
Impact of Translocator Protein 18 kDa (TSPO) Deficiency on Mitochondrial Function and the Inflammatory State of Human C20 Microglia Cells
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
2.2. CRISPR/Cas9-Mediated TSPO Knockout
2.3. Western Blotting
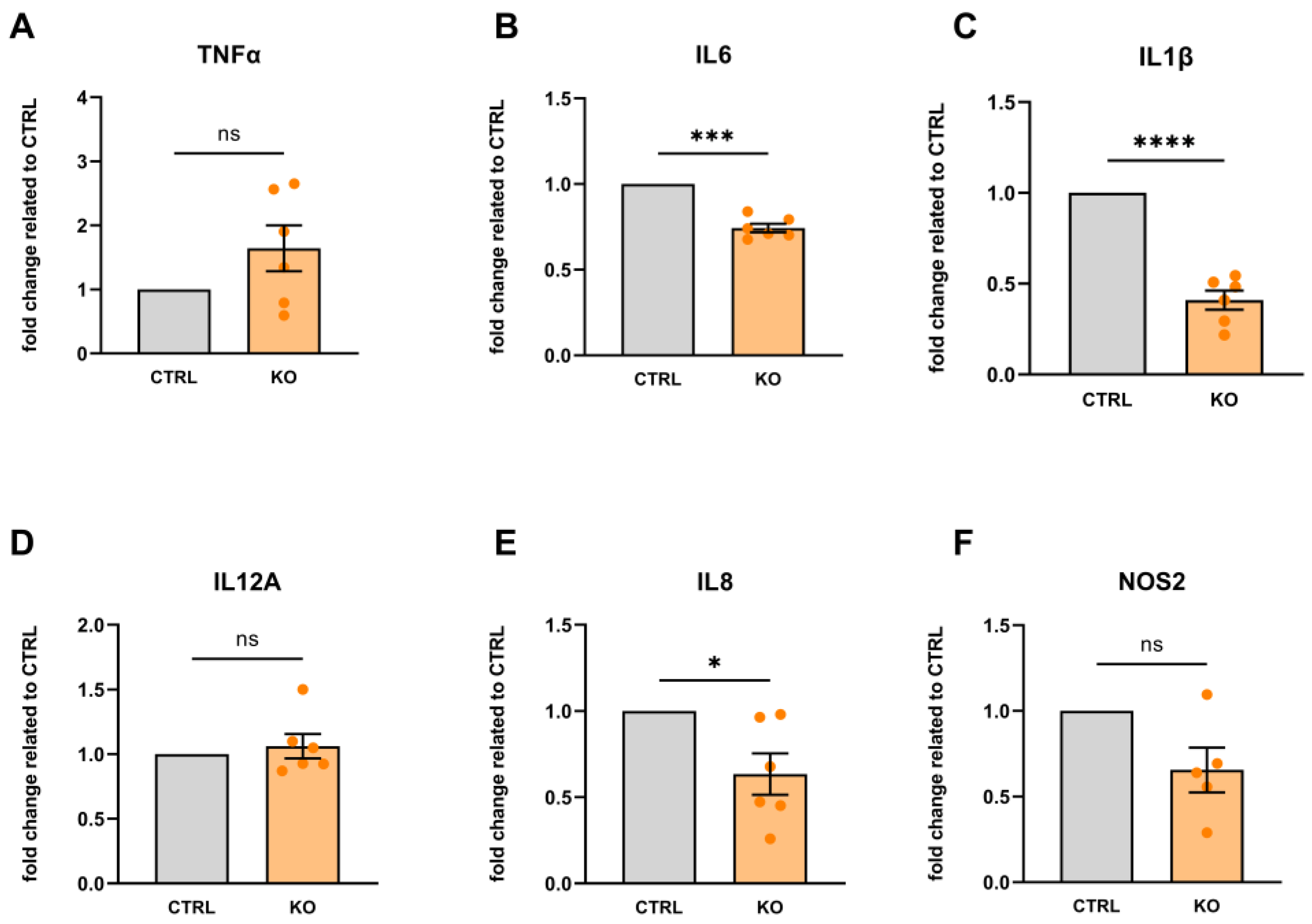
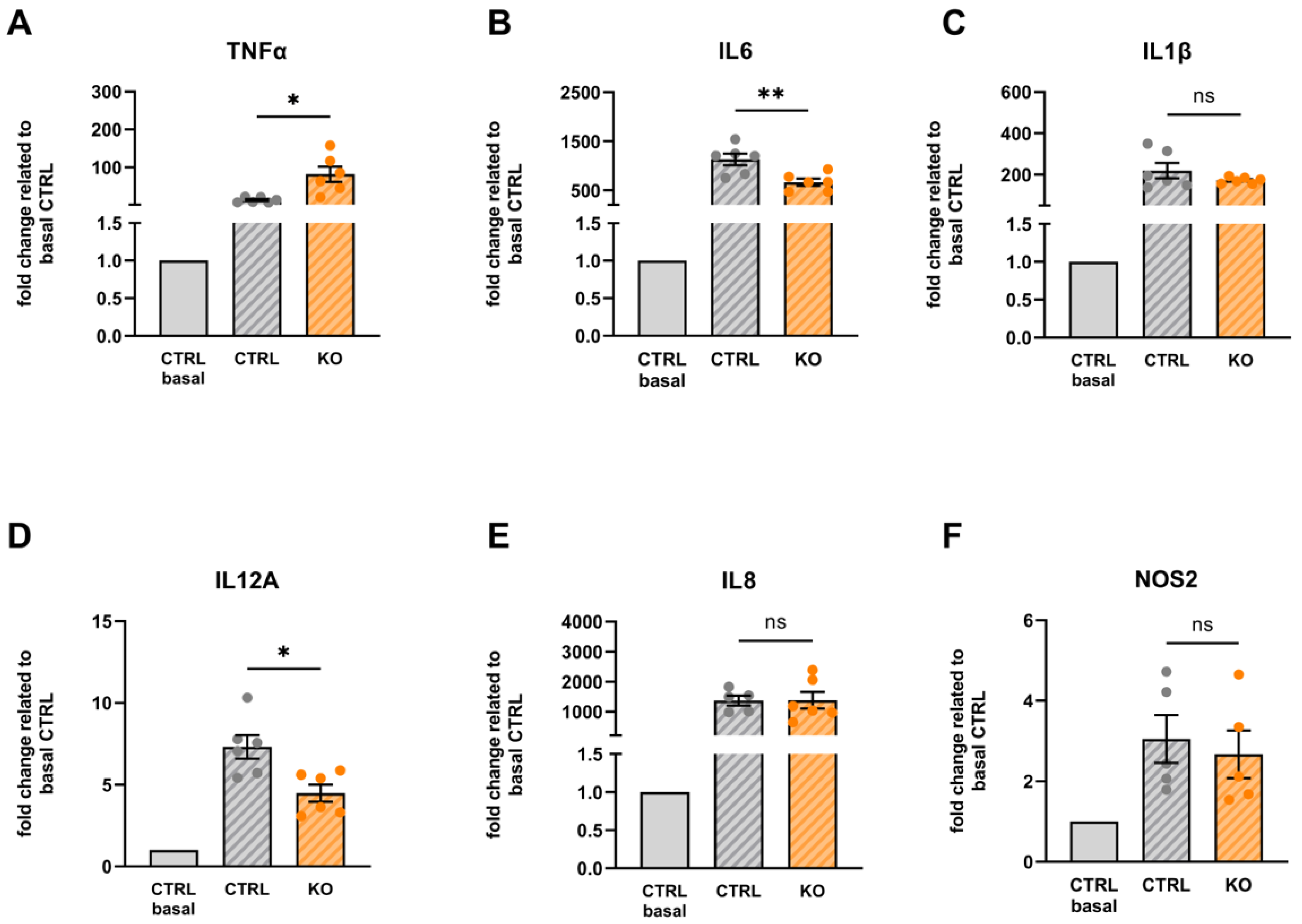
2.4. RNA Isolation, Reverse Transcription, and Quantitative Real-Time RT-PCR of Inflammatory Markers
2.5. mtDNA Copy Number
2.6. ATP Content
2.7. Mitochondrial Membrane Potential
2.8. Cytosolic Ca2+ and Mitochondrial Ca2+ Levels
2.9. Pharmacology/Pro-Inflammatory Stimulation
2.10. Metabolic Profile
2.11. Statistical Analysis
3. Results
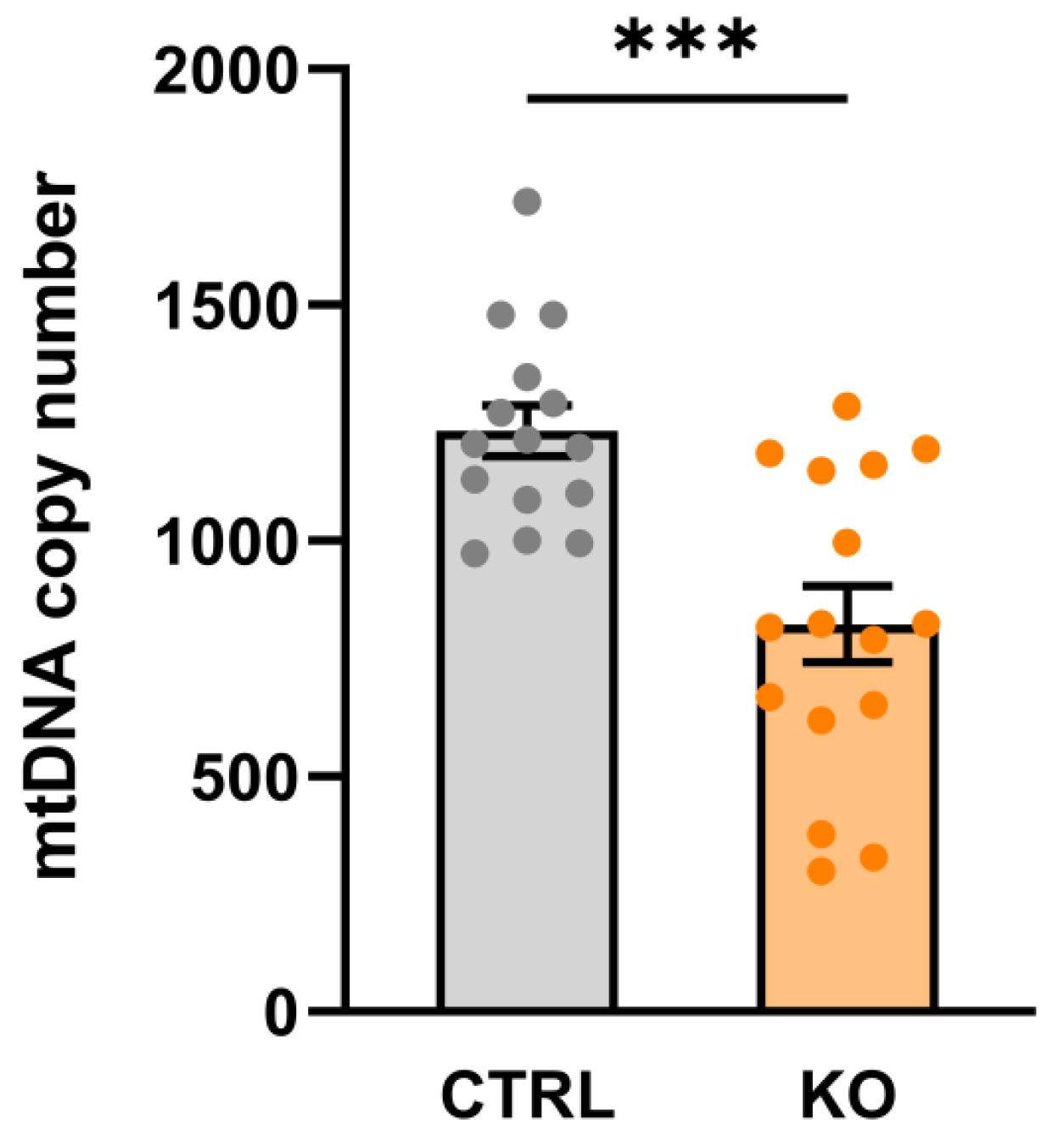
3.1. Mitochondrial DNA Copy Number
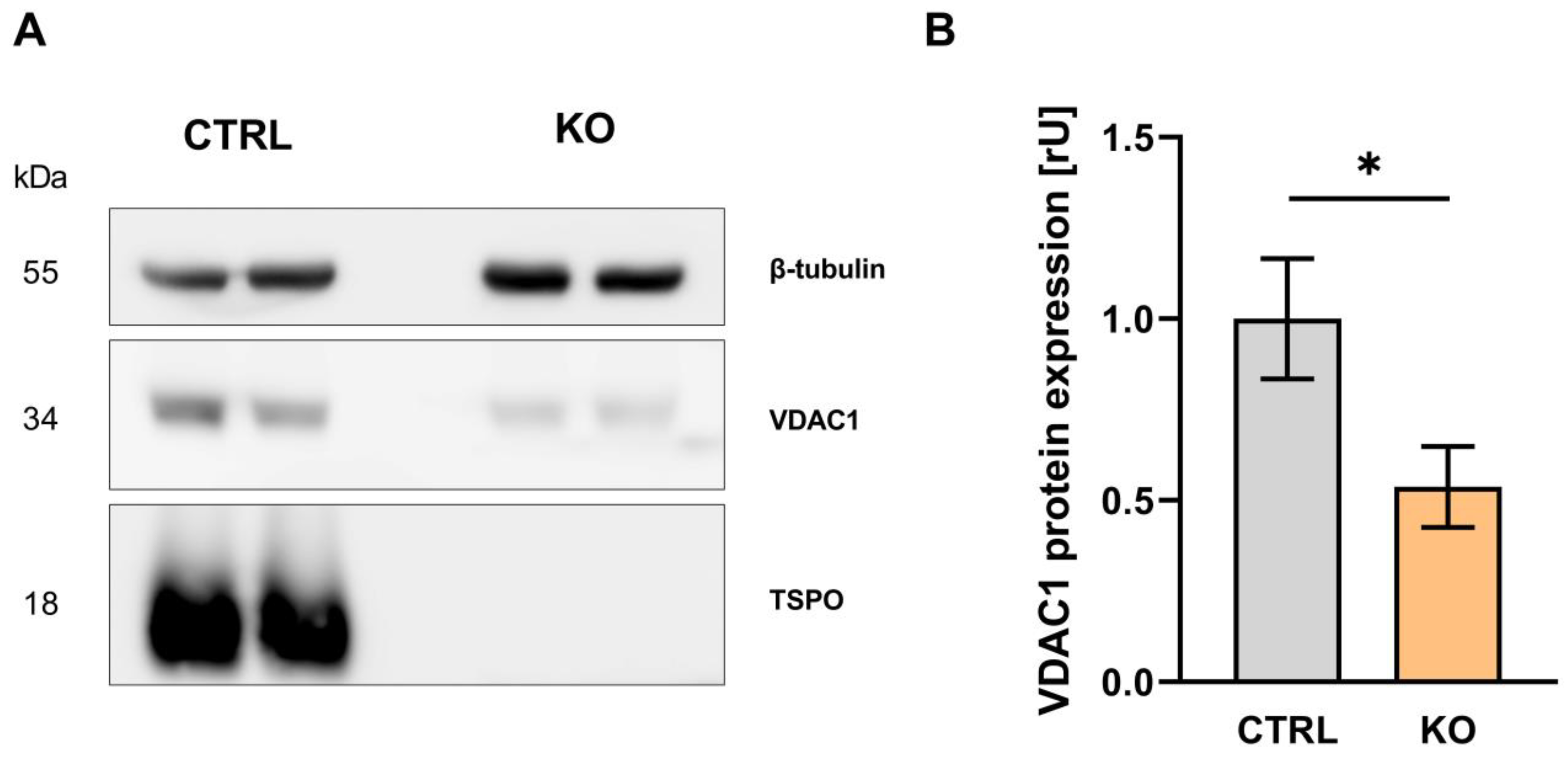
3.2. VDAC1 Expression
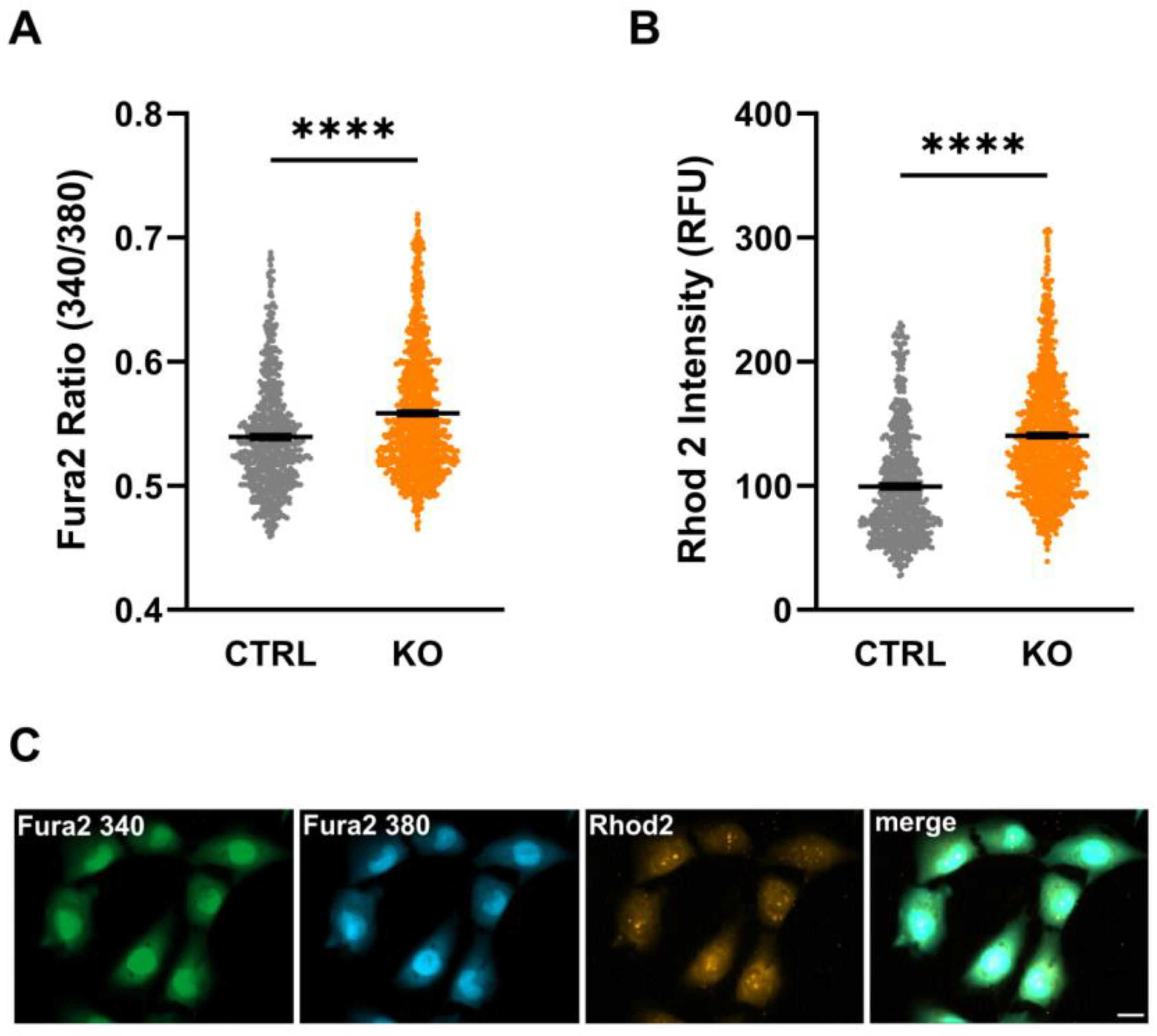
3.3. Intracellular Ca2+ Levels
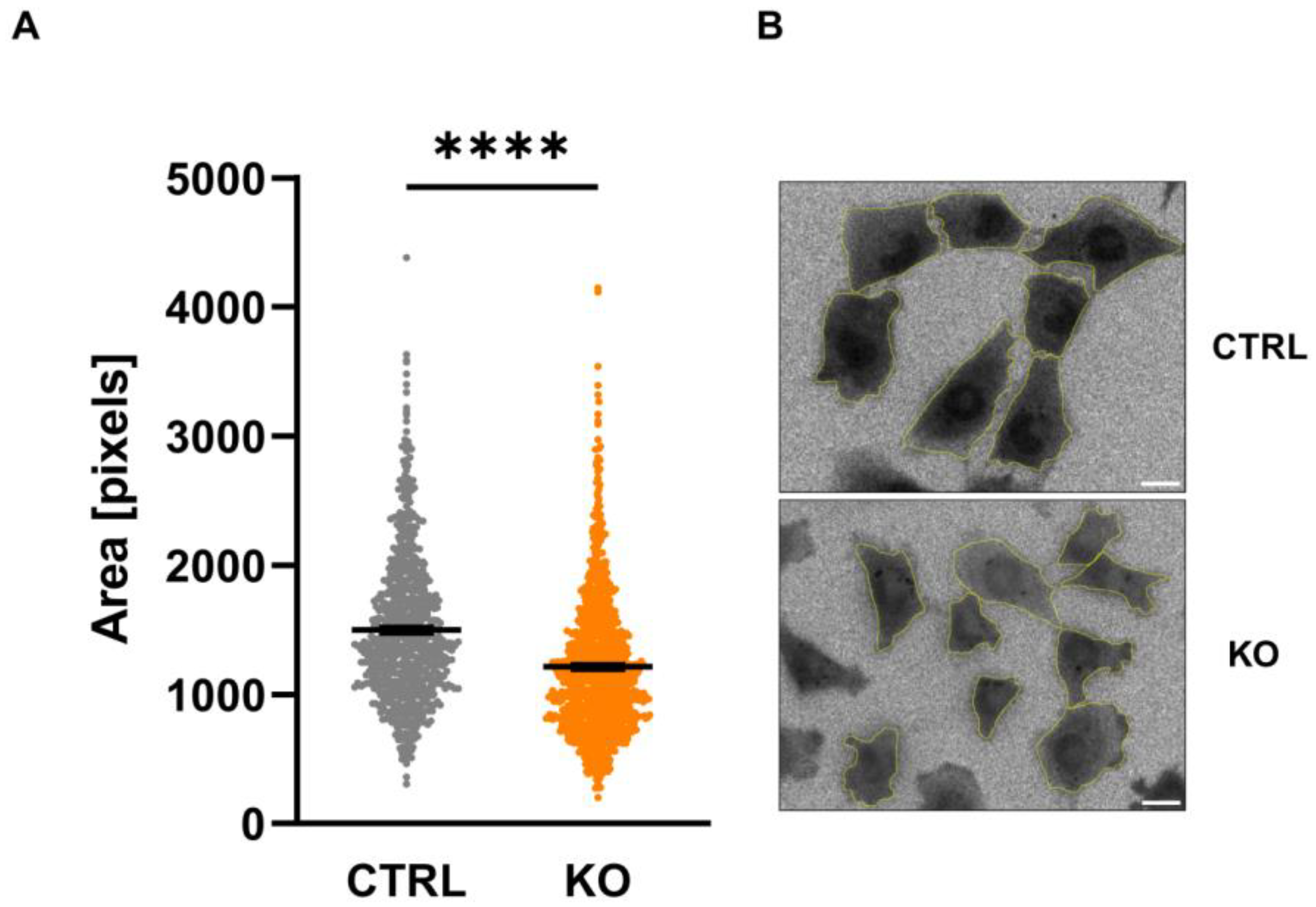
3.4. Size of C20 Microglia Cells
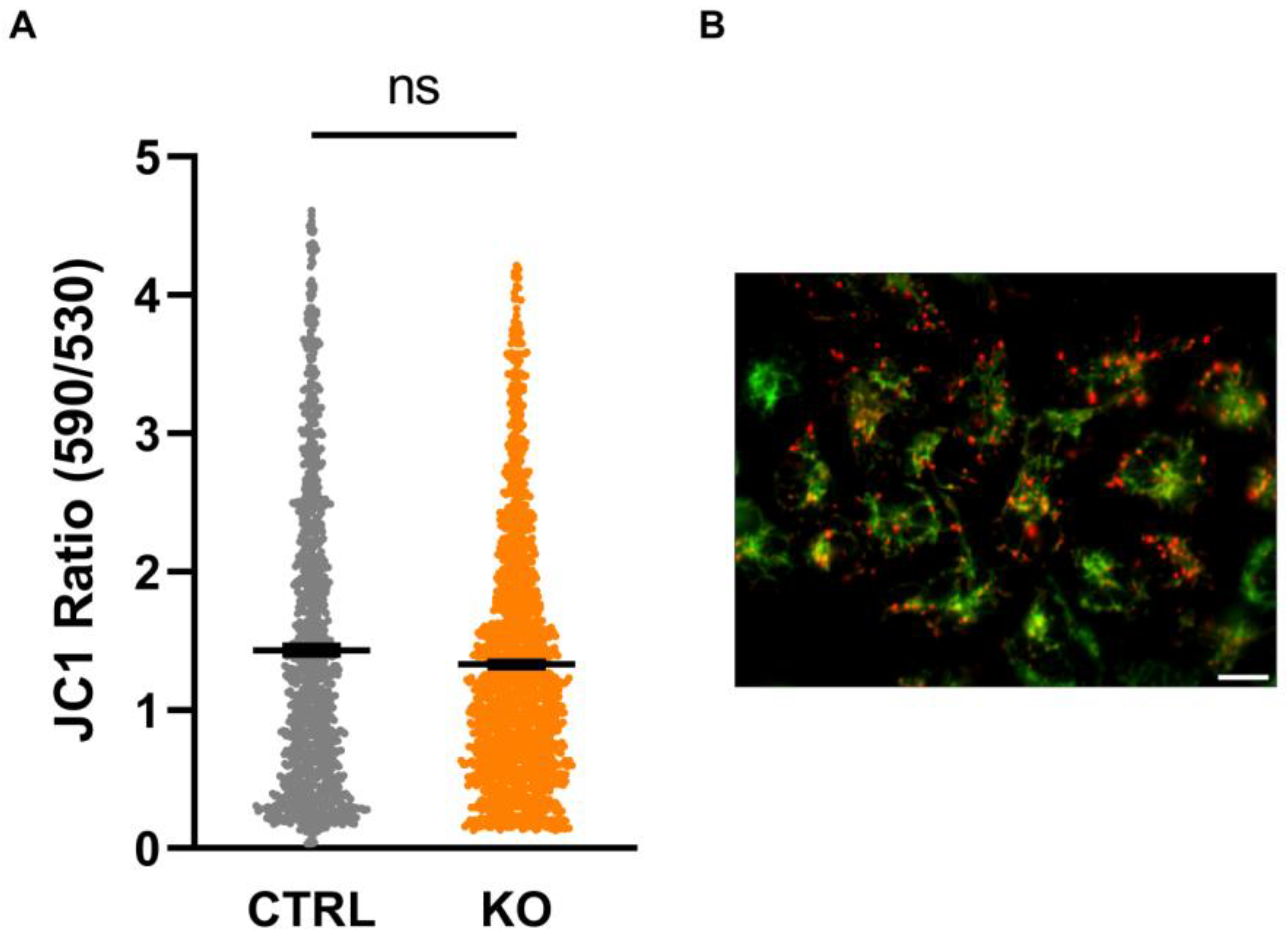
3.5. Mitochondrial Membrane Potential
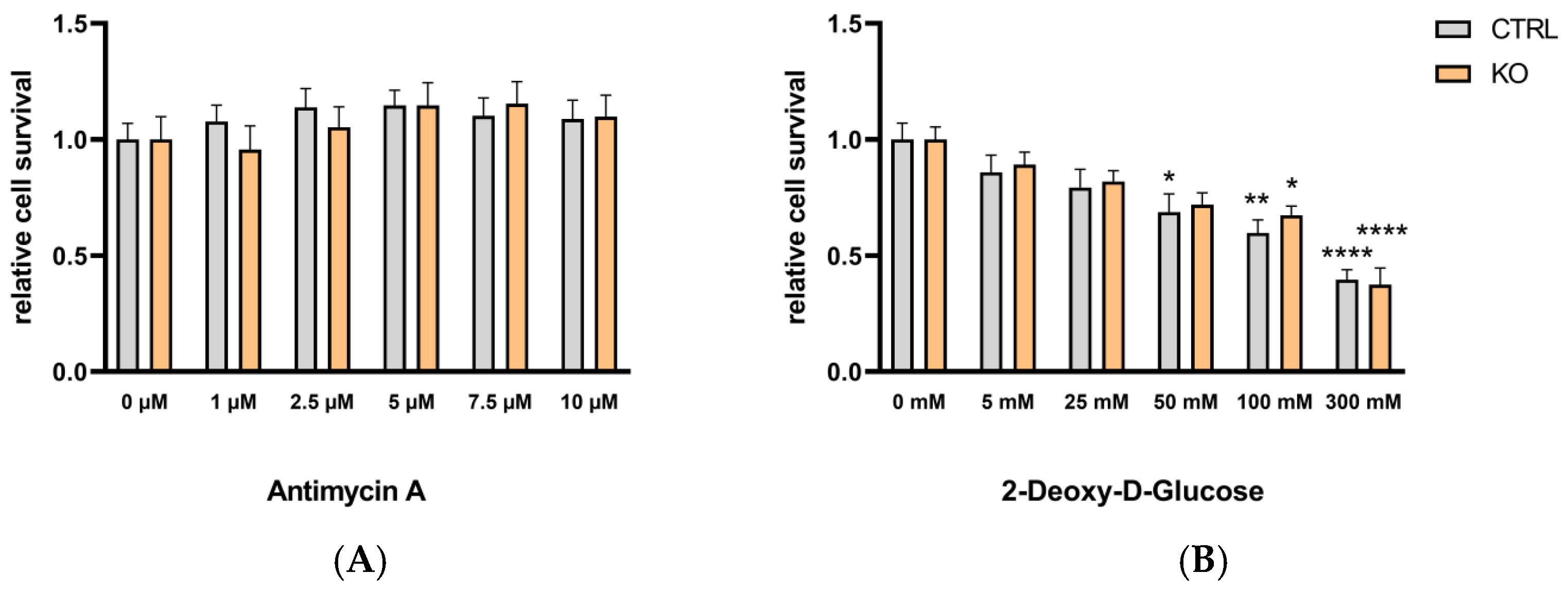
3.6. Metabolic Profile
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Prinz, M.; Priller, J. Microglia and brain macrophages in the molecular age: From origin to neuropsychiatric disease. Nat. Rev. Neurosci. 2014, 15, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Sarma, S.D.; Chatterjee, K.; Dinda, H.; Chatterjee, D.; Sarma, J.D. Cytomorphological and Cytochemical Identification of Microglia. Int. Sch. Res. Not. 2013, 2013, 205431. [Google Scholar]
- Jang, E.; Kim, J.H.; Lee, S.; Kim, J.H.; Seo, J.W.; Jin, M.; Lee, M.G.; Jang, I.S.; Lee, W.H.; Suk, K. Phenotypic polarization of activated astrocytes: The critical role of lipocalin-2 in the classical inflammatory activation of astrocytes. J. Immunol. 2013, 191, 5204–5219. [Google Scholar] [CrossRef] [Green Version]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef]
- Yao, R.; Pan, R.; Shang, C.; Li, X.; Cheng, J.; Xu, J.; Li, Y. Translocator Protein 18 kDa (TSPO) Deficiency Inhibits Microglial Activation and Impairs Mitochondrial Function. Front. Pharmacol. 2020, 11, 986. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Sobotka, K.S.; Joshi, P.; Gressens, P.; Fleiss, B.; Thornton, C.; Mallard, C.; Hagberg, H. Lipopolysaccharide-induced alteration of mitochondrial morphology induces a metabolic shift in microglia modulating the inflammatory response in vitro and in vivo. Glia 2019, 67, 1047–1061. [Google Scholar] [CrossRef] [Green Version]
- Papadopoulos, V.; Baraldi, M.; Guilarte, T.R.; Knudsen, T.B.; Lacapere, J.J.; Lindemann, P.; Norenberg, M.D.; Nutt, D.; Weizman, A.; Zhang, M.R.; et al. Translocator protein (18kDa): New nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol. Sci. 2006, 27, 402–409. [Google Scholar] [CrossRef]
- Cosenza-Nashat, M.; Zhao, M.L.; Suh, H.S.; Morgan, J.; Natividad, R.; Morgello, S.; Lee, S.C. Expression of the translocator protein of 18 kDa by microglia, macrophages and astrocytes based on immunohistochemical localization in abnormal human brain. Neuropathol. Appl. Neurobiol. 2009, 35, 306–328. [Google Scholar] [CrossRef] [Green Version]
- Rupprecht, R.; Papadopoulos, V.; Rammes, G.; Baghai, T.C.; Fan, J.; Akula, N.; Groyer, G.; Adams, D.; Schumacher, M. Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat. Rev. Drug Discov. 2010, 9, 971. [Google Scholar] [CrossRef]
- Rupprecht, R.; Wetzel, C.H.; Dorostkar, M.; Herms, J.; Albert, N.L.; Schwarzbach, J.; Schumacher, M.; Neumann, I.D. Translocator protein (18kDa) TSPO: A new diagnostic or therapeutic target for stress-related disorders? Mol. Psychiatry 2022, 27, 2918–2926. [Google Scholar] [CrossRef]
- Banati, R.B.; Middleton, R.J.; Chan, R.; Hatty, C.R.; Kam, W.W.; Quin, C.; Graeber, M.B.; Parmar, A.; Zahra, D.; Callaghan, P.; et al. Positron emission tomography and functional characterization of a complete PBR/TSPO knockout. Nat. Commun. 2014, 5, 5452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morohaku, K.; Pelton, S.H.; Daugherty, D.J.; Butler, W.R.; Deng, W.; Selvaraj, V. Translocator protein/peripheral benzodiazepine receptor is not required for steroid hormone biosynthesis. Endocrinology 2014, 155, 89–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, L.N.; Morohaku, K.; Manna, P.R.; Pelton, S.H.; Butler, W.R.; Stocco, D.M.; Selvaraj, V. Peripheral benzodiazepine receptor/translocator protein global knock-out mice are viable with no effects on steroid hormone biosynthesis. J. Biol. Chem. 2014, 289, 27444–27454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadopoulos, V.; Fan, J.; Zirkin, B. Translocator protein (18 kDa): An update on its function in steroidogenesis. J. Neuroendocrinol. 2018, 30, e12500. [Google Scholar] [CrossRef]
- Bader, S.; Wolf, L.; Milenkovic, V.M.; Gruber, M.; Nothdurfter, C.; Rupprecht, R.; Wetzel, C.H. Differential effects of TSPO ligands on mitochondrial function in mouse microglia cells. Psychoneuroendocrinology 2019, 106, 65–76. [Google Scholar] [CrossRef]
- Milenkovic, V.M.; Slim, D.; Bader, S.; Koch, V.; Heinl, E.S.; Alvarez-Carbonell, D.; Nothdurfter, C.; Rupprecht, R.; Wetzel, C.H. CRISPR-Cas9 Mediated TSPO Gene Knockout alters Respiration and Cellular Metabolism in Human Primary Microglia Cells. Int. J. Mol. Sci. 2019, 20, 3359. [Google Scholar] [CrossRef] [Green Version]
- Germelli, L.; Da Pozzo, E.; Giacomelli, C.; Tremolanti, C.; Marchetti, L.; Wetzel, C.H.; Barresi, E.; Taliani, S.; Da Settimo, F.; Martini, C.; et al. De novo neurosteroidogenesis in human microglia: Involvement of the 18 kDa translocator protein. Int. J. Mol. Sci. 2021, 22, 3115. [Google Scholar] [CrossRef]
- Wolf, L.; Bauer, A.; Melchner, D.; Hallof-Buestrich, H.; Stoertebecker, P.; Haen, E.; Kreutz, M.; Sarubin, N.; Milenkovic, V.M.; Wetzel, C.H.; et al. Enhancing neurosteroid synthesis--relationship to the pharmacology of translocator protein (18 kDa) (TSPO) ligands and benzodiazepines. Pharmacopsychiatry 2015, 48, 72. [Google Scholar] [CrossRef]
- Owen, D.R.; Fan, J.; Campioli, E.; Venugopal, S.; Midzak, A.; Daly, E.; Harlay, A.; Issop, L.; Libri, V.; Kalogiannopoulou, D.; et al. TSPO mutations in rats and a human polymorphism impair the rate of steroid synthesis. Biochem. J. 2017, 474, 3985. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.J.; Middleton, R.J.; Kam, W.W.; Chin, D.Y.; Hatty, C.R.; Chan, R.H.; Banati, R.B. Functional gains in energy and cell metabolism after TSPO gene insertion. Cell Cycle 2017, 16, 436–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, L.N.; Zhao, A.H.; Hussein, M.; Stocco, D.M.; Selvaraj, V. Translocator Protein (TSPO) Affects Mitochondrial Fatty Acid Oxidation in Steroidogenic Cells. Endocrinology 2016, 157, 1110–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatliff, J.; East, D.; Crosby, J.; Abeti, R.; Harvey, R.; Craigen, W.; Parker, P.; Campanella, M. TSPO interacts with VDAC1 and triggers a ROS-mediated inhibition of mitochondrial quality control. Autophagy 2014, 10, 2279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatliff, J.; East, D.A.; Singh, A.; Alvarez, M.S.; Frison, M.; Matic, I.; Ferraina, C.; Sampson, N.; Turkheimer, F.; Campanella, M. A role for TSPO in mitochondrial Ca(2+) homeostasis and redox stress signaling. Cell Death Dis. 2017, 8, e2896. [Google Scholar] [CrossRef] [Green Version]
- Papadopoulos, V.; Lecanu, L. Translocator protein (18 kDa) TSPO: An emerging therapeutic target in neurotrauma. Exp. Neurol. 2009, 219, 53–57. [Google Scholar] [CrossRef] [Green Version]
- Shoshan-Barmatz, V.; Pittala, S.; Mizrachi, D. VDAC1 and the TSPO: Expression, interactions, and associated functions in health and disease states. Int. J. Mol. Sci. 2019, 20, 3348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, R.L.; Buck, D.J.; McCracken, K.; Cox, G.W.; Das, S. Interleukin-1β-induced inflammatory signaling in C20 human microglial cells. Neuroimmunol. Neuroinflamm. 2018, 5, 50. [Google Scholar] [CrossRef]
- Garcia-Mesa, Y.; Jay, T.R.; Checkley, M.A.; Luttge, B.; Dobrowolski, C.; Valadkhan, S.; Landreth, G.E.; Karn, J.; Alvarez-Carbonell, D. Immortalization of primary microglia: A new platform to study HIV regulation in the central nervous system. J. Neurovirol. 2017, 23, 47–66. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Milenkovic, V.M.; Bader, S.; Sudria-Lopez, D.; Siebert, R.; Brandl, C.; Nothdurfter, C.; Weber, B.H.F.; Rupprecht, R.; Wetzel, C.H. Effects of genetic variants in the TSPO gene on protein structure and stability. PLoS ONE 2018, 13, e0195627. [Google Scholar] [CrossRef] [PubMed]
- Arif, T.; Krelin, Y.; Shoshan-Barmatz, V. Reducing VDAC1 expression induces a non-apoptotic role for pro-apoptotic proteins in cancer cell differentiation. Biochim. Biophys. Acta 2016, 1857, 1228–1242. [Google Scholar] [CrossRef] [PubMed]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gellerich, F.N.; Gizatullina, Z.; Trumbeckaite, S.; Nguyen, H.P.; Pallas, T.; Arandarcikaite, O.; Vielhaber, S.; Seppet, E.; Striggow, F. The regulation of OXPHOS by extramitochondrial calcium. Biochim. Biophys. Acta 2010, 1797, 1018–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betlazar, C.; Middleton, R.J.; Banati, R.; Liu, G.J. The Translocator Protein (TSPO) in Mitochondrial Bioenergetics and Immune Processes. Cells 2020, 9, 512. [Google Scholar] [CrossRef] [Green Version]
- Kelly, R.D.; Mahmud, A.; McKenzie, M.; Trounce, I.A.; St John, J.C. Mitochondrial DNA copy number is regulated in a tissue specific manner by DNA methylation of the nuclear-encoded DNA polymerase gamma A. Nucleic Acids Res. 2012, 40, 10124–10138. [Google Scholar] [CrossRef] [Green Version]
- Jeng, J.Y.; Yeh, T.S.; Lee, J.W.; Lin, S.H.; Fong, T.H.; Hsieh, R.H. Maintenance of mitochondrial DNA copy number and expression are essential for preservation of mitochondrial function and cell growth. J. Cell Biochem. 2008, 103, 347–357. [Google Scholar] [CrossRef]
- Castellani, C.A.; Longchamps, R.J.; Sun, J.; Guallar, E.; Arking, D.E. Thinking outside the nucleus: Mitochondrial DNA copy number in health and disease. Mitochondrion 2020, 53, 214–223. [Google Scholar] [CrossRef]
- Yang, S.; Qin, C.; Hu, Z.W.; Zhou, L.Q.; Yu, H.H.; Chen, M.; Bosco, D.B.; Wang, W.; Wu, L.J.; Tian, D.S. Microglia reprogram metabolic profiles for phenotype and function changes in central nervous system. Neurobiol. Dis. 2021, 152, 105290. [Google Scholar] [CrossRef]
- Ghosh, S.; Castillo, E.; Frias, E.S.; Swanson, R.A. Bioenergetic regulation of microglia. Glia 2018, 66, 1200–1212. [Google Scholar] [CrossRef]
- Missiroli, S.; Perrone, M.; Genovese, I.; Pinton, P.; Giorgi, C. Cancer metabolism and mitochondria: Finding novel mechanisms to fight tumours. EBioMedicine 2020, 59, 102943. [Google Scholar] [CrossRef] [PubMed]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Wang, D.; Wang, H.; Cai, M.; Li, C.; Zhang, X.; Chen, H.; Hu, Y.; Zhang, X.; Ying, M.; et al. TSPO deficiency induces mitochondrial dysfunction, leading to hypoxia, angiogenesis and a growth-promoting metabolic shift towards glycolysis in glioblastoma. Neuro Oncol. 2019, 22, 240–252. [Google Scholar] [CrossRef] [PubMed]
- Pozzo, E.D.; Tremolanti, C.; Costa, B.; Giacomelli, C.; Milenkovic, V.M.; Bader, S.; Wetzel, C.H.; Rupprecht, R.; Taliani, S.; Settimo, F.D.; et al. Microglial Pro-Inflammatory and Anti-Inflammatory Phenotypes Are Modulated by Translocator Protein Activation. Int. J. Mol. Sci. 2019, 20, 4467. [Google Scholar] [CrossRef] [Green Version]
- Scheidereit, C. IkappaB kinase complexes: Gateways to NF-kappaB activation and transcription. Oncogene 2006, 25, 6685–6705. [Google Scholar] [CrossRef] [Green Version]
- Bae, K.R.; Shim, H.J.; Balu, D.; Kim, S.R.; Yu, S.W. Translocator protein 18 kDa negatively regulates inflammation in microglia. J. Neuroimmune Pharmacol. 2014, 9, 424–437. [Google Scholar] [CrossRef]
- Owen, D.R.; Narayan, N.; Wells, L.; Healy, L.; Smyth, E.; Rabiner, E.A.; Galloway, D.; Williams, J.B.; Lehr, J.; Mandhair, H.; et al. Pro-inflammatory activation of primary microglia and macrophages increases 18 kDa translocator protein expression in rodents but not humans. J. Cereb. Blood Flow Metab. 2017, 37, 2679–2690. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, A.; Kann, O.; Ohlemeyer, C.; Hanisch, U.K.; Kettenmann, H. Elevation of basal intracellular calcium as a central element in the activation of brain macrophages (microglia): Suppression of receptor-evoked calcium signaling and control of release function. J. Neurosci. 2003, 23, 4410–4419. [Google Scholar] [CrossRef] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Forward Primer (5′-3′) | Reverse Primer (5′-3′) |
|---|---|---|
| TSPO-ex1 | GAGGTGGCTTTGAGGAGTGA | GCAGGGTTGTCCAAGTTTTC |
| TSPO-ex2 | CTGGAAATGCGTTCACTCAG | GCCTGGAGAAGACCCTCTGT |
| TSPO-ex3 | GAAGCACTGCCAATGTGCTA | GCTTCGTGTGGGTTTTCCTA |
| TSPO-ex4 | AGTTGGGCAGTGGGACAG | GCAGATCCTGCAGAGACGA |
| TNFA | CCCCAGGGACCTCTCTCTAA | CAGCTTGAGGGTTTGCTACA |
| IL1B | CTGTCCTGCGTGTTGAAAGA | TGAAGACAAATCGCTTTTCCA |
| TSPO IL6 IL12A NOS2 CXCL8 | TCTTTGGTGCCCGACAAAT CTCAGCCCTGAGAAAGGAGA CATGCTGGCAGTTATTGATGA GTATCCTGGAGCGAGTGGTG TGCGCCAACACAGAAATTAT | GGTACCAGGCCACGGTAGT AGGTTGTTTTCTGCCAGTGC TCAAGGGAGGATTTTTGTGG GACCCAGTAGCTGCCACTCT TGAATTCTCAGCCCTCTTCAA |
| HPRT1 Mt-TL1 B2M | TTGCTTTCCTTGGTCAGGCA CACCCAAGAACAGGGTTTGT TGCTGTCTCCATGTTTGATGTATCT | ATCCAACACTTCGTGGGGTC TGGCCATGGGTATGTTGTTA TCTCTGCTCCCCACCTCTAAGT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bader, S.; Würfel, T.; Jahner, T.; Nothdurfter, C.; Rupprecht, R.; Milenkovic, V.M.; Wetzel, C.H. Impact of Translocator Protein 18 kDa (TSPO) Deficiency on Mitochondrial Function and the Inflammatory State of Human C20 Microglia Cells. Cells 2023, 12, 954. https://doi.org/10.3390/cells12060954
Bader S, Würfel T, Jahner T, Nothdurfter C, Rupprecht R, Milenkovic VM, Wetzel CH. Impact of Translocator Protein 18 kDa (TSPO) Deficiency on Mitochondrial Function and the Inflammatory State of Human C20 Microglia Cells. Cells. 2023; 12(6):954. https://doi.org/10.3390/cells12060954
Chicago/Turabian StyleBader, Stefanie, Thea Würfel, Tatjana Jahner, Caroline Nothdurfter, Rainer Rupprecht, Vladimir M. Milenkovic, and Christian H. Wetzel. 2023. "Impact of Translocator Protein 18 kDa (TSPO) Deficiency on Mitochondrial Function and the Inflammatory State of Human C20 Microglia Cells" Cells 12, no. 6: 954. https://doi.org/10.3390/cells12060954