede, F.; Barclay, W.; Fodor, E.; et al. RIG-I detects viral genomic RNA during negative-strand RNA virus infection. Cell 2010, 140, 397–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, M.; Gawanbacht, A.; Habjan, M.; Rang, A.; Borner, C.; Schmidt, A.M.; Veitinger, S.; Jacob, R.; Devignot, S.; Kochs, G.; et al. Incoming RNA virus nucleocapsids containing a 5′-triphosphorylated genome activate RIG-I and antiviral signaling. Cell Host Microbe 2013, 13, 336–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Park, H.S.; Pyo, H.M.; Liu, Q.; Zhou, Y. Influenza A Virus Panhandle Structure Is Directly Involved in RIG-I Activation and Interferon Induction. J. Virol. 2015, 89, 6067–6079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Te Velthuis, A.J.W.; Long, J.C.; Bauer, D.L.V.; Fan, R.L.Y.; Yen, H.L.; Sharps, J.; Siegers, J.Y.; Killip, M.J.; French, H.; Oliva-Martin, M.J.; et al. Mini viral RNAs act as innate immune agonists during influenza virus infection. Nat. Microbiol. 2018, 3, 1234–1242. [Google Scholar] [CrossRef] [PubMed]
- Chazal, M.; Beauclair, G.; Gracias, S.; Najburg, V.; Simon-Loriere, E.; Tangy, F.; Komarova, A.V.; Jouvenet, N. RIG-I Recognizes the 5′ Region of Dengue and Zika Virus Genomes. Cell Rep. 2018, 24, 320–328. [Google Scholar] [CrossRef] [Green Version]
- Sprokholt, J.K.; Kaptein, T.M.; van Hamme, J.L.; Overmars, R.J.; Gringhuis, S.I.; Geijtenbeek, T.B.H. RIG-I-like receptor activation by dengue virus drives follicular T helper cell formation and antibody production. PLoS Pathog. 2017, 13, e1006738. [Google Scholar] [CrossRef] [Green Version]
- Sprokholt, J.K.; Kaptein, T.M.; van Hamme, J.L.; Overmars, R.J.; Gringhuis, S.I.; Geijtenbeek, T.B.H. RIG-I-like Receptor Triggering by Dengue Virus Drives Dendritic Cell Immune Activation and T(H)1 Differentiation. J. Immunol. 2017, 198, 4764–4771. [Google Scholar] [CrossRef] [Green Version]
- Riedl, W.; Acharya, D.; Lee, J.H.; Liu, G.; Serman, T.; Chiang, C.; Chan, Y.K.; Diamond, M.S.; Gack, M.U. Zika Virus NS3 Mimics a Cellular 14-3-3-Binding Motif to Antagonize RIG-I- and MDA5-Mediated Innate Immunity. Cell Host Microbe 2019, 26, 493–503 e496. [Google Scholar] [CrossRef]
- Errett, J.S.; Suthar, M.S.; McMillan, A.; Diamond, M.S.; Gale, M., Jr. The essential, nonredundant roles of RIG-I and MDA5 in detecting and controlling West Nile virus infection. J. Virol. 2013, 87, 11416–11425. [Google Scholar] [CrossRef] [Green Version]
- Fredericksen, B.L.; Gale, M., Jr. West Nile virus evades activation of interferon regulatory factor 3 through RIG-I-dependent and -independent pathways without antagonizing host defense signaling. J. Virol. 2006, 80, 2913–2923. [Google Scholar] [CrossRef] [Green Version]
- Fredericksen, B.L.; Keller, B.C.; Fornek, J.; Katze, M.G.; Gale, M., Jr. Establishment and maintenance of the innate antiviral response to West Nile Virus involves both RIG-I and MDA5 signaling through IPS-1. J. Virol. 2008, 82, 609–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.P.; Cerny, A.; Asher, D.R.; Kurt-Jones, E.A.; Bronson, R.T.; Finberg, R.W. MDA5 and MAVS mediate type I interferon responses to coxsackie B virus. J. Virol. 2010, 84, 254–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, X.; Ding, Q.; Lu, J.; Tao, W.; Huang, B.; Zhao, Y.; Niu, J.; Liu, Y.J.; Zhong, J. MDA5 plays a critical role in interferon response during hepatitis C virus infection. J. Hepatol. 2015, 62, 771–778. [Google Scholar] [CrossRef]
- Du, X.; Pan, T.; Xu, J.; Zhang, Y.; Song, W.; Yi, Z.; Yuan, Z. Hepatitis C virus replicative double-stranded RNA is a potent interferon inducer that triggers interferon production through MDA5. J. Gen. Virol. 2016, 97, 2868–2882. [Google Scholar] [CrossRef] [PubMed]
- Arai, Y.; Yamanaka, I.; Okamoto, T.; Isobe, A.; Nakai, N.; Kamimura, N.; Suzuki, T.; Daidoji, T.; Ono, T.; Nakaya, T.; et al. Stimulation of interferon-beta responses by aberrant SARS-CoV-2 small viral RNAs acting as retinoic acid-inducible gene-I agonists. iScience 2023, 26, 105742. [Google Scholar] [CrossRef]
- Sampaio, N.G.; Chauveau, L.; Hertzog, J.; Bridgeman, A.; Fowler, G.; Moonen, J.P.; Dupont, M.; Russell, R.A.; Noerenberg, M.; Rehwinkel, J. The RNA sensor MDA5 detects SARS-CoV-2 infection. Sci Rep. 2021, 11, 13638. [Google Scholar] [CrossRef]
- Gack, M.U.; Nistal-Villan, E.; Inn, K.S.; Garcia-Sastre, A.; Jung, J.U. Phosphorylation-mediated negative regulation of RIG-I antiviral activity. J. Virol. 2010, 84, 3220–3229. [Google Scholar] [CrossRef] [Green Version]
- Maharaj, N.P.; Wies, E.; Stoll, A.; Gack, M.U. Conventional protein kinase C-alpha (PKC-alpha) and PKC-beta negatively regulate RIG-I antiviral signal transduction. J. Virol. 2012, 86, 1358–1371. [Google Scholar] [CrossRef] [Green Version]
- Nistal-Villan, E.; Gack, M.U.; Martinez-Delgado, G.; Maharaj, N.P.; Inn, K.S.; Yang, H.; Wang, R.; Aggarwal, A.K.; Jung, J.U.; Garcia-Sastre, A. Negative role of RIG-I serine 8 phosphorylation in the regulation of interferon-beta production. J. Biol. Chem. 2010, 285, 20252–20261. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Ren, H.; Liu, Y.; Teeling, J.L.; Gu, J. Phosphorylation of RIG-I by casein kinase II inhibits its antiviral response. J. Virol. 2011, 85, 1036–1047. [Google Scholar] [CrossRef] [Green Version]
- Willemsen, J.; Wicht, O.; Wolanski, J.C.; Baur, N.; Bastian, S.; Haas, D.A.; Matula, P.; Knapp, B.; Meyniel-Schicklin, L.; Wang, C.; et al. Phosphorylation-Dependent Feedback Inhibition of RIG-I by DAPK1 Identified by Kinome-wide siRNA Screening. Mol. Cell 2017, 65, 403–415. [Google Scholar] [CrossRef] [Green Version]
- Takashima, K.; Oshiumi, H.; Takaki, H.; Matsumoto, M.; Seya, T. RIOK3-mediated phosphorylation of MDA5 interferes with its assembly and attenuates the innate immune response. Cell Rep. 2015, 11, 192–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wies, E.; Wang, M.K.; Maharaj, N.P.; Chen, K.; Zhou, S.; Finberg, R.W.; Gack, M.U. Dephosphorylation of the RNA sensors RIG-I and MDA5 by the phosphatase PP1 is essential for innate immune signaling. Immunity 2013, 38, 437–449. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.E.; Wang, M.K.; Rennick, L.J.; Full, F.; Gableske, S.; Mesman, A.W.; Gringhuis, S.I.; Geijtenbeek, T.B.; Duprex, W.P.; Gack, M.U. Antagonism of the phosphatase PP1 by the measles virus V protein is required for innate immune escape of MDA5. Cell Host Microbe 2014, 16, 19–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.J.; Lee, H.C.; Kim, J.H.; Park, S.Y.; Kim, T.H.; Lee, W.K.; Jang, D.J.; Yoon, J.E.; Choi, Y.I.; Kim, S.; et al. HDAC6 regulates cellular viral RNA sensing by deacetylation of RIG-I. EMBO J. 2016, 35, 429–442. [Google Scholar] [CrossRef]
- Liu, H.M.; Jiang, F.; Loo, Y.M.; Hsu, S.; Hsiang, T.Y.; Marcotrigiano, J.; Gale, M., Jr. Regulation of Retinoic Acid Inducible Gene-I (RIG-I) Activation by the Histone Deacetylase 6. EBioMedicine 2016, 9, 195–206. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.M.; Liao, C.Y.; Yang, Q.; Xie, X.Q.; Shu, H.B. Innate immunity to RNA virus is regulated by temporal and reversible sumoylation of RIG-I and MDA5. J. Exp. Med. 2017, 214, 973–989. [Google Scholar] [CrossRef] [Green Version]
- He, S.; Zhao, J.; Song, S.; He, X.; Minassian, A.; Zhou, Y.; Zhang, J.; Brulois, K.; Wang, Y.; Cabo, J.; et al. Viral pseudo-enzymes activate RIG-I via deamidation to evade cytokine production. Mol. Cell 2015, 58, 134–146. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.J.; Hwang, S.Y.; Imaizumi, T.; Yoo, J.Y. Negative feedback regulation of RIG-I-mediated antiviral signaling by interferon-induced ISG15 conjugation. J. Virol. 2008, 82, 1474–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfaller, C.K.; George, C.X.; Samuel, C.E. Adenosine Deaminases Acting on RNA (ADARs) and Viral Infections. Annu. Rev. Virol. 2021, 8, 239–264. [Google Scholar] [CrossRef] [PubMed]
- Quin, J.; Sedmik, J.; Vukic, D.; Khan, A.; Keegan, L.P.; O’Connell, M.A. ADAR RNA Modifications, the Epitranscriptome and Innate Immunity. Trends Biochem. Sci. 2021, 46, 758–771. [Google Scholar] [CrossRef] [PubMed]
- Samuel, C.E. Adenosine deaminase acting on RNA (ADAR1), a suppressor of double-stranded RNA-triggered innate immune responses. J. Biol. Chem. 2019, 294, 1710–1720. [Google Scholar] [CrossRef] [Green Version]
- Gack, M.U.; Shin, Y.C.; Joo, C.H.; Urano, T.; Liang, C.; Sun, L.; Takeuchi, O.; Akira, S.; Chen, Z.; Inoue, S.; et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 2007, 446, 916–920. [Google Scholar] [CrossRef]
- Gao, D.; Yang, Y.K.; Wang, R.P.; Zhou, X.; Diao, F.C.; Li, M.D.; Zhai, Z.H.; Jiang, Z.F.; Chen, D.Y. REUL is a novel E3 ubiquitin ligase and stimulator of retinoic-acid-inducible gene-I. PLoS ONE 2009, 4, e5760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oshiumi, H.; Matsumoto, M.; Hatakeyama, S.; Seya, T. Riplet/RNF135, a RING finger protein, ubiquitinates RIG-I to promote interferon-beta induction during the early phase of viral infection. J. Biol. Chem. 2009, 284, 807–817. [Google Scholar] [CrossRef] [Green Version]
- Oshiumi, H.; Miyashita, M.; Inoue, N.; Okabe, M.; Matsumoto, M.; Seya, T. The ubiquitin ligase Riplet is essential for RIG-I-dependent innate immune responses to RNA virus infection. Cell Host Microbe 2010, 8, 496–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oshiumi, H.; Miyashita, M.; Matsumoto, M.; Seya, T. A distinct role of Riplet-mediated K63-Linked polyubiquitination of the RIG-I repressor domain in human antiviral innate immune responses. PLoS Pathog. 2013, 9, e1003533. [Google Scholar] [CrossRef] [Green Version]
- Kuniyoshi, K.; Takeuchi, O.; Pandey, S.; Satoh, T.; Iwasaki, H.; Akira, S.; Kawai, T. Pivotal role of RNA-binding E3 ubiquitin ligase MEX3C in RIG-I-mediated antiviral innate immunity. Proc. Natl. Acad. Sci. USA 2014, 111, 5646–5651. [Google Scholar] [CrossRef] [Green Version]
- Arimoto, K.; Takahashi, H.; Hishiki, T.; Konishi, H.; Fujita, T.; Shimotohno, K. Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc. Natl. Acad. Sci. USA 2007, 104, 7500–7505. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Han, C.; Xie, B.; Hu, X.; Yu, Q.; Shi, L.; Wang, Q.; Li, D.; Wang, J.; Zheng, P.; et al. Induction of Siglec-G by RNA viruses inhibits the innate immune response by promoting RIG-I degradation. Cell 2013, 152, 467–478. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Jiang, M.; Liu, S.; Zhang, S.; Liu, W.; Ma, Y.; Zhang, L.; Zhang, J.; Cao, X. RNF122 suppresses antiviral type I interferon production by targeting RIG-I CARDs to mediate RIG-I degradation. Proc. Natl. Acad. Sci. USA 2016, 113, 9581–9586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauli, E.K.; Chan, Y.K.; Davis, M.E.; Gableske, S.; Wang, M.K.; Feister, K.F.; Gack, M.U. The ubiquitin-specific protease USP15 promotes RIG-I-mediated antiviral signaling by deubiquitylating TRIM25. Sci. Signal. 2014, 7, ra3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Zhao, W.; Zhang, M.; Wang, P.; Zhao, K.; Zhao, X.; Yang, S.; Gao, C. USP4 positively regulates RIG-I-mediated antiviral response through deubiquitination and stabilization of RIG-I. J. Virol. 2013, 87, 4507–4515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, J.; Song, Y.; Li, Y.; Zhu, Q.; Tan, P.; Qin, Y.; Wang, H.Y.; Wang, R.F. USP3 inhibits type I interferon signaling by deubiquitinating RIG-I-like receptors. Cell Res. 2014, 24, 400–416. [Google Scholar] [CrossRef] [Green Version]
- Friedman, C.S.; O’Donnell, M.A.; Legarda-Addison, D.; Ng, A.; Cardenas, W.B.; Yount, J.S.; Moran, T.M.; Basler, C.F.; Komuro, A.; Horvath, C.M.; et al. The tumour suppressor CYLD is a negative regulator of RIG-I-mediated antiviral response. EMBO Rep. 2008, 9, 930–936. [Google Scholar] [CrossRef]
- Lang, X.; Tang, T.; Jin, T.; Ding, C.; Zhou, R.; Jiang, W. TRIM65-catalized ubiquitination is essential for MDA5-mediated antiviral innate immunity. J. Exp. Med. 2017, 214, 459–473. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Zhang, L.; Zhong, B.; Tan, B.; Liu, Y.; Shu, H.B. The ubiquitin-specific protease 17 is involved in virus-triggered type I IFN signaling. Cell Res. 2010, 20, 802–811. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.; Zhou, Z.; Zhu, J.; Liu, X.; Ouyang, G.; Wang, J.; Li, Z.; Li, X.; Zha, H.; Zhu, C.; et al. Opposing effects of deubiquitinase OTUD3 in innate immunity against RNA and DNA viruses. Cell Rep. 2022, 39, 110920. [Google Scholar] [CrossRef]
- Zeng, W.; Sun, L.; Jiang, X.; Chen, X.; Hou, F.; Adhikari, A.; Xu, M.; Chen, Z.J. Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell 2010, 141, 315–330. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Kinch, L.N.; Brautigam, C.A.; Chen, X.; Du, F.; Grishin, N.V.; Chen, Z.J. Ubiquitin-induced oligomerization of the RNA sensors RIG-I and MDA5 activates antiviral innate immune response. Immunity 2012, 36, 959–973. [Google Scholar] [CrossRef] [Green Version]
- Peisley, A.; Wu, B.; Xu, H.; Chen, Z.J.; Hur, S. Structural basis for ubiquitin-mediated antiviral signal activation by RIG-I. Nature 2014, 509, 110–114. [Google Scholar] [CrossRef]
- Peisley, A.; Wu, B.; Yao, H.; Walz, T.; Hur, S. RIG-I forms signaling-competent filaments in an ATP-dependent, ubiquitin-independent manner. Mol. Cell 2013, 51, 573–583. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Peisley, A.; Richards, C.; Yao, H.; Zeng, X.; Lin, C.; Chu, F.; Walz, T.; Hur, S. Structural basis for dsRNA recognition, filament formation, and antiviral signal activation by MDA5. Cell 2013, 152, 276–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peisley, A.; Lin, C.; Wu, B.; Orme-Johnson, M.; Liu, M.; Walz, T.; Hur, S. Cooperative assembly and dynamic disassembly of MDA5 filaments for viral dsRNA recognition. Proc. Natl. Acad. Sci. USA 2011, 108, 21010–21015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peisley, A.; Jo, M.H.; Lin, C.; Wu, B.; Orme-Johnson, M.; Walz, T.; Hohng, S.; Hur, S. Kinetic mechanism for viral dsRNA length discrimination by MDA5 filaments. Proc. Natl. Acad. Sci. USA 2012, 109, E3340–E3349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berke, I.C.; Modis, Y. MDA5 cooperatively forms dimers and ATP-sensitive filaments upon binding double-stranded RNA. EMBO J. 2012, 31, 1714–1726. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Peisley, A.; Tetrault, D.; Li, Z.; Egelman, E.H.; Magor, K.E.; Walz, T.; Penczek, P.A.; Hur, S. Molecular imprinting as a signal-activation mechanism of the viral RNA sensor RIG-I. Mol. Cell 2014, 55, 511–523. [Google Scholar] [CrossRef] [Green Version]
- Patel, J.R.; Jain, A.; Chou, Y.Y.; Baum, A.; Ha, T.; Garcia-Sastre, A. ATPase-driven oligomerization of RIG-I on RNA allows optimal activation of type-I interferon. EMBO Rep. 2013, 14, 780–787. [Google Scholar] [CrossRef] [Green Version]
- Dixit, E.; Boulant, S.; Zhang, Y.; Lee, A.S.; Odendall, C.; Shum, B.; Hacohen, N.; Chen, Z.J.; Whelan, S.P.; Fransen, M.; et al. Peroxisomes are signaling platforms for antiviral innate immunity. Cell 2010, 141, 668–681. [Google Scholar] [CrossRef] [Green Version]
- Horner, S.M.; Liu, H.M.; Park, H.S.; Briley, J.; Gale, M., Jr. Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc. Natl. Acad. Sci. USA 2011, 108, 14590–14595. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.M.; Loo, Y.M.; Horner, S.M.; Zornetzer, G.A.; Katze, M.G.; Gale, M., Jr. The mitochondrial targeting chaperone 14-3-3epsilon regulates a RIG-I translocon that mediates membrane association and innate antiviral immunity. Cell Host Microbe 2012, 11, 528–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pothlichet, J.; Burtey, A.; Kubarenko, A.V.; Caignard, G.; Solhonne, B.; Tangy, F.; Ben-Ali, M.; Quintana-Murci, L.; Heinzmann, A.; Chiche, J.D.; et al. Study of human RIG-I polymorphisms identifies two variants with an opposite impact on the antiviral immune response. PLoS ONE 2009, 4, e7582. [Google Scholar] [CrossRef] [PubMed]
- Shigemoto, T.; Kageyama, M.; Hirai, R.; Zheng, J.; Yoneyama, M.; Fujita, T. Identification of loss of function mutations in human genes encoding RIG-I and MDA5: Implications for resistance to type I diabetes. J. Biol. Chem. 2009, 284, 13348–13354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Nistal-Villan, E.; Voho, A.; Ganee, A.; Kumar, M.; Ding, Y.; Garcia-Sastre, A.; Wetmur, J.G. A common polymorphism in the caspase recruitment domain of RIG-I modifies the innate immune response of human dendritic cells. J. Immunol. 2010, 185, 424–432. [Google Scholar] [CrossRef] [Green Version]
- Lamborn, I.T.; Jing, H.; Zhang, Y.; Drutman, S.B.; Abbott, J.K.; Munir, S.; Bade, S.; Murdock, H.M.; Santos, C.P.; Brock, L.G.; et al. Recurrent rhinovirus infections in a child with inherited MDA5 deficiency. J. Exp. Med. 2017, 214, 1949–1972. [Google Scholar] [CrossRef] [Green Version]
- Dou, Y.; Yim, H.C.; Kirkwood, C.D.; Williams, B.R.; Sadler, A.J. The innate immune receptor MDA5 limits rotavirus infection but promotes cell death and pancreatic inflammation. EMBO J. 2017, 36, 2742–2757. [Google Scholar] [CrossRef]
- Morishita, H.; Mizushima, N. Diverse Cellular Roles of Autophagy. Annu. Rev. Cell Dev. Biol. 2019, 35, 453–475. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, H.; Zhang, S.; Mizushima, N. Autophagy genes in biology and disease. Nat. Rev. Genet. 2023, 1–19. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in major human diseases. EMBO J. 2021, 40, e108863. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B. Autophagy in Human Diseases. N. Engl. J. Med. 2020, 383, 1564–1576. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. A brief history of autophagy from cell biology to physiology and disease. Nat. Cell Biol. 2018, 20, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Klionsky, D.J. Eaten alive: A history of macroautophagy. Nat. Cell Biol. 2010, 12, 814–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef]
- Mijaljica, D.; Prescott, M.; Devenish, R.J. Microautophagy in mammalian cells: Revisiting a 40-year-old conundrum. Autophagy 2011, 7, 673–682. [Google Scholar] [CrossRef] [Green Version]
- Oku, M.; Sakai, Y. Three Distinct Types of Microautophagy Based on Membrane Dynamics and Molecular Machineries. Bioessays 2018, 40, e1800008. [Google Scholar] [CrossRef] [Green Version]
- Schuck, S. Microautophagy—Distinct molecular mechanisms handle cargoes of many sizes. J. Cell Sci. 2020, 133, jcs246322. [Google Scholar] [CrossRef]
- Wang, L.; Klionsky, D.J.; Shen, H.M. The emerging mechanisms and functions of microautophagy. Nat. Rev. Mol. Cell Biol. 2022, 24, 186–203. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef]
- Tekirdag, K.; Cuervo, A.M. Chaperone-mediated autophagy and endosomal microautophagy: Joint by a chaperone. J. Biol. Chem. 2018, 293, 5414–5424. [Google Scholar] [CrossRef] [Green Version]
- Cuervo, A.M.; Wong, E. Chaperone-mediated autophagy: Roles in disease and aging. Cell Res. 2014, 24, 92–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawabata, T.; Yoshimori, T. Autophagosome biogenesis and human health. Cell Discov. 2020, 6, 33. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef]
- Itakura, E.; Mizushima, N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 2010, 6, 764–776. [Google Scholar] [CrossRef] [Green Version]
- Jewell, J.L.; Russell, R.C.; Guan, K.L. Amino acid signalling upstream of mTOR. Nat. Rev. Mol. Cell Biol. 2013, 14, 133–139. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr. Opin. Cell Biol. 2010, 22, 132–139. [Google Scholar] [CrossRef]
- Gomez-Sanchez, R.; Tooze, S.A.; Reggiori, F. Membrane supply and remodeling during autophagosome biogenesis. Curr. Opin. Cell Biol. 2021, 71, 112–119. [Google Scholar] [CrossRef]
- Melia, T.J.; Lystad, A.H.; Simonsen, A. Autophagosome biogenesis: From membrane growth to closure. J. Cell Biol. 2020, 219, e202002085. [Google Scholar] [CrossRef]
- Matsunaga, K.; Morita, E.; Saitoh, T.; Akira, S.; Ktistakis, N.T.; Izumi, T.; Noda, T.; Yoshimori, T. Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J. Cell Biol. 2010, 190, 511–521. [Google Scholar] [CrossRef]
- Matsunaga, K.; Saitoh, T.; Tabata, K.; Omori, H.; Satoh, T.; Kurotori, N.; Maejima, I.; Shirahama-Noda, K.; Ichimura, T.; Isobe, T.; et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 2009, 11, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef] [Green Version]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polson, H.E.; de Lartigue, J.; Rigden, D.J.; Reedijk, M.; Urbe, S.; Clague, M.J.; Tooze, S.A. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy 2010, 6, 506–522. [Google Scholar] [CrossRef] [Green Version]
- Molejon, M.I.; Ropolo, A.; Re, A.L.; Boggio, V.; Vaccaro, M.I. The VMP1-Beclin 1 interaction regulates autophagy induction. Sci. Rep. 2013, 3, 1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ropolo, A.; Grasso, D.; Pardo, R.; Sacchetti, M.L.; Archange, C.; Lo Re, A.; Seux, M.; Nowak, J.; Gonzalez, C.D.; Iovanna, J.L.; et al. The pancreatitis-induced vacuole membrane protein 1 triggers autophagy in mammalian cells. J. Biol. Chem. 2007, 282, 37124–37133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.G.; Chen, Y.; Miao, G.; Zhao, H.; Qu, W.; Li, D.; Wang, Z.; Liu, N.; Li, L.; Chen, S.; et al. The ER-Localized Transmembrane Protein EPG-3/VMP1 Regulates SERCA Activity to Control ER-Isolation Membrane Contacts for Autophagosome Formation. Mol. Cell 2017, 67, 974–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, H.; Kakuta, S.; Watanabe, T.M.; Kitamura, A.; Sekito, T.; Kondo-Kakuta, C.; Ichikawa, R.; Kinjo, M.; Ohsumi, Y. Atg9 vesicles are an important membrane source during early steps of autophagosome formation. J. Cell Biol. 2012, 198, 219–233. [Google Scholar] [CrossRef] [Green Version]
- Orsi, A.; Razi, M.; Dooley, H.C.; Robinson, D.; Weston, A.E.; Collinson, L.M.; Tooze, S.A. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol. Biol. Cell 2012, 23, 1860–1873. [Google Scholar] [CrossRef]
- Mari, M.; Griffith, J.; Rieter, E.; Krishnappa, L.; Klionsky, D.J.; Reggiori, F. An Atg9-containing compartment that functions in the early steps of autophagosome biogenesis. J. Cell Biol. 2010, 190, 1005–1022. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Backues, S.K.; Baba, M.; Heo, J.M.; Harper, J.W.; Klionsky, D.J. Phosphorylation of Atg9 regulates movement to the phagophore assembly site and the rate of autophagosome formation. Autophagy 2016, 12, 648–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mailler, E.; Guardia, C.M.; Bai, X.; Jarnik, M.; Williamson, C.D.; Li, Y.; Maio, N.; Golden, A.; Bonifacino, J.S. The autophagy protein ATG9A enables lipid mobilization from lipid droplets. Nat. Commun. 2021, 12, 6750. [Google Scholar] [CrossRef]
- Hayashi-Nishino, M.; Fujita, N.; Noda, T.; Yamaguchi, A.; Yoshimori, T.; Yamamoto, A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell Biol. 2009, 11, 1433–1437. [Google Scholar] [CrossRef]
- Yla-Anttila, P.; Vihinen, H.; Jokitalo, E.; Eskelinen, E.L. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy 2009, 5, 1180–1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reggiori, F.; Shintani, T.; Nair, U.; Klionsky, D.J. Atg9 cycles between mitochondria and the pre-autophagosomal structure in yeasts. Autophagy 2005, 1, 101–109. [Google Scholar] [CrossRef] [Green Version]
- Yen, W.L.; Shintani, T.; Nair, U.; Cao, Y.; Richardson, B.C.; Li, Z.; Hughson, F.M.; Baba, M.; Klionsky, D.J. The conserved oligomeric Golgi complex is involved in double-membrane vesicle formation during autophagy. J. Cell Biol. 2010, 188, 101–114. [Google Scholar] [CrossRef] [Green Version]
- Puri, C.; Vicinanza, M.; Ashkenazi, A.; Gratian, M.J.; Zhang, Q.; Bento, C.F.; Renna, M.; Menzies, F.M.; Rubinsztein, D.C. The RAB11A-Positive Compartment Is a Primary Platform for Autophagosome Assembly Mediated by WIPI2 Recognition of PI3P-RAB11A. Dev. Cell 2018, 45, 114–131.e118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knaevelsrud, H.; Soreng, K.; Raiborg, C.; Haberg, K.; Rasmuson, F.; Brech, A.; Liestol, K.; Rusten, T.E.; Stenmark, H.; Neufeld, T.P.; et al. Membrane remodeling by the PX-BAR protein SNX18 promotes autophagosome formation. J. Cell Biol. 2013, 202, 331–349. [Google Scholar] [CrossRef]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef]
- Ravikumar, B.; Moreau, K.; Jahreiss, L.; Puri, C.; Rubinsztein, D.C. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat. Cell Biol. 2010, 12, 747–757. [Google Scholar] [CrossRef]
- Mizushima, N.; Sugita, H.; Yoshimori, T.; Ohsumi, Y. A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J. Biol. Chem. 1998, 273, 33889–33892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Noda, T.; Yoshimori, T.; Tanaka, Y.; Ishii, T.; George, M.D.; Klionsky, D.J.; Ohsumi, M.; Ohsumi, Y. A protein conjugation system essential for autophagy. Nature 1998, 395, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Kirisako, T.; Kamada, Y.; Mizushima, N.; Noda, T.; Ohsumi, Y. The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J. 2001, 20, 5971–5981. [Google Scholar] [CrossRef] [PubMed]
- Nakatogawa, H.; Ichimura, Y.; Ohsumi, Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 2007, 130, 165–178. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.G.; Codogno, P.; Zhang, H. Machinery, regulation and pathophysiological implications of autophagosome maturation. Nat. Rev. Mol. Cell Biol. 2021, 22, 733–750. [Google Scholar] [CrossRef]
- Fujita, N.; Itoh, T.; Omori, H.; Fukuda, M.; Noda, T.; Yoshimori, T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell 2008, 19, 2092–2100. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Noda, T.; Ohsumi, Y. Apg16p is required for the function of the Apg12p-Apg5p conjugate in the yeast autophagy pathway. EMBO J. 1999, 18, 3888–3896. [Google Scholar] [CrossRef] [Green Version]
- Kuma, A.; Mizushima, N.; Ishihara, N.; Ohsumi, Y. Formation of the approximately 350-kDa Apg12-Apg5.Apg16 multimeric complex, mediated by Apg16 oligomerization, is essential for autophagy in yeast. J. Biol. Chem. 2002, 277, 18619–18625. [Google Scholar] [CrossRef] [Green Version]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Yamamoto, A.; Oshitani-Okamoto, S.; Ohsumi, Y.; Yoshimori, T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 2004, 117 Pt 13, 2805–2812. [Google Scholar] [CrossRef] [Green Version]
- Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Mizushima, N.; Tanida, I.; Kominami, E.; Ohsumi, M.; et al. A ubiquitin-like system mediates protein lipidation. Nature 2000, 408, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Nair, U.; Klionsky, D.J. Atg8 controls phagophore expansion during autophagosome formation. Mol. Biol. Cell 2008, 19, 3290–3298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieto-Torres, J.L.; Leidal, A.M.; Debnath, J.; Hansen, M. Beyond Autophagy: The Expanding Roles of ATG8 Proteins. Trends Biochem. Sci. 2021, 46, 673–686. [Google Scholar] [CrossRef] [PubMed]
- Hanada, T.; Noda, N.N.; Satomi, Y.; Ichimura, Y.; Fujioka, Y.; Takao, T.; Inagaki, F.; Ohsumi, Y. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J. Biol. Chem. 2007, 282, 37298–37302. [Google Scholar] [CrossRef] [Green Version]
- Romanov, J.; Walczak, M.; Ibiricu, I.; Schuchner, S.; Ogris, E.; Kraft, C.; Martens, S. Mechanism and functions of membrane binding by the Atg5-Atg12/Atg16 complex during autophagosome formation. EMBO J. 2012, 31, 4304–4317. [Google Scholar] [CrossRef]
- Liang, C.; Lee, J.S.; Inn, K.S.; Gack, M.U.; Li, Q.; Roberts, E.A.; Vergne, I.; Deretic, V.; Feng, P.; Akazawa, C.; et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat. Cell Biol. 2008, 10, 776–787. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Y.; Wang, Q.J.; Li, X.; Yan, Y.; Backer, J.M.; Chait, B.T.; Heintz, N.; Yue, Z. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat. Cell Biol. 2009, 11, 468–476. [Google Scholar] [CrossRef]
- Kriegenburg, F.; Ungermann, C.; Reggiori, F. Coordination of Autophagosome-Lysosome Fusion by Atg8 Family Members. Curr. Biol. 2018, 28, R512–R518. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Melia, T.J. The coordination of membrane fission and fusion at the end of autophagosome maturation. Curr. Opin. Cell Biol. 2017, 47, 92–98. [Google Scholar] [CrossRef]
- Nakamura, S.; Yoshimori, T. New insights into autophagosome-lysosome fusion. J. Cell Sci. 2017, 130, 1209–1216. [Google Scholar] [CrossRef] [Green Version]
- Reggiori, F.; Ungermann, C. Autophagosome Maturation and Fusion. J. Mol. Biol. 2017, 429, 486–496. [Google Scholar] [CrossRef]
- Lorincz, P.; Juhasz, G. Autophagosome-Lysosome Fusion. J. Mol. Biol. 2020, 432, 2462–2482. [Google Scholar] [CrossRef] [PubMed]
- Jager, S.; Bucci, C.; Tanida, I.; Ueno, T.; Kominami, E.; Saftig, P.; Eskelinen, E.L. Role for Rab7 in maturation of late autophagic vacuoles. J. Cell Sci. 2004, 117 Pt 20, 4837–4848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez, M.G.; Munafo, D.B.; Beron, W.; Colombo, M.I. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J. Cell Sci. 2004, 117 Pt 13, 2687–2697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pankiv, S.; Johansen, T. FYCO1: Linking autophagosomes to microtubule plus end-directing molecular motors. Autophagy 2010, 6, 550–552. [Google Scholar] [CrossRef] [Green Version]
- Pankiv, S.; Alemu, E.A.; Brech, A.; Bruun, J.A.; Lamark, T.; Overvatn, A.; Bjorkoy, G.; Johansen, T. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J. Cell Biol. 2010, 188, 253–269. [Google Scholar] [CrossRef] [Green Version]
- Jordens, I.; Fernandez-Borja, M.; Marsman, M.; Dusseljee, S.; Janssen, L.; Calafat, J.; Janssen, H.; Wubbolts, R.; Neefjes, J. The Rab7 effector protein RILP controls lysosomal transport by inducing the recruitment of dynein-dynactin motors. Curr. Biol. 2001, 11, 1680–1685. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Koga, H.; Kawaguchi, Y.; Tang, W.; Wong, E.; Gao, Y.S.; Pandey, U.B.; Kaushik, S.; Tresse, E.; Lu, J.; et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010, 29, 969–980. [Google Scholar] [CrossRef] [Green Version]
- Jiang, P.; Nishimura, T.; Sakamaki, Y.; Itakura, E.; Hatta, T.; Natsume, T.; Mizushima, N. The HOPS complex mediates autophagosome-lysosome fusion through interaction with syntaxin 17. Mol. Biol. Cell 2014, 25, 1327–1337. [Google Scholar] [CrossRef]
- McEwan, D.G.; Popovic, D.; Gubas, A.; Terawaki, S.; Suzuki, H.; Stadel, D.; Coxon, F.P.; Miranda de Stegmann, D.; Bhogaraju, S.; Maddi, K.; et al. PLEKHM1 regulates autophagosome-lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol. Cell 2015, 57, 39–54. [Google Scholar] [CrossRef] [Green Version]
- Takats, S.; Pircs, K.; Nagy, P.; Varga, A.; Karpati, M.; Hegedus, K.; Kramer, H.; Kovacs, A.L.; Sass, M.; Juhasz, G. Interaction of the HOPS complex with Syntaxin 17 mediates autophagosome clearance in Drosophila. Mol. Biol. Cell 2014, 25, 1338–1354. [Google Scholar] [CrossRef] [PubMed]
- Diao, J.; Li, L.; Lai, Y.; Zhong, Q. In Vitro Reconstitution of Autophagosome-Lysosome Fusion. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2017; Volume 587, pp. 365–376. [Google Scholar]
- Diao, J.; Liu, R.; Rong, Y.; Zhao, M.; Zhang, J.; Lai, Y.; Zhou, Q.; Wilz, L.M.; Li, J.; Vivona, S.; et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature 2015, 520, 563–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.N.; Padman, B.S.; Usher, J.; Oorschot, V.; Ramm, G.; Lazarou, M. Atg8 family LC3/GABARAP proteins are crucial for autophagosome-lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J. Cell Biol. 2016, 215, 857–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Su, Q.P.; Sun, Y.; Yu, L. Visualizing Autophagic Lysosome Reformation in Cells Using In Vitro Reconstitution Systems. Curr. Protoc. Cell Biol. 2018, 78, 11–24. [Google Scholar] [CrossRef]
- Chen, Y.; Su, Q.P.; Yu, L. Studying Autophagic Lysosome Reformation in Cells and by an In Vitro Reconstitution System. Methods Mol. Biol. 2019, 1880, 163–172. [Google Scholar]
- Chen, Y.; Yu, L. Autophagic lysosome reformation. Exp. Cell Res. 2013, 319, 142–146. [Google Scholar] [CrossRef]
- Chen, Y.; Yu, L. Recent progress in autophagic lysosome reformation. Traffic 2017, 18, 358–361. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Yu, L. Development of Research into Autophagic Lysosome Reformation. Mol. Cells 2018, 41, 45–49. [Google Scholar]
- Eramo, M.J.; Gurung, R.; Mitchell, C.A.; McGrath, M.J. Bidirectional interconversion between PtdIns4P and PtdIns(4,5)P(2) is required for autophagic lysosome reformation and protection from skeletal muscle disease. Autophagy 2021, 17, 1287–1289. [Google Scholar] [CrossRef]
- Liu, X.; Klionsky, D.J. Regulation of autophagic lysosome reformation by kinesin 1, clathrin and phosphatidylinositol-4,5-bisphosphate. Autophagy 2018, 14, 1–2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanayakkara, R.; Gurung, R.; Rodgers, S.J.; Eramo, M.J.; Ramm, G.; Mitchell, C.A.; McGrath, M.J. Autophagic lysosome reformation in health and disease. Autophagy 2022, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.; Liu, M.; Ma, L.; Du, W.; Zhang, H.; Tian, Y.; Cao, Z.; Li, Y.; Ren, H.; Zhang, C.; et al. Clathrin and phosphatidylinositol-4,5-bisphosphate regulate autophagic lysosome reformation. Nat. Cell Biol. 2012, 14, 924–934. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.; McPhee, C.K.; Deng, S.; Huang, L.; Chen, L.; Liu, M.; Tracy, K.; Baehrecke, E.H.; Yu, L.; Lenardo, M.J. Spinster is required for autophagic lysosome reformation and mTOR reactivation following starvation. Proc. Natl. Acad. Sci. USA 2011, 108, 7826–7831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.C.; Lin, Y.C.; Chen, Y.H.; Chen, C.M.; Pang, L.Y.; Chen, H.A.; Wu, P.R.; Lin, M.Y.; Jiang, S.T.; Tsai, T.F.; et al. Cul3-KLHL20 Ubiquitin Ligase Governs the Turnover of ULK1 and VPS34 Complexes to Control Autophagy Termination. Mol. Cell 2016, 61, 84–97. [Google Scholar] [CrossRef]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef]
- Zaffagnini, G.; Martens, S. Mechanisms of Selective Autophagy. J. Mol. Biol. 2016, 428 Pt 9, 1714–1724. [Google Scholar] [CrossRef] [Green Version]
- Anding, A.L.; Baehrecke, E.H. Cleaning House: Selective Autophagy of Organelles. Dev. Cell 2017, 41, 10–22. [Google Scholar] [CrossRef] [Green Version]
- Kirkin, V.; Rogov, V.V. A Diversity of Selective Autophagy Receptors Determines the Specificity of the Autophagy Pathway. Mol. Cell 2019, 76, 268–285. [Google Scholar] [CrossRef]
- Vargas, J.N.S.; Hamasaki, M.; Kawabata, T.; Youle, R.J.; Yoshimori, T. The mechanisms and roles of selective autophagy in mammals. Nat. Rev. Mol. Cell Biol. 2022, 24, 167–185. [Google Scholar] [CrossRef]
- Stolz, A.; Ernst, A.; Dikic, I. Cargo recognition and trafficking in selective autophagy. Nat. Cell Biol. 2014, 16, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Rogov, V.; Dotsch, V.; Johansen, T.; Kirkin, V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol. Cell 2014, 53, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Birgisdottir, A.B.; Lamark, T.; Johansen, T. The LIR motif—Crucial for selective autophagy. J. Cell Sci. 2013, 126 Pt 15, 3237–3247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansen, T.; Lamark, T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011, 7, 279–296. [Google Scholar] [CrossRef] [PubMed]
- Johansen, T.; Lamark, T. Selective Autophagy: ATG8 Family Proteins, LIR Motifs and Cargo Receptors. J. Mol. Biol. 2020, 432, 80–103. [Google Scholar] [CrossRef] [PubMed]
- Kirkin, V.; McEwan, D.G.; Novak, I.; Dikic, I. A role for ubiquitin in selective autophagy. Mol. Cell 2009, 34, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, E.; Ferrari, L.; Martens, S. Orchestration of selective autophagy by cargo receptors. Curr. Biol. 2022, 32, R1357–R1371. [Google Scholar] [CrossRef]
- Rogov, V.V.; Stolz, A.; Ravichandran, A.C.; Rios-Szwed, D.O.; Suzuki, H.; Kniss, A.; Lohr, F.; Wakatsuki, S.; Dotsch, V.; Dikic, I.; et al. Structural and functional analysis of the GABARAP interaction motif (GIM). EMBO Rep. 2017, 18, 1382–1396. [Google Scholar] [CrossRef]
- Fracchiolla, D.; Sawa-Makarska, J.; Martens, S. Beyond Atg8 binding: The role of AIM/LIR motifs in autophagy. Autophagy 2017, 13, 978–979. [Google Scholar] [CrossRef] [Green Version]
- Schaaf, M.B.; Keulers, T.G.; Vooijs, M.A.; Rouschop, K.M. LC3/GABARAP family proteins: Autophagy-(un)related functions. FASEB J. 2016, 30, 3961–3978. [Google Scholar] [CrossRef] [Green Version]
- Jacomin, A.C.; Samavedam, S.; Promponas, V.; Nezis, I.P. iLIR database: A web resource for LIR motif-containing proteins in eukaryotes. Autophagy 2016, 12, 1945–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, K. Organellophagy: Eliminating cellular building blocks via selective autophagy. J. Cell Biol. 2014, 205, 435–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickles, S.; Vigie, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef] [Green Version]
- Meissner, C.; Lorenz, H.; Weihofen, A.; Selkoe, D.J.; Lemberg, M.K. The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. J. Neurochem. 2011, 117, 856–867. [Google Scholar] [CrossRef]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; de Vries, R.L.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [Green Version]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [Green Version]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014, 510, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 2014, 205, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.Y.; Yang, W.Y. Bit-by-bit autophagic removal of parkin-labelled mitochondria. Nat. Commun. 2013, 4, 2428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Heo, J.M.; Ordureau, A.; Paulo, J.A.; Rinehart, J.; Harper, J.W. The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol. Cell 2015, 60, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Richter, B.; Sliter, D.A.; Herhaus, L.; Stolz, A.; Wang, C.; Beli, P.; Zaffagnini, G.; Wild, P.; Martens, S.; Wagner, S.A.; et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. USA 2016, 113, 4039–4044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, G.; Shimogori, T.; Hattori, N.; Nukina, N. TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum. Mol. Genet. 2015, 24, 4429–4442. [Google Scholar] [CrossRef] [Green Version]
- Hoshino, A.; Wang, W.J.; Wada, S.; McDermott-Roe, C.; Evans, C.S.; Gosis, B.; Morley, M.P.; Rathi, K.S.; Li, J.; Li, K.; et al. The ADP/ATP translocase drives mitophagy independent of nucleotide exchange. Nature 2019, 575, 375–379. [Google Scholar] [CrossRef]
- Lu, K.; Psakhye, I.; Jentsch, S. Autophagic clearance of polyQ proteins mediated by ubiquitin-Atg8 adaptors of the conserved CUET protein family. Cell 2014, 158, 549–563. [Google Scholar] [CrossRef] [Green Version]
- Ryan, T.A.; Phillips, E.O.; Collier, C.L.; Jb Robinson, A.; Routledge, D.; Wood, R.E.; Assar, E.A.; Tumbarello, D.A. Tollip coordinates Parkin-dependent trafficking of mitochondrial-derived vesicles. EMBO J. 2020, 39, e102539. [Google Scholar] [CrossRef]
- Di Rienzo, M.; Romagnoli, A.; Ciccosanti, F.; Refolo, G.; Consalvi, V.; Arena, G.; Valente, E.M.; Piacentini, M.; Fimia, G.M. AMBRA1 regulates mitophagy by interacting with ATAD3A and promoting PINK1 stability. Autophagy 2022, 18, 1752–1762. [Google Scholar] [CrossRef] [PubMed]
- Strappazzon, F.; Nazio, F.; Corrado, M.; Cianfanelli, V.; Romagnoli, A.; Fimia, G.M.; Campello, S.; Nardacci, R.; Piacentini, M.; Campanella, M.; et al. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ. 2015, 22, 517. [Google Scholar] [CrossRef] [PubMed]
- Van Humbeeck, C.; Cornelissen, T.; Hofkens, H.; Mandemakers, W.; Gevaert, K.; De Strooper, B.; Vandenberghe, W. Parkin interacts with Ambra1 to induce mitophagy. J. Neurosci. 2011, 31, 10249–10261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zachari, M.; Gudmundsson, S.R.; Li, Z.; Manifava, M.; Cugliandolo, F.; Shah, R.; Smith, M.; Stronge, J.; Karanasios, E.; Piunti, C.; et al. Selective Autophagy of Mitochondria on a Ubiquitin-Endoplasmic-Reticulum Platform. Dev. Cell 2019, 50, 627–643.e5. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, C.W.; Yang, W.Y. Omegasome-proximal PtdIns(4,5)P2 couples F-actin mediated mitoaggregate disassembly with autophagosome formation during mitophagy. Nat. Commun. 2019, 10, 969. [Google Scholar] [CrossRef] [PubMed]
- Bingol, B.; Tea, J.S.; Phu, L.; Reichelt, M.; Bakalarski, C.E.; Song, Q.; Foreman, O.; Kirkpatrick, D.S.; Sheng, M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 2014, 510, 370–375. [Google Scholar] [CrossRef]
- Wang, Y.; Serricchio, M.; Jauregui, M.; Shanbhag, R.; Stoltz, T.; Di Paolo, C.T.; Kim, P.K.; McQuibban, G.A. Deubiquitinating enzymes regulate PARK2-mediated mitophagy. Autophagy 2015, 11, 595–606. [Google Scholar] [CrossRef] [Green Version]
- Ding, W.X.; Ni, H.M.; Li, M.; Liao, Y.; Chen, X.; Stolz, D.B.; Dorn, G.W., 2nd; Yin, X.M. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J. Biol. Chem. 2010, 285, 27879–27890. [Google Scholar] [CrossRef] [Green Version]
- Novak, I.; Kirkin, V.; McEwan, D.G.; Zhang, J.; Wild, P.; Rozenknop, A.; Rogov, V.; Lohr, F.; Popovic, D.; Occhipinti, A.; et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010, 11, 45–51. [Google Scholar] [CrossRef] [Green Version]
- Quinsay, M.N.; Thomas, R.L.; Lee, Y.; Gustafsson, A.B. Bnip3-mediated mitochondrial autophagy is independent of the mitochondrial permeability transition pore. Autophagy 2010, 6, 855–862. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef]
- Wu, W.; Tian, W.; Hu, Z.; Chen, G.; Huang, L.; Li, W.; Zhang, X.; Xue, P.; Zhou, C.; Liu, L.; et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 2014, 15, 566–575. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Chiang, W.C.; Sumpter, R., Jr.; Mishra, P.; Levine, B. Prohibitin 2 Is an Inner Mitochondrial Membrane Mitophagy Receptor. Cell 2017, 168, 224–238 e210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, C.; Gong, L.; Chen, L.; Xu, M.; Abou-Hamdan, H.; Tang, M.; Desaubry, L.; Song, Z. PHB2 (prohibitin 2) promotes PINK1-PRKN/Parkin-dependent mitophagy by the PARL-PGAM5-PINK1 axis. Autophagy 2020, 16, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Hou, H.; Ma, G.; Ma, Q.; Li, N.; Zhang, L.; Dong, C.; Cao, M.; Tam, K.Y.; Ying, Z.; et al. The interaction between E3 ubiquitin ligase Parkin and mitophagy receptor PHB2 links inner mitochondrial membrane ubiquitination to efficient mitophagy. J. Biol. Chem. 2022, 298, 102704. [Google Scholar] [CrossRef] [PubMed]
- Bernales, S.; Schuck, S.; Walter, P. ER-phagy: Selective autophagy of the endoplasmic reticulum. Autophagy 2007, 3, 285–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mochida, K.; Nakatogawa, H. ER-phagy: Selective autophagy of the endoplasmic reticulum. EMBO Rep. 2022, 23, e55192. [Google Scholar] [CrossRef]
- Chino, H.; Mizushima, N. ER-Phagy: Quality Control and Turnover of Endoplasmic Reticulum. Trends Cell Biol. 2020, 30, 384–398. [Google Scholar] [CrossRef]
- Mochida, K.; Oikawa, Y.; Kimura, Y.; Kirisako, H.; Hirano, H.; Ohsumi, Y.; Nakatogawa, H. Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature 2015, 522, 359–362. [Google Scholar] [CrossRef]
- Khaminets, A.; Heinrich, T.; Mari, M.; Grumati, P.; Huebner, A.K.; Akutsu, M.; Liebmann, L.; Stolz, A.; Nietzsche, S.; Koch, N.; et al. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 2015, 522, 354–358. [Google Scholar] [CrossRef]
- Grumati, P.; Morozzi, G.; Holper, S.; Mari, M.; Harwardt, M.I.; Yan, R.; Muller, S.; Reggiori, F.; Heilemann, M.; Dikic, I. Full length RTN3 regulates turnover of tubular endoplasmic reticulum via selective autophagy. eLife 2017, 6, e25555. [Google Scholar] [CrossRef] [PubMed]
- Forrester, A.; De Leonibus, C.; Grumati, P.; Fasana, E.; Piemontese, M.; Staiano, L.; Fregno, I.; Raimondi, A.; Marazza, A.; Bruno, G.; et al. A selective ER-phagy exerts procollagen quality control via a Calnexin-FAM134B complex. EMBO J. 2019, 38, e99847. [Google Scholar] [CrossRef] [PubMed]
- An, H.; Ordureau, A.; Paulo, J.A.; Shoemaker, C.J.; Denic, V.; Harper, J.W. TEX264 Is an Endoplasmic Reticulum-Resident ATG8-Interacting Protein Critical for ER Remodeling during Nutrient Stress. Mol. Cell 2019, 74, 891–908. [Google Scholar] [CrossRef]
- Delorme-Axford, E.; Popelka, H.; Klionsky, D.J. TEX264 is a major receptor for mammalian reticulophagy. Autophagy 2019, 15, 1677–1681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Teng, J.; Chen, J. ATL3, a cargo receptor for reticulophagy. Autophagy 2019, 15, 1465–1466. [Google Scholar] [CrossRef]
- Chen, Q.; Xiao, Y.; Chai, P.; Zheng, P.; Teng, J.; Chen, J. ATL3 Is a Tubular ER-Phagy Receptor for GABARAP-Mediated Selective Autophagy. Curr. Biol. 2019, 29, 846–855. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.D.; Harley, M.E.; Kemp, A.J.; Wills, J.; Lee, M.; Arends, M.; von Kriegsheim, A.; Behrends, C.; Wilkinson, S. CCPG1 Is a Non-canonical Autophagy Cargo Receptor Essential for ER-Phagy and Pancreatic ER Proteostasis. Dev. Cell 2018, 44, 217–232. [Google Scholar] [CrossRef] [Green Version]
- Fumagalli, F.; Noack, J.; Bergmann, T.J.; Cebollero, E.; Pisoni, G.B.; Fasana, E.; Fregno, I.; Galli, C.; Loi, M.; Solda, T.; et al. Translocon component Sec62 acts in endoplasmic reticulum turnover during stress recovery. Nat. Cell Biol. 2016, 18, 1173–1184. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.J.; Knupp, J.; Arunagiri, A.; Haataja, L.; Arvan, P.; Tsai, B. PGRMC1 acts as a size-selective cargo receptor to drive ER-phagic clearance of mutant prohormones. Nat. Commun. 2021, 12, 5991. [Google Scholar] [CrossRef]
- Meguro, S.; Zhuang, X.; Kirisako, H.; Nakatogawa, H. Pex3 confines pexophagy receptor activity of Atg36 to peroxisomes by regulating Hrr25-mediated phosphorylation and proteasomal degradation. J. Biol. Chem. 2020, 295, 16292–16298. [Google Scholar] [CrossRef]
- Motley, A.M.; Nuttall, J.M.; Hettema, E.H. Pex3-anchored Atg36 tags peroxisomes for degradation in Saccharomyces cerevisiae. EMBO J. 2012, 31, 2852–2868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Kamber, R.A.; Denic, V. The peroxisomal exportomer directly inhibits phosphoactivation of the pexophagy receptor Atg36 to suppress pexophagy in yeast. eLife 2022, 11, e74531. [Google Scholar] [CrossRef] [PubMed]
- Deosaran, E.; Larsen, K.B.; Hua, R.; Sargent, G.; Wang, Y.; Kim, S.; Lamark, T.; Jauregui, M.; Law, K.; Lippincott-Schwartz, J.; et al. NBR1 acts as an autophagy receptor for peroxisomes. J. Cell Sci. 2013, 126 Pt 4, 939–952. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Tripathi, D.N.; Jing, J.; Alexander, A.; Kim, J.; Powell, R.T.; Dere, R.; Tait-Mulder, J.; Lee, J.H.; Paull, T.T.; et al. ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat. Cell Biol. 2015, 17, 1259–1269. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, C.; Tan, L.J.; Mochida, K.; Kirisako, H.; Koizumi, M.; Asai, E.; Sakoh-Nakatogawa, M.; Ohsumi, Y.; Nakatogawa, H. Hrr25 triggers selective autophagy-related pathways by phosphorylating receptor proteins. J. Cell Biol. 2014, 207, 91–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sargent, G.; van Zutphen, T.; Shatseva, T.; Zhang, L.; Di Giovanni, V.; Bandsma, R.; Kim, P.K. PEX2 is the E3 ubiquitin ligase required for pexophagy during starvation. J. Cell Biol. 2016, 214, 677–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.H.; Chang, Y.J.; Lin, S.; Yang, W.Y. Hsc70/Stub1 promotes the removal of individual oxidatively stressed peroxisomes. Nat. Commun. 2020, 11, 5267. [Google Scholar] [CrossRef] [PubMed]
- Maejima, I.; Takahashi, A.; Omori, H.; Kimura, T.; Takabatake, Y.; Saitoh, T.; Yamamoto, A.; Hamasaki, M.; Noda, T.; Isaka, Y.; et al. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J. 2013, 32, 2336–2347. [Google Scholar] [CrossRef] [Green Version]
- Hung, Y.H.; Chen, L.M.; Yang, J.Y.; Yang, W.Y. Spatiotemporally controlled induction of autophagy-mediated lysosome turnover. Nat. Commun. 2013, 4, 2111. [Google Scholar] [CrossRef] [Green Version]
- Papadopoulos, C.; Kravic, B.; Meyer, H. Repair or Lysophagy: Dealing with Damaged Lysosomes. J. Mol. Biol. 2020, 432, 231–239. [Google Scholar] [CrossRef]
- Papandreou, M.E.; Tavernarakis, N. Nucleophagy: From homeostasis to disease. Cell Death Differ. 2019, 26, 630–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyant, G.A.; Abu-Remaileh, M.; Frenkel, E.M.; Laqtom, N.N.; Dharamdasani, V.; Lewis, C.A.; Chan, S.H.; Heinze, I.; Ori, A.; Sabatini, D.M. NUFIP1 is a ribosome receptor for starvation-induced ribophagy. Science 2018, 360, 751–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, H.; Harper, J.W. Systematic analysis of ribophagy in human cells reveals bystander flux during selective autophagy. Nat. Cell Biol. 2018, 20, 135–143. [Google Scholar] [CrossRef] [PubMed]
- An, H.; Harper, J.W. Ribosome Abundance Control Via the Ubiquitin-Proteasome System and Autophagy. J. Mol. Biol. 2020, 432, 170–184. [Google Scholar] [CrossRef]
- Seibenhener, M.L.; Babu, J.R.; Geetha, T.; Wong, H.C.; Krishna, N.R.; Wooten, M.W. Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol. Cell Biol. 2004, 24, 8055–8068. [Google Scholar] [CrossRef] [Green Version]
- Olzmann, J.A.; Chin, L.S. Parkin-mediated K63-linked polyubiquitination: A signal for targeting misfolded proteins to the aggresome-autophagy pathway. Autophagy 2008, 4, 85–87. [Google Scholar] [CrossRef] [Green Version]
- Olzmann, J.A.; Li, L.; Chudaev, M.V.; Chen, J.; Perez, F.A.; Palmiter, R.D.; Chin, L.S. Parkin-mediated K63-linked polyubiquitination targets misfolded DJ-1 to aggresomes via binding to HDAC6. J. Cell Biol. 2007, 178, 1025–1038. [Google Scholar] [CrossRef] [Green Version]
- Mauthe, M.; Kampinga, H.H.; Hipp, M.S.; Reggiori, F. Digest it all: The lysosomal turnover of cytoplasmic aggregates. Trends Biochem. Sci. 2022, 48, 216–228. [Google Scholar] [CrossRef]
- Shibata, M.; Yoshimura, K.; Furuya, N.; Koike, M.; Ueno, T.; Komatsu, M.; Arai, H.; Tanaka, K.; Kominami, E.; Uchiyama, Y. The MAP1-LC3 conjugation system is involved in lipid droplet formation. Biochem. Biophys. Res. Commun. 2009, 382, 419–423. [Google Scholar] [CrossRef]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [Green Version]
- Schott, M.B.; Rozeveld, C.N.; Weller, S.G.; McNiven, M.A. Lipophagy at a glance. J. Cell Sci. 2022, 135, jcs259402. [Google Scholar] [CrossRef]
- Zhang, S.; Peng, X.; Yang, S.; Li, X.; Huang, M.; Wei, S.; Liu, J.; He, G.; Zheng, H.; Yang, L.; et al. The regulation, function, and role of lipophagy, a form of selective autophagy, in metabolic disorders. Cell Death Dis. 2022, 13, 132. [Google Scholar] [CrossRef] [PubMed]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J., 3rd; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef] [PubMed]
- Bellelli, R.; Federico, G.; Matte, A.; Colecchia, D.; Iolascon, A.; Chiariello, M.; Santoro, M.; De Franceschi, L.; Carlomagno, F. NCOA4 Deficiency Impairs Systemic Iron Homeostasis. Cell Rep. 2016, 14, 411–421. [Google Scholar] [CrossRef] [Green Version]
- Mancias, J.D.; Pontano Vaites, L.; Nissim, S.; Biancur, D.E.; Kim, A.J.; Wang, X.; Liu, Y.; Goessling, W.; Kimmelman, A.C.; Harper, J.W. Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. eLife 2015, 4, e10308. [Google Scholar] [CrossRef]
- Cong, Y.; Dinesh Kumar, N.; Mauthe, M.; Verlhac, P.; Reggiori, F. Manipulation of selective macroautophagy by pathogens at a glance. J. Cell Sci. 2020, 133, jcs240440. [Google Scholar] [CrossRef]
- Pao, K.C.; Rape, M. Tug of War in the Xenophagy World. Trends Cell Biol. 2019, 29, 767–769. [Google Scholar] [CrossRef]
- Gomes, L.C.; Dikic, I. Autophagy in antimicrobial immunity. Mol. Cell 2014, 54, 224–233. [Google Scholar] [CrossRef] [Green Version]
- Goodall, E.A.; Kraus, F.; Harper, J.W. Mechanisms underlying ubiquitin-driven selective mitochondrial and bacterial autophagy. Mol. Cell 2022, 82, 1501–1513. [Google Scholar] [CrossRef]
- Ravenhill, B.J.; Boyle, K.B.; von Muhlinen, N.; Ellison, C.J.; Masson, G.R.; Otten, E.G.; Foeglein, A.; Williams, R.; Randow, F. The Cargo Receptor NDP52 Initiates Selective Autophagy by Recruiting the ULK Complex to Cytosol-Invading Bacteria. Mol. Cell 2019, 74, 320–329.e326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wild, P.; Farhan, H.; McEwan, D.G.; Wagner, S.; Rogov, V.V.; Brady, N.R.; Richter, B.; Korac, J.; Waidmann, O.; Choudhary, C.; et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 2011, 333, 228–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, I.; Amano, A.; Mizushima, N.; Yamamoto, A.; Yamaguchi, H.; Kamimoto, T.; Nara, A.; Funao, J.; Nakata, M.; Tsuda, K.; et al. Autophagy defends cells against invading group A Streptococcus. Science 2004, 306, 1037–1040. [Google Scholar] [CrossRef]
- Gutierrez, M.G.; Master, S.S.; Singh, S.B.; Taylor, G.A.; Colombo, M.I.; Deretic, V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 2004, 119, 753–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orvedahl, A.; MacPherson, S.; Sumpter, R., Jr.; Talloczy, Z.; Zou, Z.; Levine, B. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe 2010, 7, 115–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichimura, Y.; Waguri, S.; Sou, Y.S.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol. Cell 2013, 51, 618–631. [Google Scholar] [CrossRef] [Green Version]
- Yamada, A.; Hikichi, M.; Nozawa, T.; Nakagawa, I. FBXO2/SCF ubiquitin ligase complex directs xenophagy through recognizing bacterial surface glycan. EMBO Rep. 2021, 22, e52584. [Google Scholar] [CrossRef]
- Chai, Q.; Wang, X.; Qiang, L.; Zhang, Y.; Ge, P.; Lu, Z.; Zhong, Y.; Li, B.; Wang, J.; Zhang, L.; et al. A Mycobacterium tuberculosis surface protein recruits ubiquitin to trigger host xenophagy. Nat. Commun. 2019, 10, 1973. [Google Scholar] [CrossRef] [Green Version]
- Tripathi-Giesgen, I.; Behrends, C.; Alpi, A.F. The ubiquitin ligation machinery in the defense against bacterial pathogens. EMBO Rep. 2021, 22, e52864. [Google Scholar] [CrossRef]
- Choi, Y.; Bowman, J.W.; Jung, J.U. Autophagy during viral infection—A double-edged sword. Nat. Rev. Microbiol. 2018, 16, 341–354. [Google Scholar] [CrossRef]
- Jackson, W.T. Viruses and the autophagy pathway. Virology 2015, 479–480, 450–456. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, T.; Hu, J.; Jin, S.; Wu, J.; Guan, X.; Wu, Y.; Cui, J. Targeting Selective Autophagy as a Therapeutic Strategy for Viral Infectious Diseases. Front. Microbiol. 2022, 13, 889835. [Google Scholar] [CrossRef]
- Talloczy, Z.; Virgin, H.W.t.; Levine, B. PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy 2006, 2, 24–29. [Google Scholar] [CrossRef] [Green Version]
- Alexander, D.E.; Leib, D.A. Xenophagy in herpes simplex virus replication and pathogenesis. Autophagy 2008, 4, 101–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, D.E.; Ward, S.L.; Mizushima, N.; Levine, B.; Leib, D.A. Analysis of the role of autophagy in replication of herpes simplex virus in cell culture. J. Virol. 2007, 81, 12128–12134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orvedahl, A.; Sumpter, R., Jr.; Xiao, G.; Ng, A.; Zou, Z.; Tang, Y.; Narimatsu, M.; Gilpin, C.; Sun, Q.; Roth, M.; et al. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature 2011, 480, 113–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaumorcel, M.; Souquere, S.; Pierron, G.; Codogno, P.; Esclatine, A. Human cytomegalovirus controls a new autophagy-dependent cellular antiviral defense mechanism. Autophagy 2008, 4, 46–53. [Google Scholar] [CrossRef] [Green Version]
- Judith, D.; Mostowy, S.; Bourai, M.; Gangneux, N.; Lelek, M.; Lucas-Hourani, M.; Cayet, N.; Jacob, Y.; Prevost, M.C.; Pierre, P.; et al. Species-specific impact of the autophagy machinery on Chikungunya virus infection. EMBO Rep. 2013, 14, 534–544. [Google Scholar] [CrossRef]
- Mohamud, Y.; Qu, J.; Xue, Y.C.; Liu, H.; Deng, H.; Luo, H. CALCOCO2/NDP52 and SQSTM1/p62 differentially regulate coxsackievirus B3 propagation. Cell Death Differ. 2019, 26, 1062–1076. [Google Scholar] [CrossRef] [Green Version]
- Orvedahl, A.; Alexander, D.; Talloczy, Z.; Sun, Q.; Wei, Y.; Zhang, W.; Burns, D.; Leib, D.A.; Levine, B. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 2007, 1, 23–35. [Google Scholar] [CrossRef] [Green Version]
- Ames, J.; Yadavalli, T.; Suryawanshi, R.; Hopkins, J.; Agelidis, A.; Patil, C.; Fredericks, B.; Tseng, H.; Valyi-Nagy, T.; Shukla, D. OPTN is a host intrinsic restriction factor against neuroinvasive HSV-1 infection. Nat. Commun. 2021, 12, 5401. [Google Scholar] [CrossRef] [PubMed]
- Jackson, W.T.; Giddings, T.H., Jr.; Taylor, M.P.; Mulinyawe, S.; Rabinovitch, M.; Kopito, R.R.; Kirkegaard, K. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 2005, 3, e156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, M.P.; Kirkegaard, K. Modification of cellular autophagy protein LC3 by poliovirus. J. Virol. 2007, 81, 12543–12553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sir, D.; Kuo, C.F.; Tian, Y.; Liu, H.M.; Huang, E.J.; Jung, J.U.; Machida, K.; Ou, J.H. Replication of hepatitis C virus RNA on autophagosomal membranes. J. Biol. Chem. 2012, 287, 18036–18043. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Kim, J.Y.; Liu, H.M.; Lai, M.M.C.; Ou, J.J. HCV-induced autophagosomes are generated via homotypic fusion of phagophores that mediate HCV RNA replication. PLoS Pathog. 2017, 13, e1006609. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Tian, Y.; Ou, J.H. HCV induces the expression of Rubicon and UVRAG to temporally regulate the maturation of autophagosomes and viral replication. PLoS Pathog. 2015, 11, e1004764. [Google Scholar] [CrossRef] [Green Version]
- Sir, D.; Ann, D.K.; Ou, J.H. Autophagy by hepatitis B virus and for hepatitis B virus. Autophagy 2010, 6, 548–549. [Google Scholar] [CrossRef] [Green Version]
- Sir, D.; Tian, Y.; Chen, W.L.; Ann, D.K.; Yen, T.S.; Ou, J.H. The early autophagic pathway is activated by hepatitis B virus and required for viral DNA replication. Proc. Natl. Acad. Sci. USA 2010, 107, 4383–4388. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.R.; Lei, H.Y.; Liu, M.T.; Wang, J.R.; Chen, S.H.; Jiang-Shieh, Y.F.; Lin, Y.S.; Yeh, T.M.; Liu, C.C.; Liu, H.S. Autophagic machinery activated by dengue virus enhances virus replication. Virology 2008, 374, 240–248. [Google Scholar] [CrossRef] [Green Version]
- Panyasrivanit, M.; Khakpoor, A.; Wikan, N.; Smith, D.R. Co-localization of constituents of the dengue virus translation and replication machinery with amphisomes. J. Gen. Virol. 2009, 90 Pt 2, 448–456. [Google Scholar] [CrossRef]
- Huang, S.C.; Chang, C.L.; Wang, P.S.; Tsai, Y.; Liu, H.S. Enterovirus 71-induced autophagy detected in vitro and in vivo promotes viral replication. J. Med. Virol. 2009, 81, 1241–1252. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Zhang, J.; Si, X.; Gao, G.; Mao, I.; McManus, B.M.; Luo, H. Autophagosome supports coxsackievirus B3 replication in host cells. J. Virol. 2008, 82, 9143–9153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, G.; Zhao, H.; Li, Y.; Ji, M.; Chen, Y.; Shi, Y.; Bi, Y.; Wang, P.; Zhang, H. ORF3a of the COVID-19 virus SARS-CoV-2 blocks HOPS complex-mediated assembly of the SNARE complex required for autolysosome formation. Dev. Cell 2021, 56, 427–442. [Google Scholar] [CrossRef]
- Stukalov, A.; Girault, V.; Grass, V.; Karayel, O.; Bergant, V.; Urban, C.; Haas, D.A.; Huang, Y.; Oubraham, L.; Wang, A.; et al. Multilevel proteomics reveals host perturbations by SARS-CoV-2 and SARS-CoV. Nature 2021, 594, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, H.; Pei, R.; Mao, B.; Zhao, Z.; Li, H.; Lin, Y.; Lu, K. The SARS-CoV-2 protein ORF3a inhibits fusion of autophagosomes with lysosomes. Cell Discov. 2021, 7, 31. [Google Scholar] [CrossRef] [PubMed]
- Mohamud, Y.; Shi, J.; Qu, J.; Poon, T.; Xue, Y.C.; Deng, H.; Zhang, J.; Luo, H. Enteroviral Infection Inhibits Autophagic Flux via Disruption of the SNARE Complex to Enhance Viral Replication. Cell Rep. 2018, 22, 3292–3303. [Google Scholar] [CrossRef] [Green Version]
- Bird, S.W.; Maynard, N.D.; Covert, M.W.; Kirkegaard, K. Nonlytic viral spread enhanced by autophagy components. Proc. Natl. Acad. Sci. USA 2014, 111, 13081–13086. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Liu, Y.; Wang, Z.; Liu, K.; Wang, Y.; Liu, J.; Ding, H.; Yuan, Z. Subversion of cellular autophagy machinery by hepatitis B virus for viral envelopment. J. Virol. 2011, 85, 6319–6333. [Google Scholar] [CrossRef] [Green Version]
- Tanida, I.; Fukasawa, M.; Ueno, T.; Kominami, E.; Wakita, T.; Hanada, K. Knockdown of autophagy-related gene decreases the production of infectious hepatitis C virus particles. Autophagy 2009, 5, 937–945. [Google Scholar] [CrossRef] [Green Version]
- Mateo, R.; Nagamine, C.M.; Spagnolo, J.; Mendez, E.; Rahe, M.; Gale, M., Jr.; Yuan, J.; Kirkegaard, K. Inhibition of cellular autophagy deranges dengue virion maturation. J. Virol. 2013, 87, 1312–1321. [Google Scholar] [CrossRef] [Green Version]
- Datan, E.; Roy, S.G.; Germain, G.; Zali, N.; McLean, J.E.; Golshan, G.; Harbajan, S.; Lockshin, R.A.; Zakeri, Z. Dengue-induced autophagy, virus replication and protection from cell death require ER stress (PERK) pathway activation. Cell Death Dis. 2016, 7, e2127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joubert, P.E.; Werneke, S.W.; de la Calle, C.; Guivel-Benhassine, F.; Giodini, A.; Peduto, L.; Levine, B.; Schwartz, O.; Lenschow, D.J.; Albert, M.L. Chikungunya virus-induced autophagy delays caspase-dependent cell death. J. Exp. Med. 2012, 209, 1029–1047. [Google Scholar] [CrossRef] [PubMed]
- Jounai, N.; Takeshita, F.; Kobiyama, K.; Sawano, A.; Miyawaki, A.; Xin, K.Q.; Ishii, K.J.; Kawai, T.; Akira, S.; Suzuki, K.; et al. The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc. Natl. Acad. Sci. USA 2007, 104, 14050–14055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tal, M.C.; Sasai, M.; Lee, H.K.; Yordy, B.; Shadel, G.S.; Iwasaki, A. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 2770–2775. [Google Scholar] [CrossRef] [Green Version]
- Ke, P.Y.; Chen, S.S. Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro. J. Clin. Invest. 2011, 121, 37–56. [Google Scholar] [CrossRef] [Green Version]
- Ke, P.Y.; Chen, S.S. Autophagy: A novel guardian of HCV against innate immune response. Autophagy 2011, 7, 533–535. [Google Scholar] [CrossRef] [Green Version]
- Shrivastava, S.; Raychoudhuri, A.; Steele, R.; Ray, R.; Ray, R.B. Knockdown of autophagy enhances the innate immune response in hepatitis C virus-infected hepatocytes. Hepatology 2011, 53, 406–414. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.T.; Lee, J.; Narula, M.; Ou, J.J. Suppression of Host Innate Immune Response by Hepatitis C Virus via Induction of Autophagic Degradation of TRAF6. J. Virol. 2016, 90, 10928–10935. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.; Duan, T.; Feng, Y.; Liu, Q.; Lin, M.; Cui, J.; Wang, R.F. LRRC25 inhibits type I IFN signaling by targeting ISG15-associated RIG-I for autophagic degradation. EMBO J. 2018, 37, 351–366. [Google Scholar] [CrossRef]
- Park, Y.J.; Oanh, N.T.K.; Heo, J.; Kim, S.G.; Lee, H.S.; Lee, H.; Lee, J.H.; Kang, H.C.; Lim, W.; Yoo, Y.S.; et al. Dual targeting of RIG-I and MAVS by MARCH5 mitochondria ubiquitin ligase in innate immunity. Cell. Signal. 2020, 67, 109520. [Google Scholar] [CrossRef]
- Bu, L.; Wang, H.; Hou, P.; Guo, S.; He, M.; Xiao, J.; Li, P.; Zhong, Y.; Jia, P.; Cao, Y.; et al. The Ubiquitin E3 Ligase Parkin Inhibits Innate Antiviral Immunity Through K48-Linked Polyubiquitination of RIG-I and MDA5. Front. Immunol. 2020, 11, 1926. [Google Scholar] [CrossRef] [PubMed]
- Glon, D.; Vilmen, G.; Perdiz, D.; Hernandez, E.; Beauclair, G.; Quignon, F.; Berlioz-Torrent, C.; Marechal, V.; Pous, C.; Lussignol, M.; et al. Essential role of hyperacetylated microtubules in innate immunity escape orchestrated by the EBV-encoded BHRF1 protein. PLoS Pathog. 2022, 18, e1010371. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.; Yang, K.; Jia, P.; Liu, L.; Lin, Y.; Li, Z.; Li, J.; Chen, S.; Guo, S.; Pan, J.; et al. A novel selective autophagy receptor, CCDC50, delivers K63 polyubiquitination-activated RIG-I/MDA5 for degradation during viral infection. Cell Res. 2021, 31, 62–79. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Tian, S.; Luo, M.; Xie, W.; Liu, T.; Duan, T.; Wu, Y.; Cui, J. Tetherin Suppresses Type I Interferon Signaling by Targeting MAVS for NDP52-Mediated Selective Autophagic Degradation in Human Cells. Mol. Cell 2017, 68, 308–322. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Zhu, Y.; Zhang, Y.; Geng, Y.; Gong, J.; Geng, J.; Zhang, P.; Zhang, X.; Liu, N.; Peng, Y.; et al. RNF34 functions in immunity and selective mitophagy by targeting MAVS for autophagic degradation. EMBO J. 2019, 38, e100978. [Google Scholar] [CrossRef]
- Wang, R.; Zhu, Y.; Ren, C.; Yang, S.; Tian, S.; Chen, H.; Jin, M.; Zhou, H. Influenza A virus protein PB1-F2 impairs innate immunity by inducing mitophagy. Autophagy 2021, 17, 496–511. [Google Scholar] [CrossRef]
- Varga, Z.T.; Ramos, I.; Hai, R.; Schmolke, M.; Garcia-Sastre, A.; Fernandez-Sesma, A.; Palese, P. The influenza virus protein PB1-F2 inhibits the induction of type I interferon at the level of the MAVS adaptor protein. PLoS Pathog. 2011, 7, e1002067. [Google Scholar] [CrossRef]
- Lei, Y.; Wen, H.; Yu, Y.; Taxman, D.J.; Zhang, L.; Widman, D.G.; Swanson, K.V.; Wen, K.W.; Damania, B.; Moore, C.B.; et al. The mitochondrial proteins NLRX1 and TUFM form a complex that regulates type I interferon and autophagy. Immunity 2012, 36, 933–946. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Y.; Xu, S.; Wei, Y.; Zhang, X.; Wang, Q.; Jia, Y.; Wang, W.; Han, L.; Chen, Z.; Wang, Z.; et al. The PB1 protein of influenza A virus inhibits the innate immune response by targeting MAVS for NBR1-mediated selective autophagic degradation. PLoS Pathog. 2021, 17, e1009300. [Google Scholar] [CrossRef]
- Liu, J.; Wu, X.; Wang, H.; Wei, J.; Wu, Q.; Wang, X.; Yan, Y.; Cui, J.; Min, J.; Wang, F.; et al. HFE inhibits type I IFNs signaling by targeting the SQSTM1-mediated MAVS autophagic degradation. Autophagy 2021, 17, 1962–1977. [Google Scholar] [CrossRef]
- Pei, J.; Deng, J.; Ye, Z.; Wang, J.; Gou, H.; Liu, W.; Zhao, M.; Liao, M.; Yi, L.; Chen, J. Absence of autophagy promotes apoptosis by modulating the ROS-dependent RLR signaling pathway in classical swine fever virus-infected cells. Autophagy 2016, 12, 1738–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, B.; Zhao, M.; Song, D.; Wu, K.; Yi, L.; Li, W.; Li, X.; Wang, K.; Chen, J. Induction of autophagy and suppression of type I IFN secretion by CSFV. Autophagy 2021, 17, 925–947. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Jin, S.; Liu, Q.; Zhang, Y.; Ma, L.; Zhao, Z.; Yang, S.; Li, Y.P.; Cui, J. Selective autophagy controls the stability of transcription factor IRF3 to balance type I interferon production and immune suppression. Autophagy 2021, 17, 1379–1392. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Jin, S.; Zhang, C.; Yang, S.; Wu, Y.; Zhao, Y.; Songyang, Z.; Cui, J. Selective autophagy controls the stability of TBK1 via NEDD4 to balance host defense. Cell Death Differ. 2022, 29, 40–53. [Google Scholar] [CrossRef]
- Sir, D.; Chen, W.L.; Choi, J.; Wakita, T.; Yen, T.S.; Ou, J.H. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 2008, 48, 1054–1061. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Syed, G.H.; Siddiqui, A. Hepatitis C virus induces the mitochondrial translocation of Parkin and subsequent mitophagy. PLoS Pathog. 2013, 9, e1003285. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Syed, G.H.; Khan, M.; Chiu, W.W.; Sohail, M.A.; Gish, R.G.; Siddiqui, A. Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc. Natl. Acad. Sci. USA 2014, 111, 6413–6418. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, A.; Morosky, S.A.; Delorme-Axford, E.; Dybdahl-Sissoko, N.; Oberste, M.S.; Wang, T.; Coyne, C.B. The coxsackievirus B 3C protease cleaves MAVS and TRIF to attenuate host type I interferon and apoptotic signaling. PLoS Pathog. 2011, 7, e1001311. [Google Scholar] [CrossRef] [Green Version]
- Feng, Q.; Langereis, M.A.; Lork, M.; Nguyen, M.; Hato, S.V.; Lanke, K.; Emdad, L.; Bhoopathi, P.; Fisher, P.B.; Lloyd, R.E.; et al. Enterovirus 2Apro targets MDA5 and MAVS in infected cells. J. Virol. 2014, 88, 3369–3378. [Google Scholar] [CrossRef] [Green Version]
- Katayama, H.; Kogure, T.; Mizushima, N.; Yoshimori, T.; Miyawaki, A. A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem. Biol. 2011, 18, 1042–1052. [Google Scholar] [CrossRef] [Green Version]
- Sun, N.; Yun, J.; Liu, J.; Malide, D.; Liu, C.; Rovira, I.I.; Holmstrom, K.M.; Fergusson, M.M.; Yoo, Y.H.; Combs, C.A.; et al. Measuring In Vivo Mitophagy. Mol. Cell 2015, 60, 685–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, S.; Noda, T.; Yoshimori, T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007, 3, 452–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamargo-Gomez, I.; Fernandez, A.F.; Marino, G. Pathogenic Single Nucleotide Polymorphisms on Autophagy-Related Genes. Int. J. Mol. Sci. 2020, 21, 8196. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Signaling pathway of retinoic acid-inducible I (RIG-I)-like receptor (RLR) antiviral immunity: (A) Domain structure of RLRs, including retinoic acid-inducible I (RIG-I), melanoma differentiation association gene 5 (MDA5), laboratory of genetics and physiology 2 (LGP2), and the downstream effector mitochondrial antiviral signaling (MAVS). Among them, RIG-I, MDA5, and LGP2 contain the central helicase domain and C-terminal domain (CTD). There are two caspase activation and recruitment domains (CARDs) located within the N-termini of RIG-I and MDA5. MAVS has one N-terminal CARD that is able to associate with the CARDs of RIG-I and MDA5 and a C-terminal transmembrane domain for targeting to the mitochondrial outer membrane. (B) Activation of RLR antiviral signaling. During virus infection, the binding of viral RNAs induces conformational changes in RIG-I and MDA5. The free CARDs of RIG-I and MDA5 undergo self-oligomerization and interact with the CARDs of MAVS on mitochondria. The formed MAVS aggregates in turn to recruit the ubiquitin E3 ligases (TNF) receptor-associated factor 2 (TRAF2), TRAF5, and TRAF6, which produce polyubiquitin chains. The polyubiquitin chain then induces the recruitment of nuclear factor kappa-light-chain-enhancer of activated B (NF-kB) essential modulator (NEMO), which subsequently activates TANK binding kinase 1 (TBK1) to phosphorylate interferon response factor 3 (IRF3). Phosphorylated IRF3 then translocates into the nucleus and subsequently activates the gene expression of type I interferons (IFNs).
Figure 1.
Signaling pathway of retinoic acid-inducible I (RIG-I)-like receptor (RLR) antiviral immunity: (A) Domain structure of RLRs, including retinoic acid-inducible I (RIG-I), melanoma differentiation association gene 5 (MDA5), laboratory of genetics and physiology 2 (LGP2), and the downstream effector mitochondrial antiviral signaling (MAVS). Among them, RIG-I, MDA5, and LGP2 contain the central helicase domain and C-terminal domain (CTD). There are two caspase activation and recruitment domains (CARDs) located within the N-termini of RIG-I and MDA5. MAVS has one N-terminal CARD that is able to associate with the CARDs of RIG-I and MDA5 and a C-terminal transmembrane domain for targeting to the mitochondrial outer membrane. (B) Activation of RLR antiviral signaling. During virus infection, the binding of viral RNAs induces conformational changes in RIG-I and MDA5. The free CARDs of RIG-I and MDA5 undergo self-oligomerization and interact with the CARDs of MAVS on mitochondria. The formed MAVS aggregates in turn to recruit the ubiquitin E3 ligases (TNF) receptor-associated factor 2 (TRAF2), TRAF5, and TRAF6, which produce polyubiquitin chains. The polyubiquitin chain then induces the recruitment of nuclear factor kappa-light-chain-enhancer of activated B (NF-kB) essential modulator (NEMO), which subsequently activates TANK binding kinase 1 (TBK1) to phosphorylate interferon response factor 3 (IRF3). Phosphorylated IRF3 then translocates into the nucleus and subsequently activates the gene expression of type I interferons (IFNs).

Figure 2.
Post-translational modifications (PTMs) of RLR signaling molecules: RIG-I and MDA5 are regulated by phosphorylation, dephosphorylation, acetylation, deacetylation, ubiquitination, and deubiquitination and modified by Lys-48- and Lys-63-linked polyubiquitin. The enzymes responsible for PTMs are illustrated, and the modified amino acid residues are specifically indicated. PKC-α, Protein kinase C-α; PKC-β, Protein kinase C-β; TRIM25, tripartite motif 25; TRIM38, tripartite motif 25; TRIM65, tripartite motif 65; CK-II, casein kinase II; DAPK1, death-associated protein kinase 1; PP1α, protein phosphatase 1α; PP1-γ, protein phosphatase 1γ; HDAC6, histone deacetylase 6; SENP2, sentrin/SUMO-specific protease 2; RNF122, RING finger protein 122; RNF125, RING finger protein 125; RNF135, RING finger protein 135; MEX3C, mex-3 RNA binding family member C; Cbl, casitas B-lineage lymphoma proto-oncogene; USP4, ubiquitin-specific protease 4; USP15, ubiquitin-specific protease 15; CYLD, CYLD lysine 63 deubiquitinase; OTUD3, ovarian tumor protease deubiquitinating enzyme 3.
Figure 2.
Post-translational modifications (PTMs) of RLR signaling molecules: RIG-I and MDA5 are regulated by phosphorylation, dephosphorylation, acetylation, deacetylation, ubiquitination, and deubiquitination and modified by Lys-48- and Lys-63-linked polyubiquitin. The enzymes responsible for PTMs are illustrated, and the modified amino acid residues are specifically indicated. PKC-α, Protein kinase C-α; PKC-β, Protein kinase C-β; TRIM25, tripartite motif 25; TRIM38, tripartite motif 25; TRIM65, tripartite motif 65; CK-II, casein kinase II; DAPK1, death-associated protein kinase 1; PP1α, protein phosphatase 1α; PP1-γ, protein phosphatase 1γ; HDAC6, histone deacetylase 6; SENP2, sentrin/SUMO-specific protease 2; RNF122, RING finger protein 122; RNF125, RING finger protein 125; RNF135, RING finger protein 135; MEX3C, mex-3 RNA binding family member C; Cbl, casitas B-lineage lymphoma proto-oncogene; USP4, ubiquitin-specific protease 4; USP15, ubiquitin-specific protease 15; CYLD, CYLD lysine 63 deubiquitinase; OTUD3, ovarian tumor protease deubiquitinating enzyme 3.

Figure 3.
Three modes of autophagy and regulatory processes: (A) Microautophagy involves the invagination and/or protrusion of the lysosomal membranous structure for the engulfment of cytosol materials, including organelles, and the transfer of these materials to the lysosome lumen for degradation. (B) CMA recognizes the cargoes containing a KFERQ (Lys-Phe-Glu-Arg-Gln) motif by heat shock cognate 70 kDa protein (HSC70). Through docking with lysosomal membrane protein 2A (LAMP2A) on the membrane of lysosomes, the cargoes are delivered to the lysosomal lumen for degradation. (C) Macroautophagy is a vacuole biogenesis process consisting of membrane rearrangement events. The signaling cascade involved in the nutrient-starvation-induced mammalian target of rapamycin (mTOR) inhibition recruits the unc-51-like kinase 1/2 ULK1/2 initiation complex, which in turn activates the class III phosphatidylinositol-3-OH kinase (PI(3)K) complex for the biosynthesis of phosphatidylinositol-3-phosphate (PtdIns(3)P). The produced PtdIns(3)P subsequently recruits double-FYVE-containing protein 1 (DFCP1) and WD-repeat domain PtdIns(3)P-interacting (WIPI, also known as ATG18) to induce emergence of the IM/phagophore from the ER-associated membranous compartment. UBL conjugation systems (ATG5-ATG12 conjugation and ATG8/LC3 conjugation systems) facilitate the expansion and transformation of IM/phagophores into autophagosomes. Subsequently, autophagosomes fuse with lysosomes to form autolysosomes via SNAREs, Ras-related protein 7 (Rab7), and the homotypic fusion and protein sorting (HOPS) complex. The acidic lysosomal proteases degrade the sequestrated materials within autolysosomes. Selective autophagy specifically eliminates various types of organelles (including mitophagy, ER-phagy, pexophagy, ribophagy, lysophagy, lipophagy, and nucleophagy), aggregated proteins (aggrephagy), invading pathogens (xenophagy), and ferritin (ferritinophagy) via the recognition of cargo receptors. The interaction between the cargo receptor and ATG8/LC3 family protein selectively targets the cargo to the autophagic process for degradation. The cargo receptors for each type of selective autophagy in yeast and mammals are shown. p62/SQSTM1, p62/sequestosome 1; NDP52/Calcoco2, calcium-binding and coiled-coil domain-containing protein 2; NBR1, the neighbor of BRCA1; OPTN, optineurin; BNIP3, BCL2/adenovirus E1B 19 kDa protein-interacting protein 3; BNIP3L/Nix, BNIP3-like; Tax1 bp1, Tax1-binding protein 1; Tollip, Toll-interacting protein; ANT, adenine nucleotide translocator; FUNDC1, FUN14 domain-containing 1; ATG32, autophagy-related gene 32; PHB2, prohibitin 2; NIPSNAP, nitrophenylphosphatase domain and nonneuronal SNAP25-like protein homolog; ATG11, autophagy-related gene 11; ATG39, autophagy-related gene 39; ATG40, autophagy-related gene 40; FAM134B, the family with sequence similarity 134; TEX264, testis-expressed sequence 264 protein; ATL3, Atlastins 3; CCPG1, cell-cycle progression gene 1; SEC62, preprotein translocation factor 62; PGRMC1, progesterone receptor membrane component 1; ATG36, autophagy-related gene 36; HDAC6, histone deacetylase 6; ALFY, autophagy linked FYVE domain protein; NCOA4, nuclear receptor coactivator 4.
Figure 3.
Three modes of autophagy and regulatory processes: (A) Microautophagy involves the invagination and/or protrusion of the lysosomal membranous structure for the engulfment of cytosol materials, including organelles, and the transfer of these materials to the lysosome lumen for degradation. (B) CMA recognizes the cargoes containing a KFERQ (Lys-Phe-Glu-Arg-Gln) motif by heat shock cognate 70 kDa protein (HSC70). Through docking with lysosomal membrane protein 2A (LAMP2A) on the membrane of lysosomes, the cargoes are delivered to the lysosomal lumen for degradation. (C) Macroautophagy is a vacuole biogenesis process consisting of membrane rearrangement events. The signaling cascade involved in the nutrient-starvation-induced mammalian target of rapamycin (mTOR) inhibition recruits the unc-51-like kinase 1/2 ULK1/2 initiation complex, which in turn activates the class III phosphatidylinositol-3-OH kinase (PI(3)K) complex for the biosynthesis of phosphatidylinositol-3-phosphate (PtdIns(3)P). The produced PtdIns(3)P subsequently recruits double-FYVE-containing protein 1 (DFCP1) and WD-repeat domain PtdIns(3)P-interacting (WIPI, also known as ATG18) to induce emergence of the IM/phagophore from the ER-associated membranous compartment. UBL conjugation systems (ATG5-ATG12 conjugation and ATG8/LC3 conjugation systems) facilitate the expansion and transformation of IM/phagophores into autophagosomes. Subsequently, autophagosomes fuse with lysosomes to form autolysosomes via SNAREs, Ras-related protein 7 (Rab7), and the homotypic fusion and protein sorting (HOPS) complex. The acidic lysosomal proteases degrade the sequestrated materials within autolysosomes. Selective autophagy specifically eliminates various types of organelles (including mitophagy, ER-phagy, pexophagy, ribophagy, lysophagy, lipophagy, and nucleophagy), aggregated proteins (aggrephagy), invading pathogens (xenophagy), and ferritin (ferritinophagy) via the recognition of cargo receptors. The interaction between the cargo receptor and ATG8/LC3 family protein selectively targets the cargo to the autophagic process for degradation. The cargo receptors for each type of selective autophagy in yeast and mammals are shown. p62/SQSTM1, p62/sequestosome 1; NDP52/Calcoco2, calcium-binding and coiled-coil domain-containing protein 2; NBR1, the neighbor of BRCA1; OPTN, optineurin; BNIP3, BCL2/adenovirus E1B 19 kDa protein-interacting protein 3; BNIP3L/Nix, BNIP3-like; Tax1 bp1, Tax1-binding protein 1; Tollip, Toll-interacting protein; ANT, adenine nucleotide translocator; FUNDC1, FUN14 domain-containing 1; ATG32, autophagy-related gene 32; PHB2, prohibitin 2; NIPSNAP, nitrophenylphosphatase domain and nonneuronal SNAP25-like protein homolog; ATG11, autophagy-related gene 11; ATG39, autophagy-related gene 39; ATG40, autophagy-related gene 40; FAM134B, the family with sequence similarity 134; TEX264, testis-expressed sequence 264 protein; ATL3, Atlastins 3; CCPG1, cell-cycle progression gene 1; SEC62, preprotein translocation factor 62; PGRMC1, progesterone receptor membrane component 1; ATG36, autophagy-related gene 36; HDAC6, histone deacetylase 6; ALFY, autophagy linked FYVE domain protein; NCOA4, nuclear receptor coactivator 4.

Figure 4.
Summary of the negative regulation of RLR antiviral signaling by autophagy: The actions of autophagy-related molecules on mitochondria and the RLR signaling components RIG-I, MDA5, MAVS, TRAF6, IRF3, and TBK1 are shown. The cargo receptors, ubiquitin E3 ligases, and other factors involved in the degradation of RLR antiviral factors are illustrated. The amino acid residues of these molecules that could be ubiquitinated by ubiquitin E3 ligases are specifically indicated. poly(I:C), polyinosinic acid-polycytidylic acid; HCV, hepatitis C virus; VSV, vesicular stomatitis virus; SeV, Sendai virus; IAV, influenza A virus; CVB3, coxsackievirus B3; CSFV, Classical swine fever virus; LRRC25, leucine-rich repeat containing protein 25; MARCH5, membrane-associated ring-CH-type finger 5; ISG15, IFN-stimulated gene 15; CCDC50, coiled-coil domain-containing protein 50; PINK1, PTEN-induced putative kinase 1; RNF34, RING finger protein 34; MARCH8, membrane-associated ring finger (C3HC4) 8; RNF5, RING finger protein 5; TUFM, Tu translation elongation factor, mitochondrial; NLRX, NLR family member X1; TRM21, tripartite motif 21; HFE, hemochromatosis gene; NEDD4, neural precursor cell-expressed developmentally downregulated gene 4.
Figure 4.
Summary of the negative regulation of RLR antiviral signaling by autophagy: The actions of autophagy-related molecules on mitochondria and the RLR signaling components RIG-I, MDA5, MAVS, TRAF6, IRF3, and TBK1 are shown. The cargo receptors, ubiquitin E3 ligases, and other factors involved in the degradation of RLR antiviral factors are illustrated. The amino acid residues of these molecules that could be ubiquitinated by ubiquitin E3 ligases are specifically indicated. poly(I:C), polyinosinic acid-polycytidylic acid; HCV, hepatitis C virus; VSV, vesicular stomatitis virus; SeV, Sendai virus; IAV, influenza A virus; CVB3, coxsackievirus B3; CSFV, Classical swine fever virus; LRRC25, leucine-rich repeat containing protein 25; MARCH5, membrane-associated ring-CH-type finger 5; ISG15, IFN-stimulated gene 15; CCDC50, coiled-coil domain-containing protein 50; PINK1, PTEN-induced putative kinase 1; RNF34, RING finger protein 34; MARCH8, membrane-associated ring finger (C3HC4) 8; RNF5, RING finger protein 5; TUFM, Tu translation elongation factor, mitochondrial; NLRX, NLR family member X1; TRM21, tripartite motif 21; HFE, hemochromatosis gene; NEDD4, neural precursor cell-expressed developmentally downregulated gene 4.


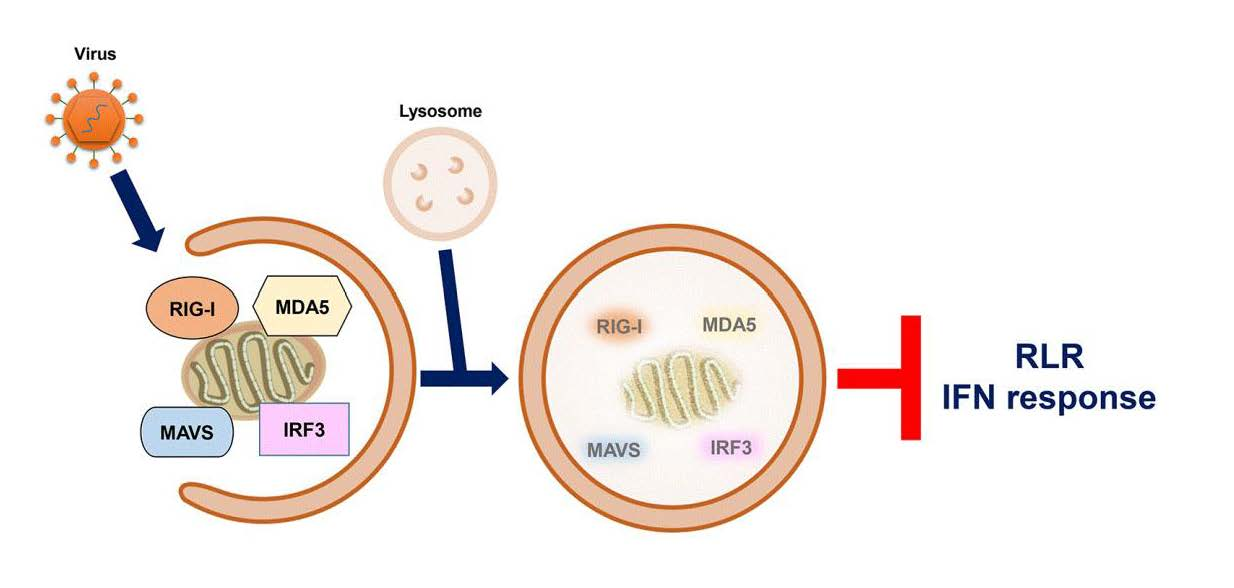{kind=link}
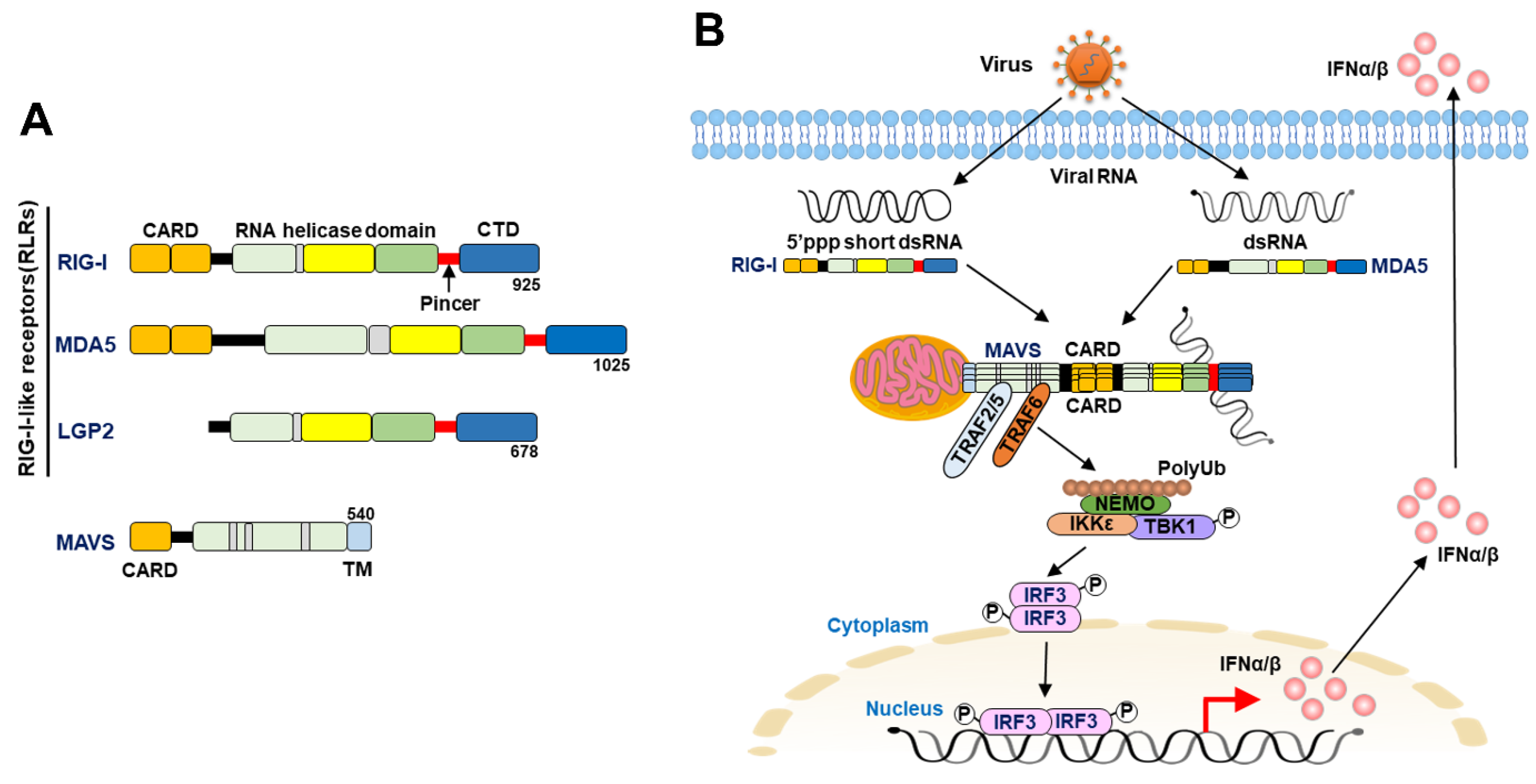{kind=link}
{kind=link}
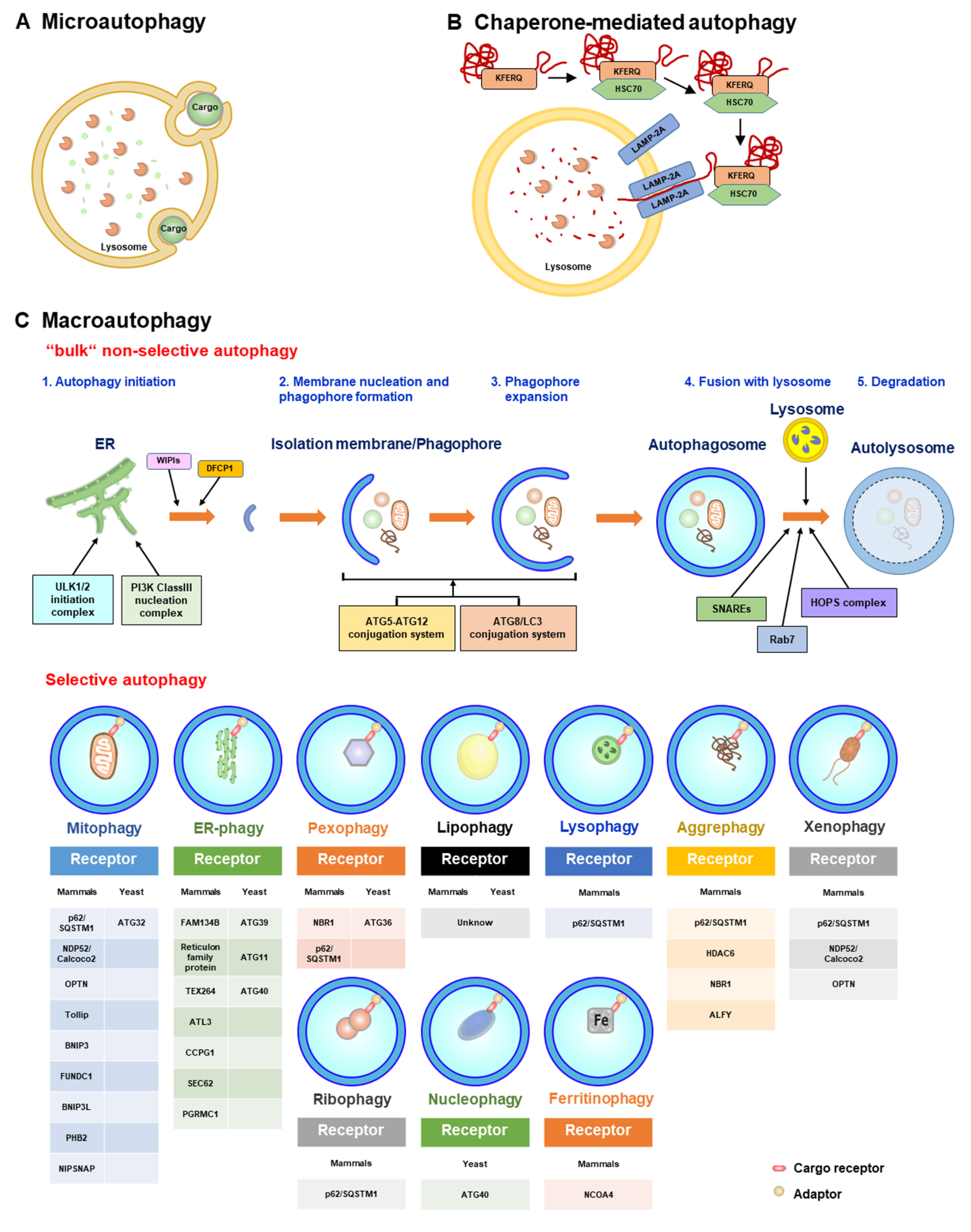{kind=link}
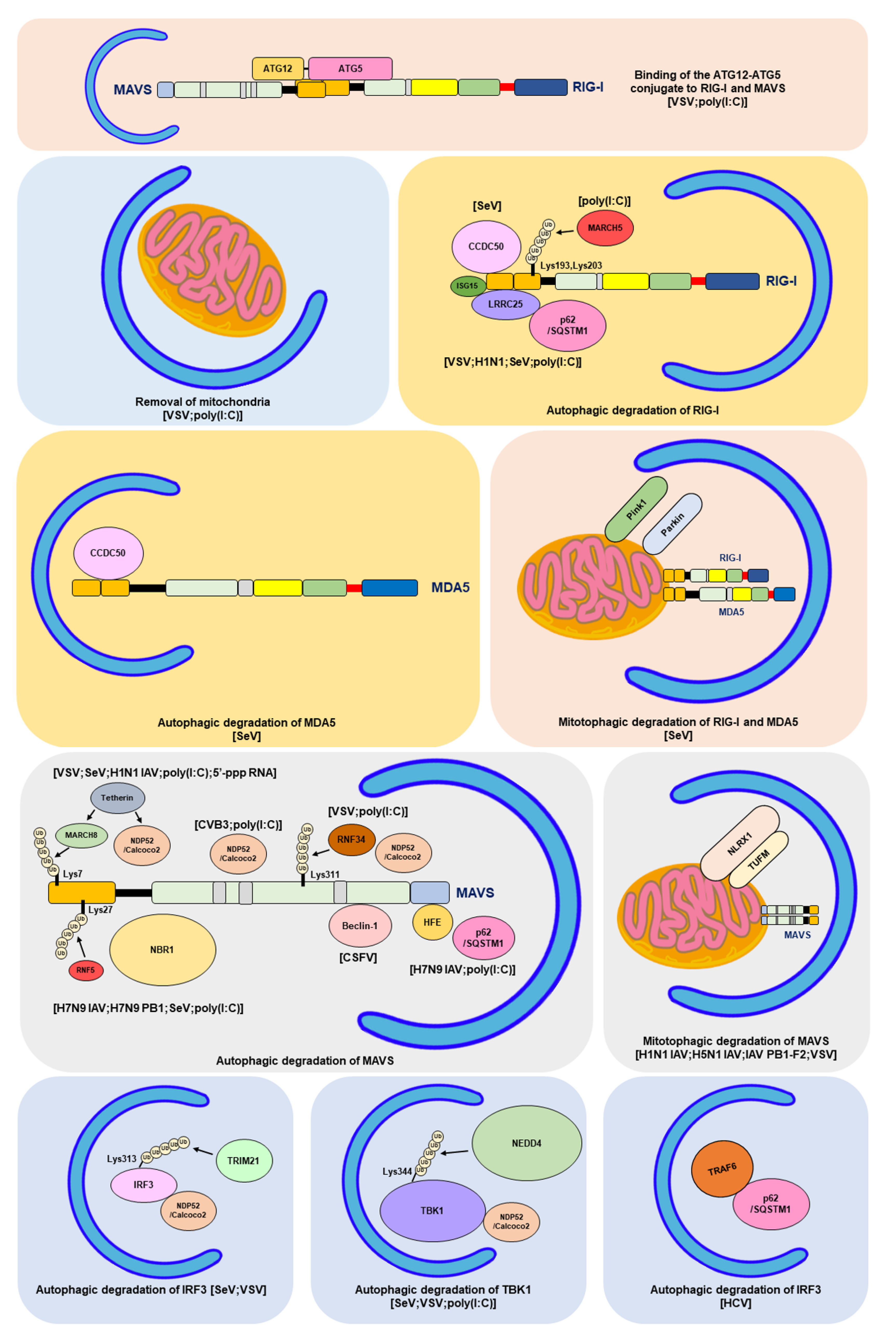{kind=link}
Table 1.
Negative regulation of RLR antiviral signaling by autophagy.
| Target in RLR Signaling | Virus/PAMP RNAs | The Biological Function of Autophagy on RLR Molecules | Reference |
|---|---|---|---|
| RIG-I; MAVS | VSV; poly(I:C) | Binding of the ATG12-ATG5 conjugate to RIG-I and MAVS | [354] |
| Mitochondria | VSV; poly(I:C) | Elimination of mitochondria through autophagy | [355] |
| Unknown | HCV PAMP; DENV PAMP | Repression of the RLR IFN response by autophagy | [356,357] |
| Unknown | HCV | Repression of the RLR innate immune response by autophagy | [358] |
| TRAF6 | HCV | Autophagic degradation of TRAF6 by p62/SQSTM1 | [359] |
| RIG-I | VSV; H1N1 IAV; SeV; poly (I:C) | Autophagic degradation of RIG-I through LRRC25 and p62/SQSTM1 | [360] |
| RIG-I | poly(I:C) | Autophagic degradation of RIG-I by MARCH5 | [361] |
| RIG-I; MDA5 | SeV; EBV | Autophagic degradation RIG-I and MDA5 by PINK1/Parkin-dependent pathway | [362,363] |
| RIG-I; MDA5 | SeV | Autophagic degradation of RIG-I and MDA5 by CCDC50 | [364] |
| MAVS | VSV; SeV; H1N1 IAV; poly(I:C); 5′-triphosphate-RNA | Autophagic degradation of MAVS by NDP52/Calcoco2 | [365] |
| MAVS | CVB3; poly(I:C) | Autophagic degradation of MAVS by NDP52/Calcoco2 | [330] |
| MAVS; mitochondria | VSV; poly(I:C) | Removal of mitochondria and autophagic degradation of MAVS by RNF34-NDP52/Calcoco2 axis | [366] |
| MAVS in mitochondria | H1N1 IAV; H5N1 IAV; IAV PB1-F2; VSV | Induction of mitophagy by IAV PB1-F2/TUFM axis and autophagic degradation of MAVS | [367,368,369] |
| MAVS | IAV H7N9; H7N9 PB1; SeV; poly(I:C) | Autophagic degradation of MAVS by NBR1 and RNF5 | [370] |
| MAVS in mitochondria and peroxisome | H7N9 IAV; poly (I:C) | Autophagic degradation of MAVS by p62/SQSTM1 and HFE | [371] |
| RIG-I; MAVS | CSFV | Removal of mitochondria through autophagy and the interaction between Beclin-1 and MAVS | [372,373] |
| IRF3 | SeV; VSV | Autophagic degradation of IRF3 by TRIM21-NDP52/Calcoco2 axis | [374] |
| TBK1 | SeV; VSV; poly (I:C) | Autophagic degradation of TBK1 through NEDD4 and NDP52/Calcoco2 | [375] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ke, P.-Y. Crosstalk between Autophagy and RLR Signaling. Cells 2023, 12, 956. https://doi.org/10.3390/cells12060956
AMA Style
Ke P-Y. Crosstalk between Autophagy and RLR Signaling. Cells. 2023; 12(6):956. https://doi.org/10.3390/cells12060956
Chicago/Turabian StyleKe, Po-Yuan. 2023. "Crosstalk between Autophagy and RLR Signaling" Cells 12, no. 6: 956. https://doi.org/10.3390/cells12060956
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.