
Understanding the Combined Effects of High Glucose Induced Hyper-Osmotic Stress and Oxygen Tension in the Progression of Tumourigenesis: From Mechanism to Anti-Cancer Therapeutics
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines, Reagents
2.2. Cell Culture Treatments with Normal, High-Glucose and Hyper-Osmotic Equivalents in Normoxia and Hypoxia Conditions
2.3. Confluence Analysis
2.4. Floating Cell Count with Trypan Blue
2.5. Proximity Ligation Assay (PLA)
2.6. Immunohistochemistry on Tissue Microarrays
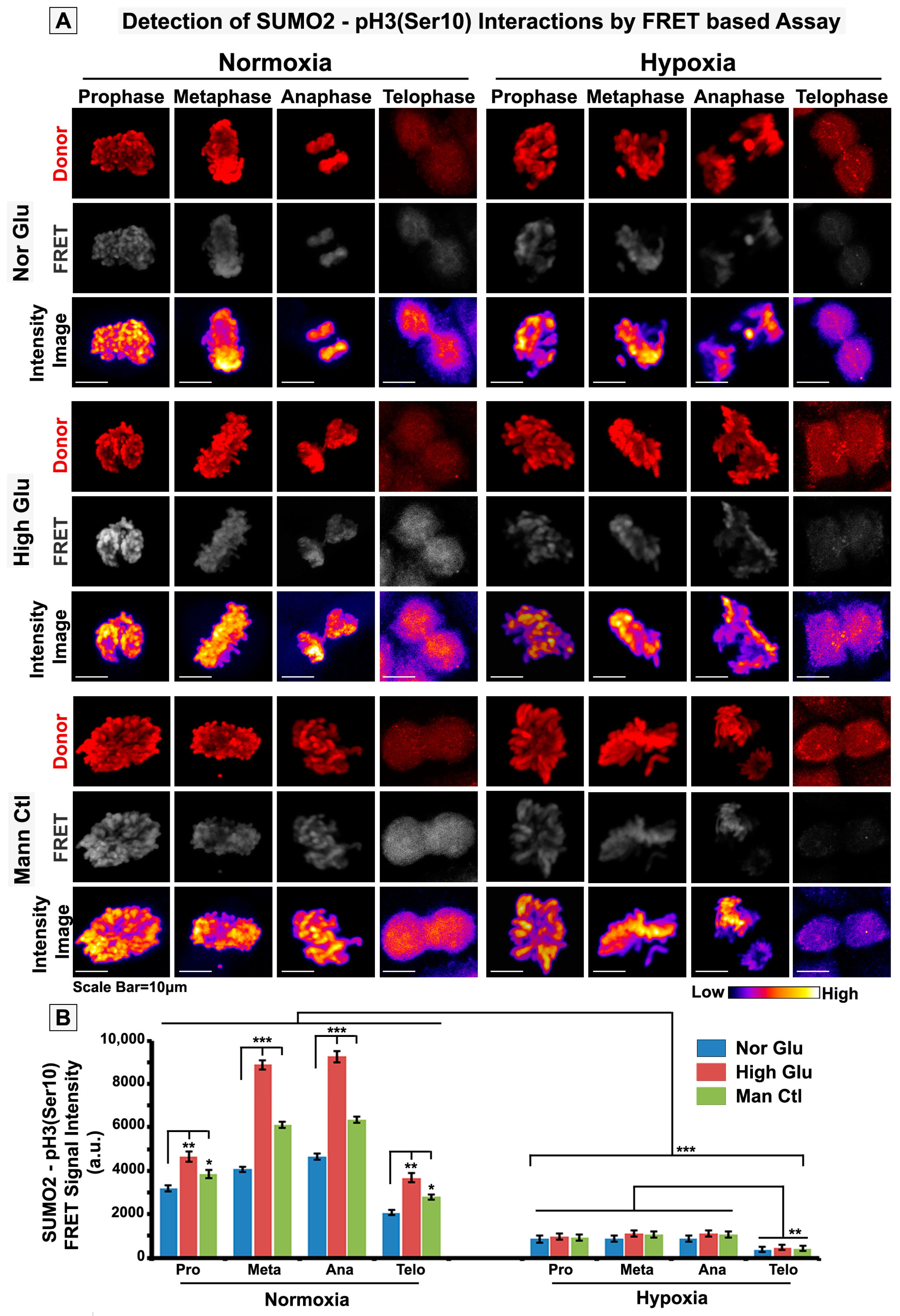
2.7. FRET Analysis
2.8. Three-Dimensional Pellet Cultures and Drug Treatments
2.9. Image Analysis
2.10. Statistical Analysis
3. Results
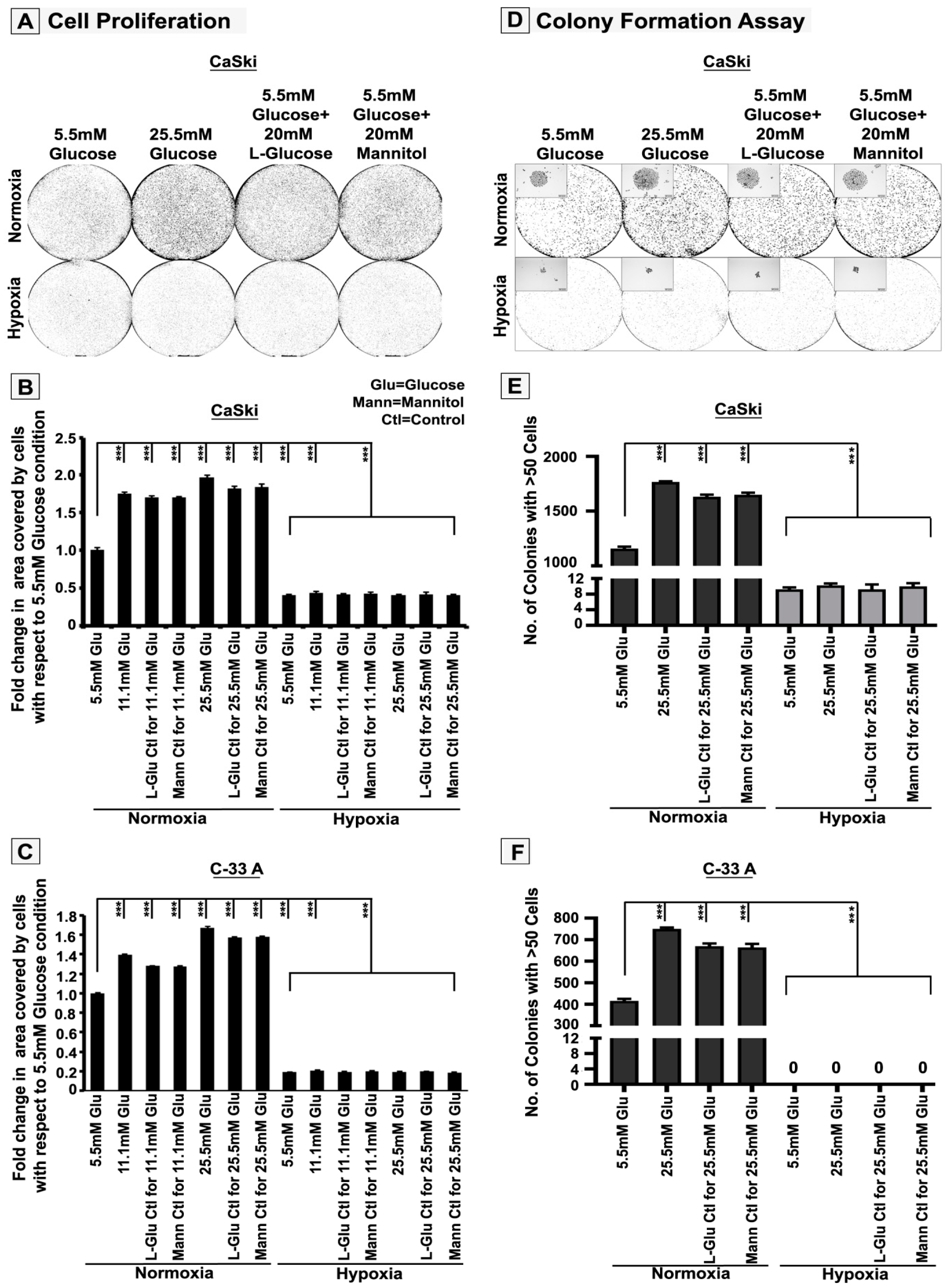
3.1. High-Glucose-Induced Hyper-Osmotic Stress Drives Tumour Cell Proliferation under Normoxia
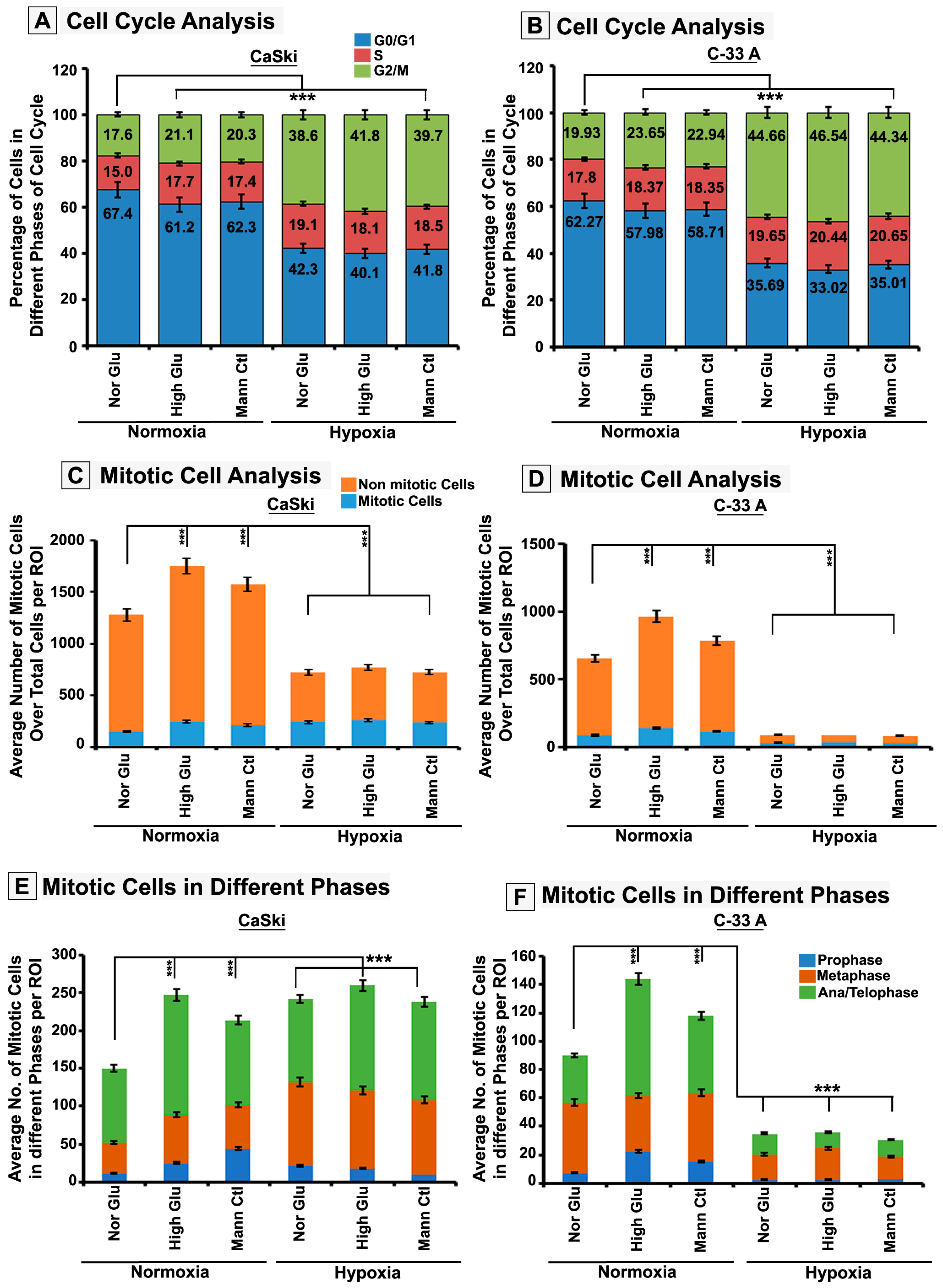
3.2. Hypoxia Retards High-Glucose Hyper-Osmotic-Stress-Induced Tumour Cell Proliferation by Triggering G2/M Cell Cycle Arrest
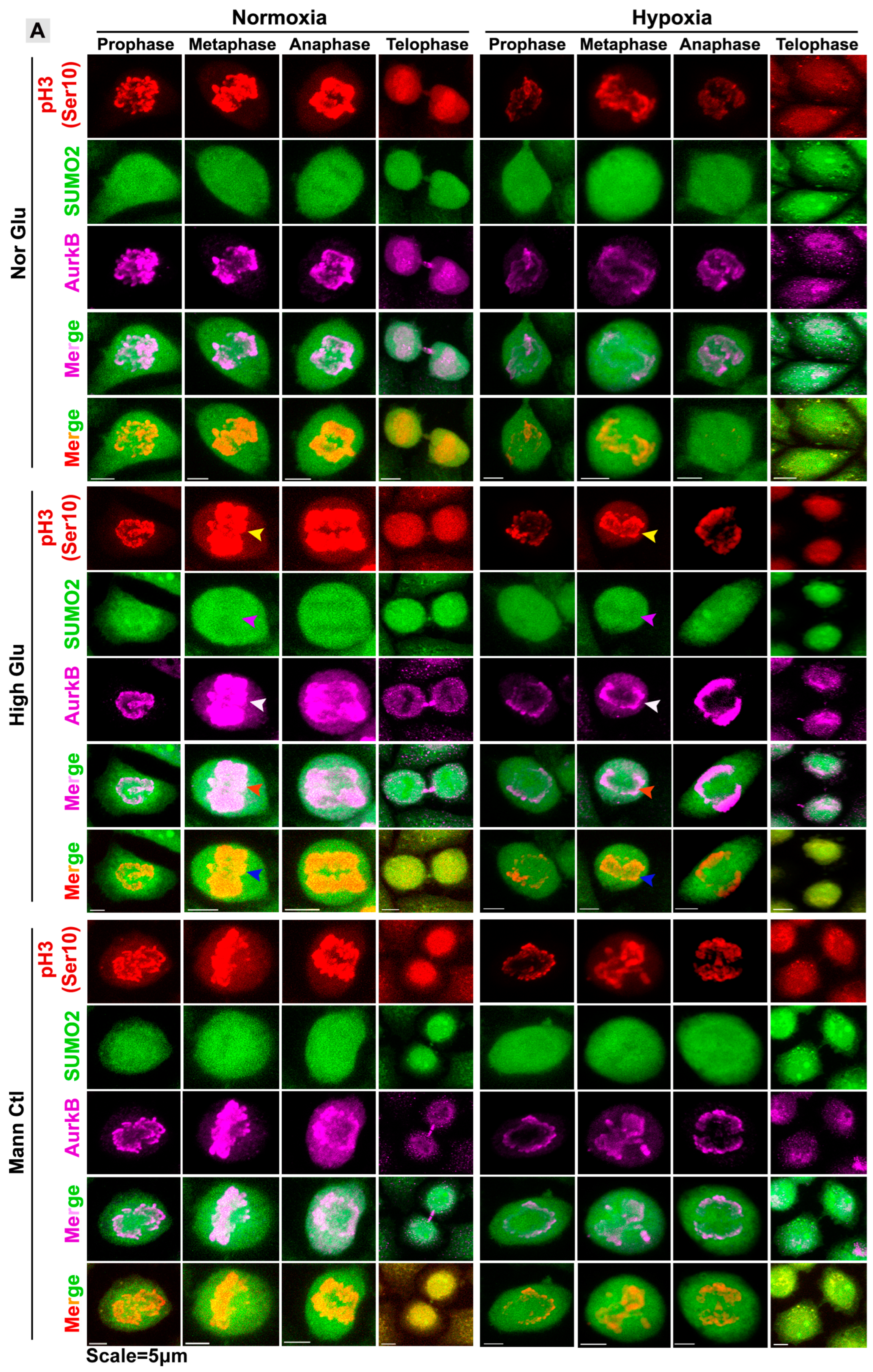
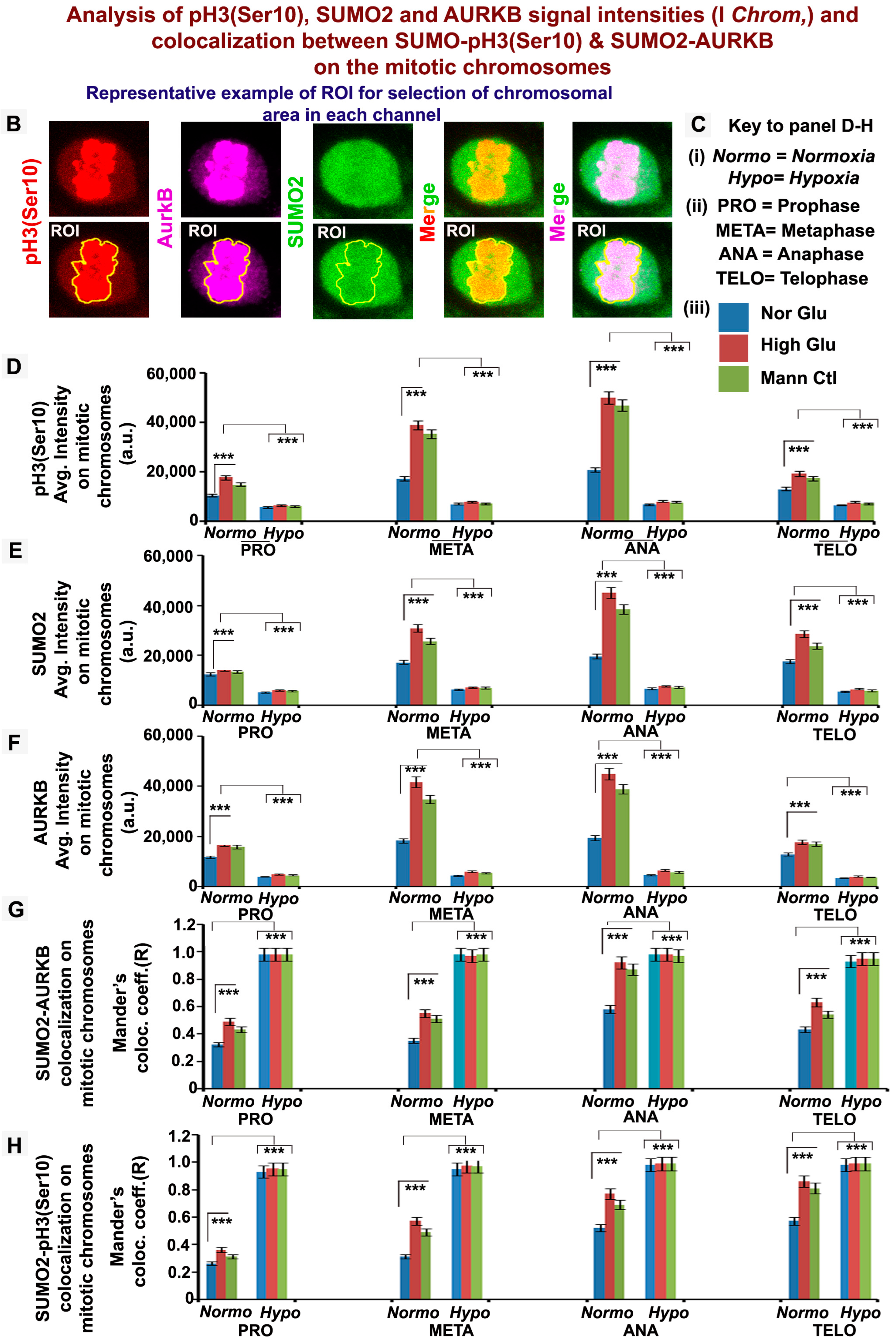
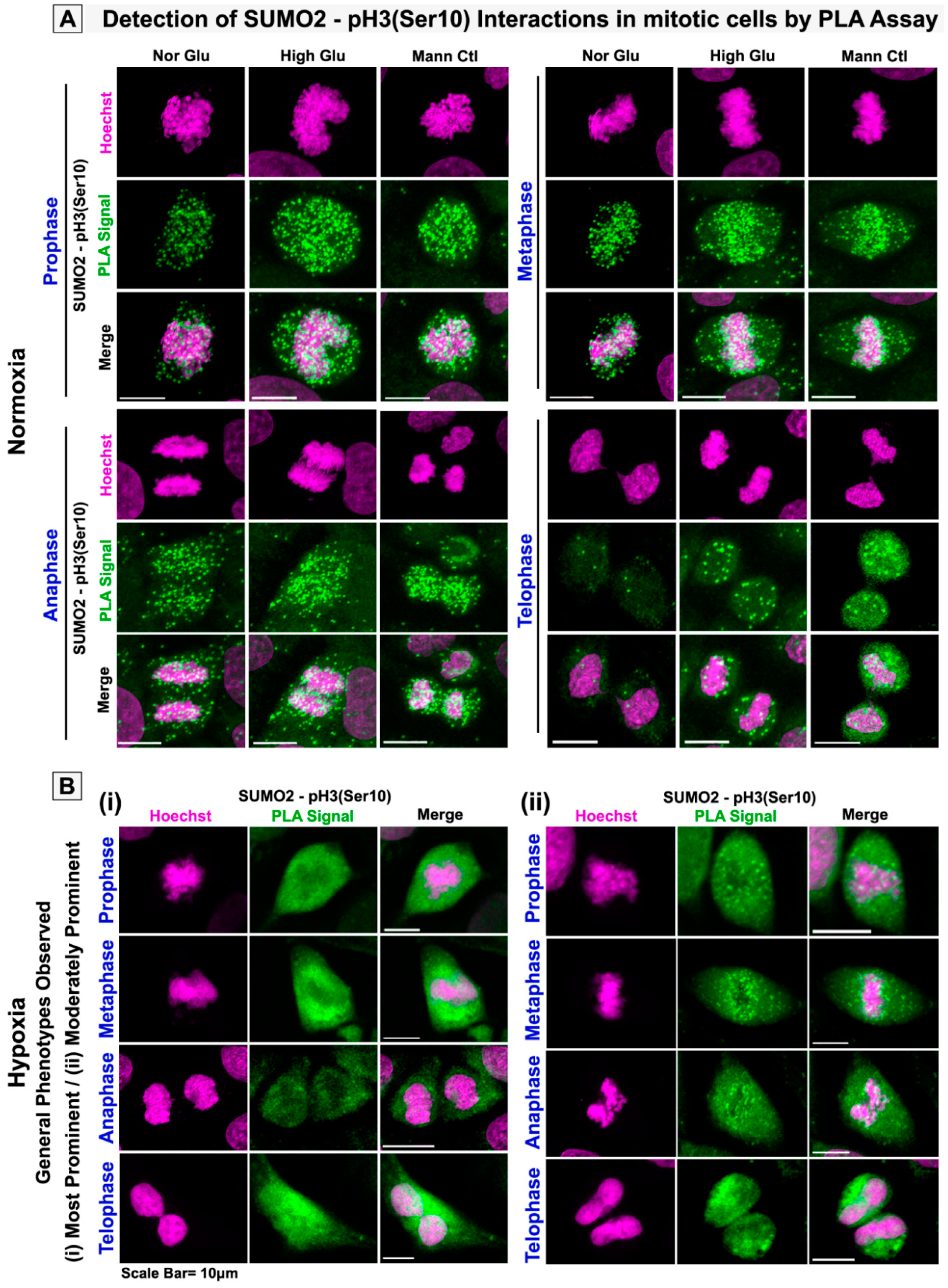
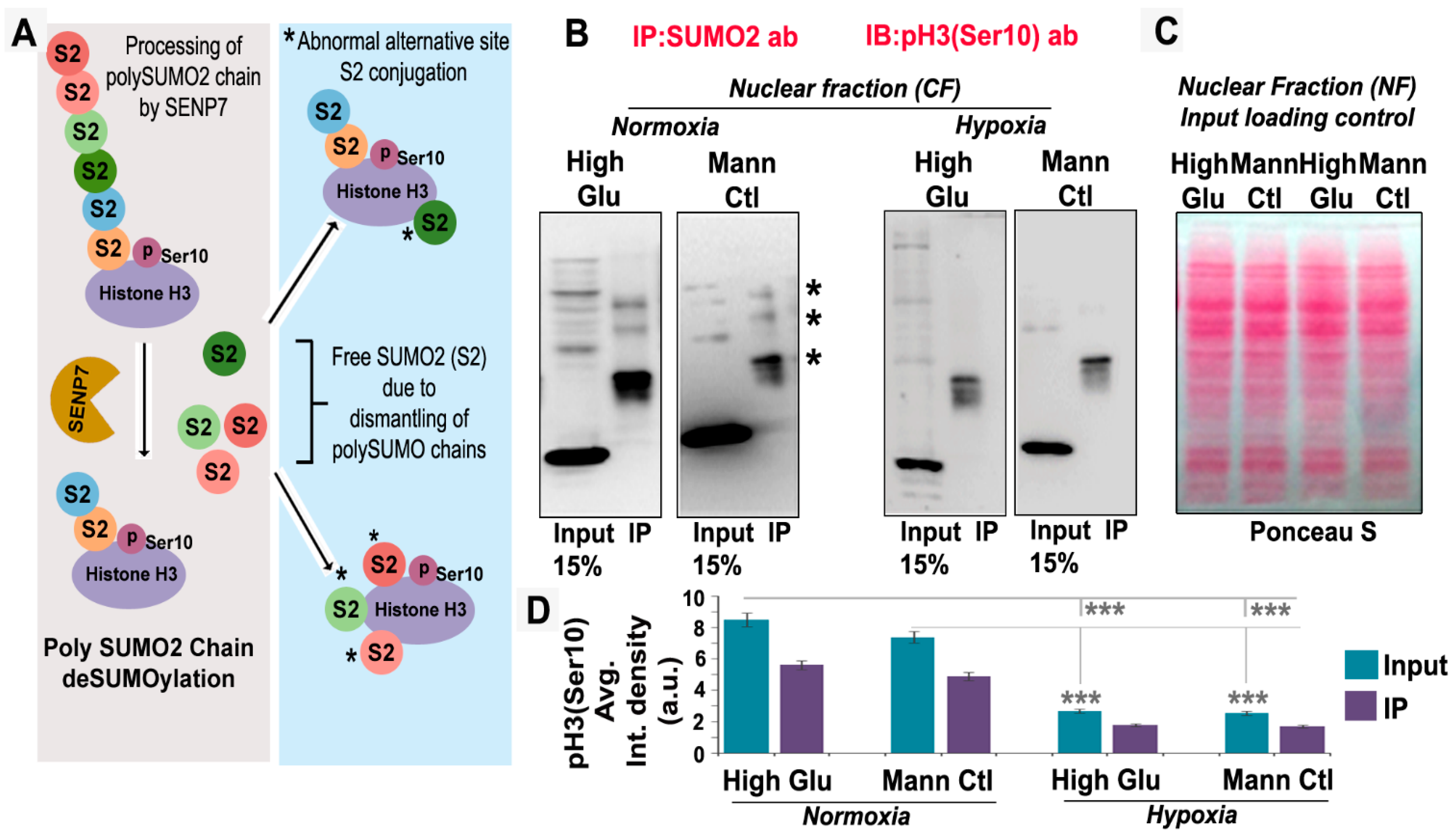
3.3. pH3(Ser10), a Master Regulator of Mitosis, Is a Target of SUMO2 and Is Abnormally SUMOylated under Acute Hypoxic Conditions
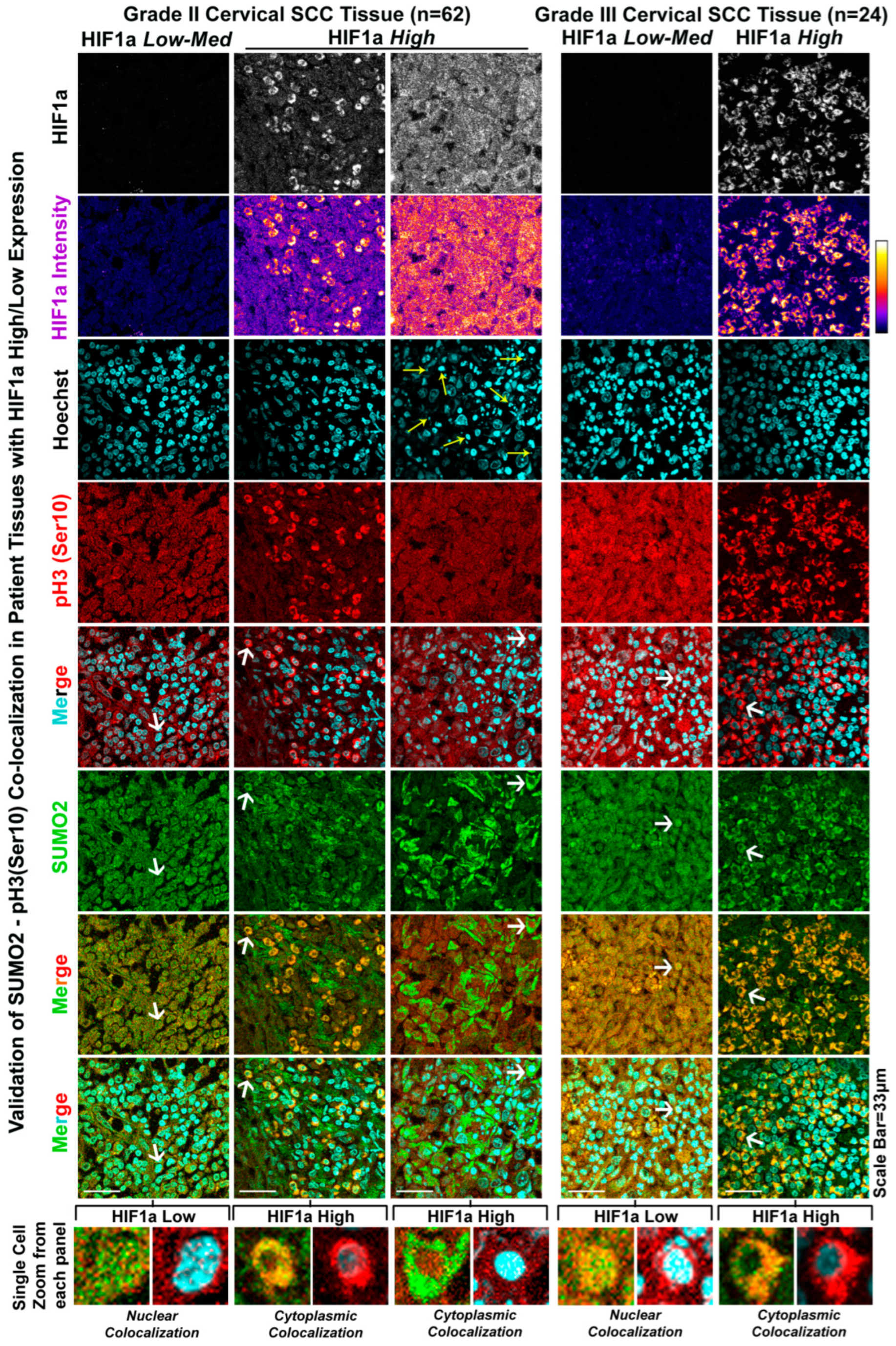
3.4. The Patient Tumour Tissue Array Validates the Extensive Cytoplasmic Sequestration of SUMO2-Modified pH3(Ser10) in Highly Hypoxic Regions
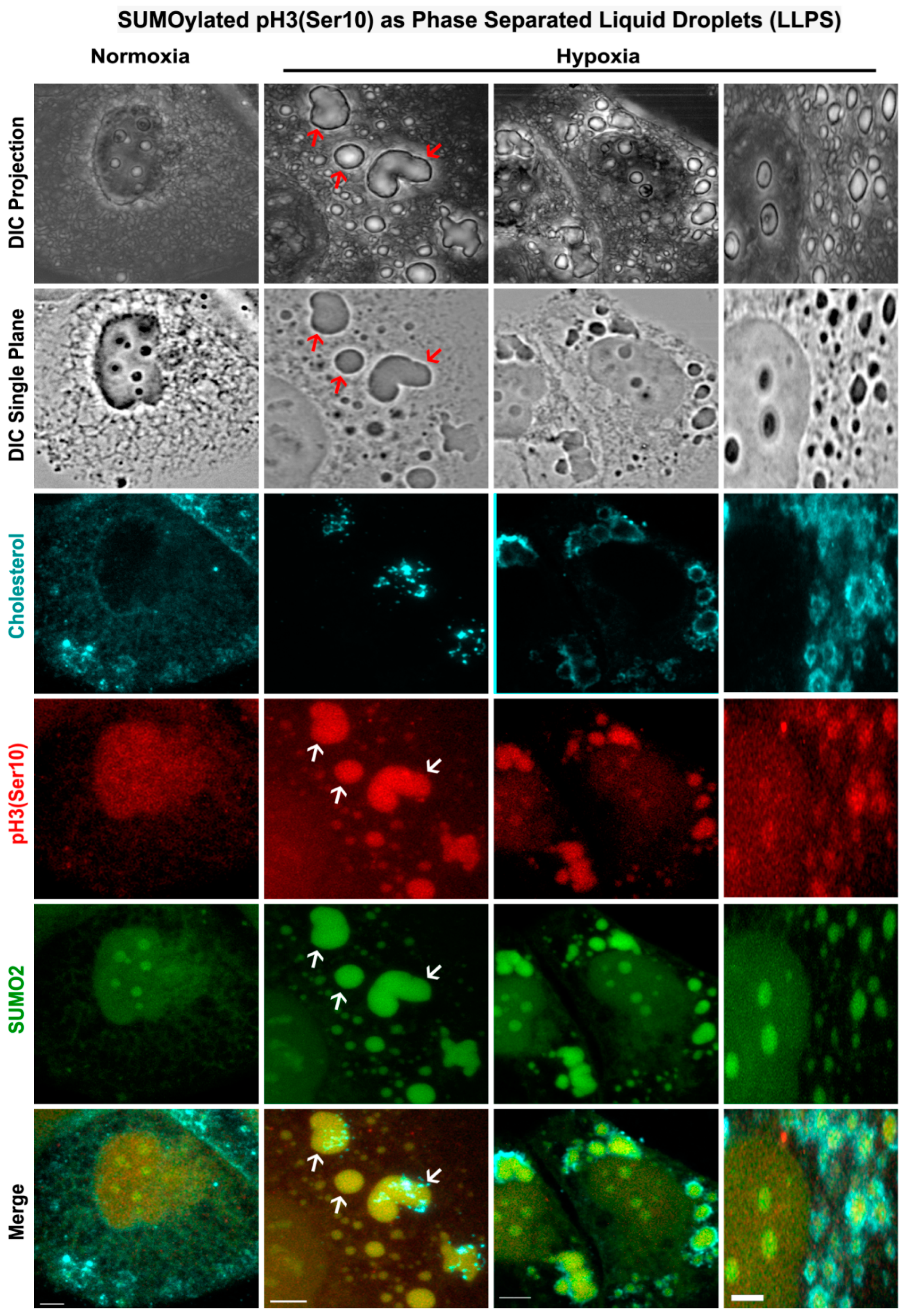
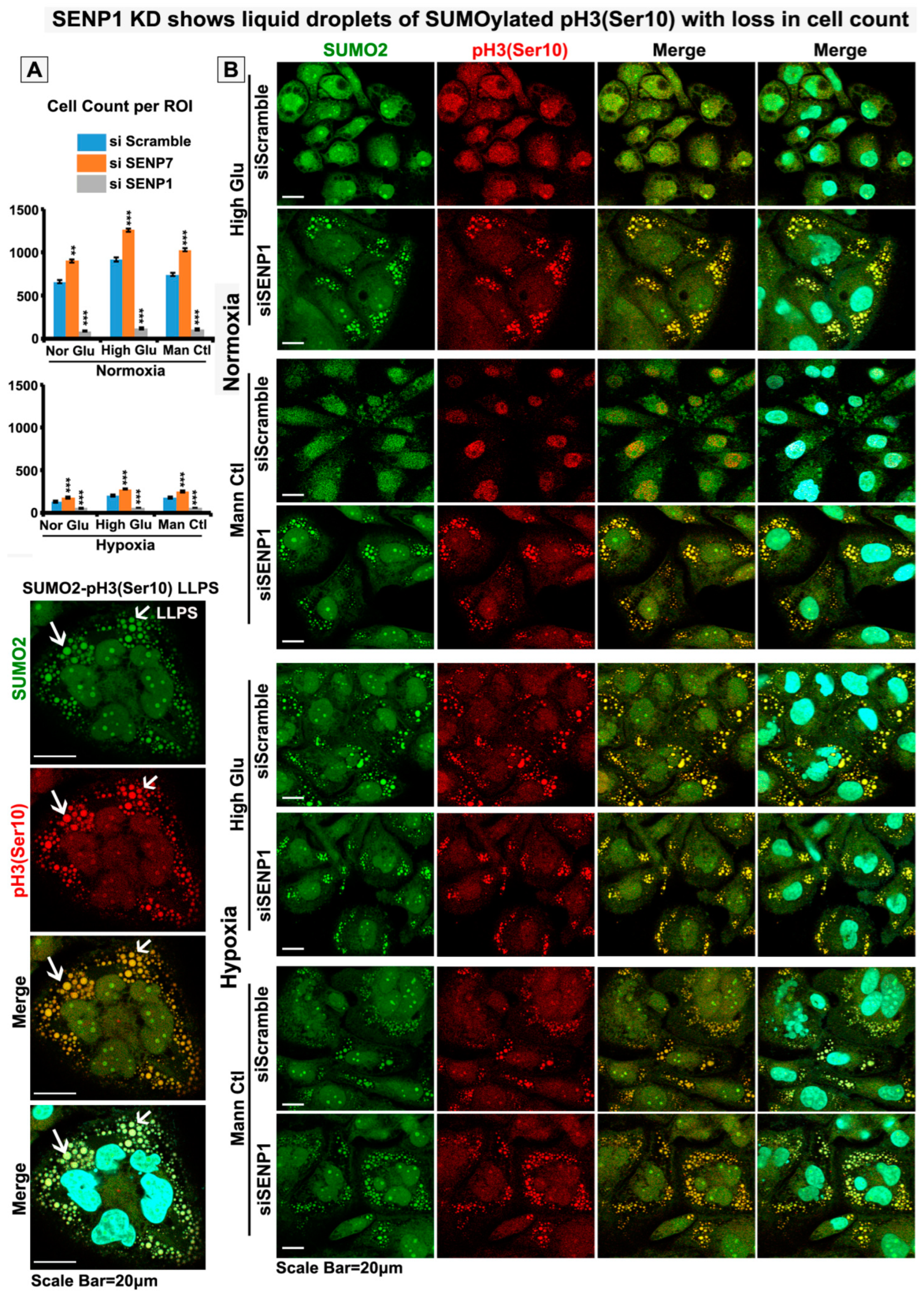
3.5. Nuclear Trafficking of SUMO2-Conjugated pH3(Ser10) Is Significantly Less in Acute Hypoxia Due to Its Phase Separation (LLPS) and Aggregation in the Cytoplasm
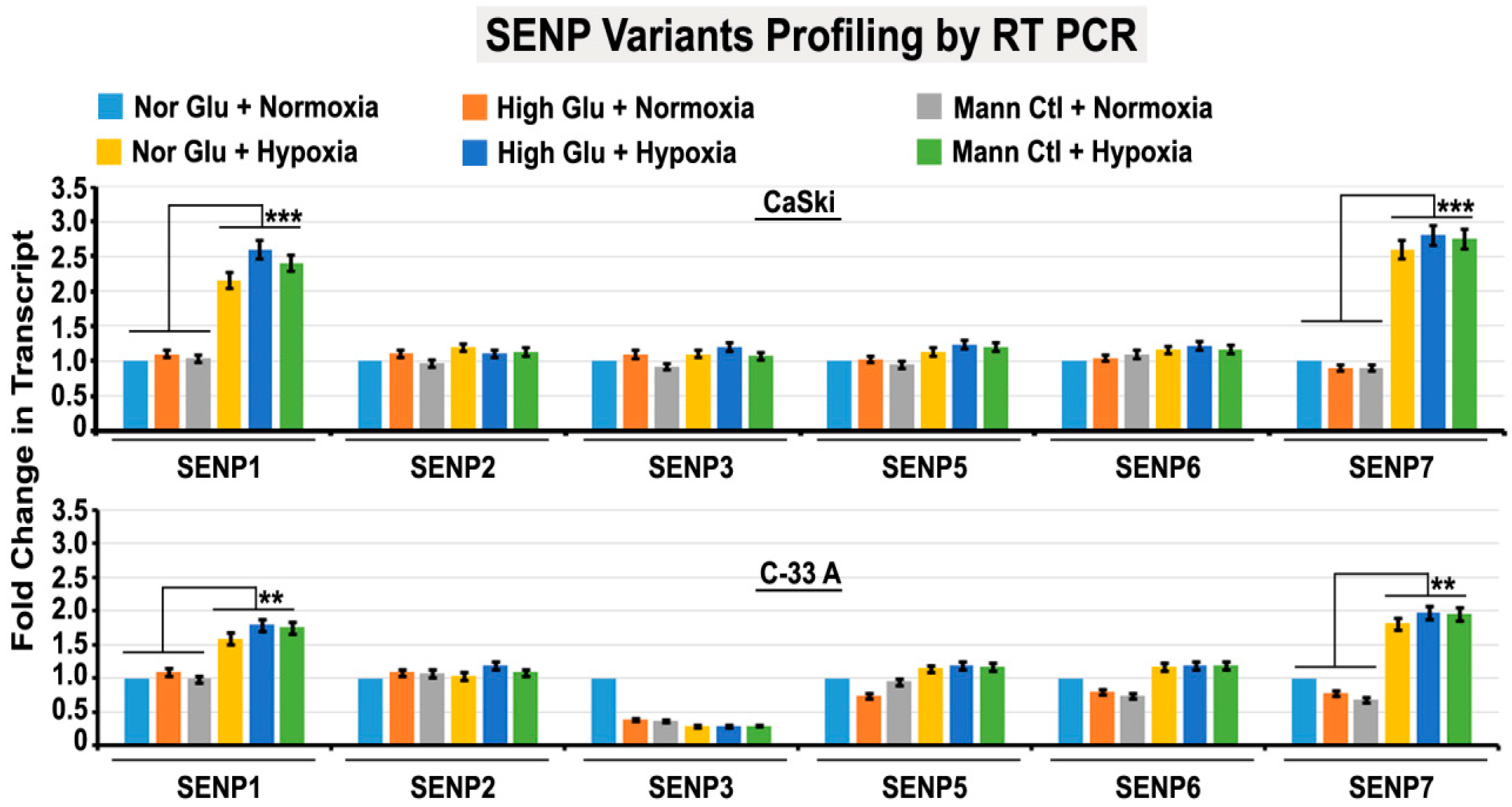
3.6. Abnormal Activity of SENPs, under Acute Hypoxia, Sequesters SUMOylated pH3(Ser10) in the Cytoplasm via LLPS, Thereby Generating Tumour Cell Growth Arrested Phenotype
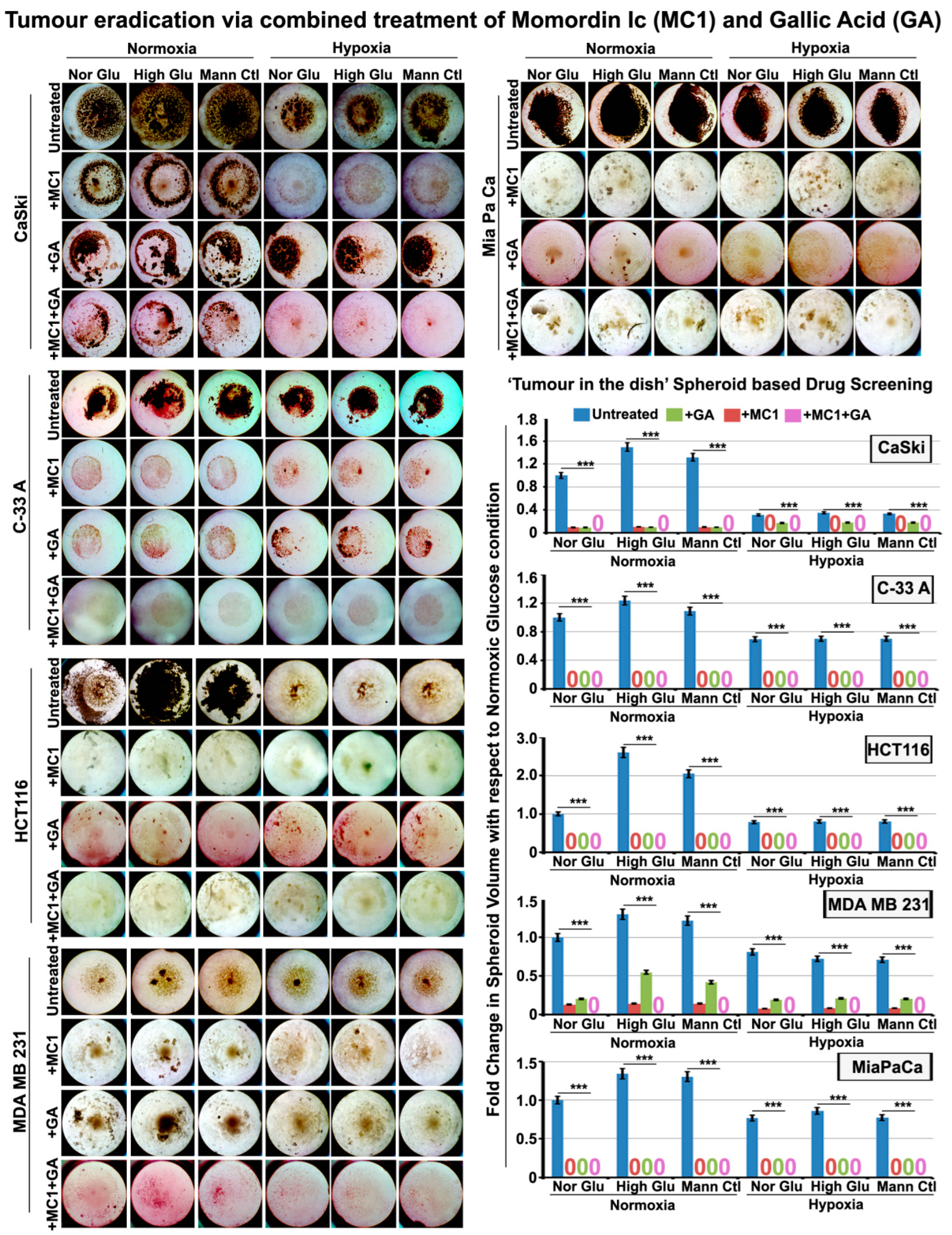
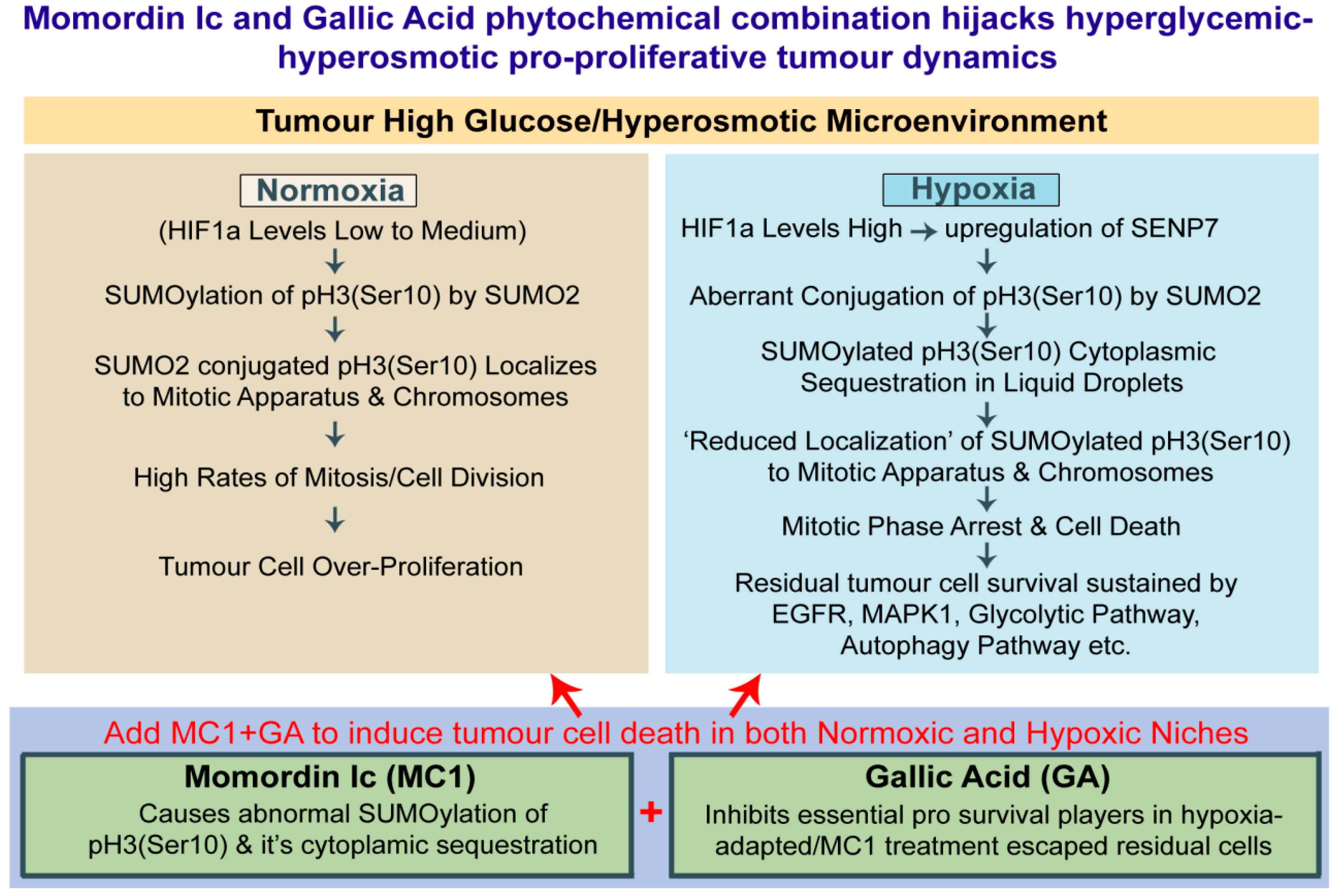
3.7. Momordin Ic (MC), an SENP1 Inhibitor, Can Mimic Hypoxia-Induced Abnormal SUMOylation of pH3(Ser10) and Thereby Retards Tumour Cell Proliferation in Normoxic High-Glucose Hyper-Osmotic Conditions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Crucitta, S.; Cucchiara, F.; Mathijssen, R.; Mateo, J.; Jager, A.; Joosse, A.; Passaro, A.; Attili, I.; Petrini, I.; van Schaik, R.; et al. Treatment-driven tumour heterogeneity and drug resistance: Lessons from solid tumours. Cancer Treat. Rev. 2022, 104, 102340. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.P.; Zheng, C.C.; Huang, Y.N.; He, M.L.; Xu, W.W.; Li, B. Molecular mechanisms of chemo- and radiotherapy resistance and the potential implications for cancer treatment. MedComm 2021, 2, 315–340. [Google Scholar] [CrossRef]
- Kumar, S.; Kushwaha, P.P.; Gupta, S. Emerging targets in cancer drug resistance. Cancer Drug Resist. 2019, 2, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Rohani, N.; Hao, L.; Alexis, M.S.; Joughin, B.A.; Krismer, K.; Moufarrej, M.N.; Soltis, A.R.; Lauffenburger, D.A.; Yaffe, M.B.; Burge, C.B.; et al. Acidification of Tumor at Stromal Boundaries Drives Transcriptome Alterations Associated with Aggressive Phenotypes. Cancer Res. 2019, 79, 1952–1966. [Google Scholar] [CrossRef]
- Wirtz, D.; Konstantopoulos, K.; Searson, P.C. The physics of cancer: The role of physical interactions and mechanical forces in metastasis. Nat. Rev. Cancer 2011, 11, 512–522. [Google Scholar] [CrossRef] [PubMed]
- De Felice, D.; Alaimo, A. Mechanosensitive Piezo Channels in Cancer: Focus on altered Calcium Signaling in Cancer Cells and in Tumor Progression. Cancers 2020, 12, 1780. [Google Scholar] [CrossRef]
- Brocker, C.; Thompson, D.C.; Vasiliou, V. The role of hyperosmotic stress in inflammation and disease. Biomol. Concepts 2012, 3, 345–364. [Google Scholar] [CrossRef]
- Chang, S.F.; Chang, C.A.; Lee, D.Y.; Lee, P.L.; Yeh, Y.M.; Yeh, C.R.; Cheng, C.K.; Chien, S.; Chiu, J.J. Tumor cell cycle arrest induced by shear stress: Roles of integrins and Smad. Proc. Natl. Acad. Sci. USA 2008, 105, 3927–3932. [Google Scholar] [CrossRef]
- Northcott, J.M.; Dean, I.S.; Mouw, J.K.; Weaver, V.M. Feeling Stress: The Mechanics of Cancer Progression and Aggression. Front. Cell Dev. Biol. 2018, 6, 17. [Google Scholar] [CrossRef]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- John, S.; Gayathri, K.G.; Krishna, A.P.; Mishra, R. Neurotherapeutic implications of sense and respond strategies generated by astrocytes and astrocytic tumours to combat pH mechanical stress. Neuropathol. Appl. Neurobiol. 2022, 48, e12774. [Google Scholar] [CrossRef] [PubMed]
- John, S.; Sivakumar, K.C.; Mishra, R. Extracellular Proton Concentrations Impacts LN229 Glioblastoma Tumor Cell Fate via Differential Modulation of Surface Lipids. Front. Oncol. 2017, 7, 20. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Zhang, Y.; Ding, T.; Ji, N.; Zhao, H. The Dual Role of Macropinocytosis in Cancers: Promoting Growth and Inducing Methuosis to Participate in Anticancer Therapies as Targets. Front. Oncol. 2021, 10, 570108. [Google Scholar] [CrossRef]
- Weaver, V.M. Cell and tissue mechanics: The new cell biology frontier. Mol. Biol. Cell. 2017, 28, 1815–1818. [Google Scholar] [CrossRef]
- Cheng, B.; Lin, M.; Huang, G.; Li, Y.; Ji, B.; Genin, G.M.; Deshpande, V.S.; Lu, T.J.; Xu, F. Cellular mechanosensing of the biophysical microenvironment: A review of mathematical models of biophysical regulation of cell responses. Phys. Life Rev. 2017, 22–23, 88–119. [Google Scholar] [CrossRef]
- Spill, F.; Bakal, C.; Mak, M. Mechanical and Systems Biology of Cancer. Comput. Struct. Biotechnol. J. 2018, 16, 237–245. [Google Scholar] [CrossRef]
- Jain, R.K.; Martin, J.D.; Stylianopoulos, T. The role of mechanical forces in tumor growth and therapy. Annu. Rev. Biomed. Eng. 2014, 16, 321–346. [Google Scholar] [CrossRef]
- Munson, J.M.; Shieh, A.C. Interstitial fluid flow in cancer: Implications for disease progression and treatment. Cancer Manag. Res. 2014, 6, 317–328. [Google Scholar] [CrossRef]
- Kang, H.; Wu, Q.; Sun, A.; Liu, X.; Fan, Y.; Deng, X. Cancer Cell Glycocalyx and Its Significance in Cancer Progression. Int. J. Mol. Sci. 2018, 19, 2484. [Google Scholar] [CrossRef]
- Goldman, J.W.; Mendenhall, M.A.; Rettinger, S.R. Hyperglycemia Associated With Targeted Oncologic Treatment: Mechanisms and Management. Oncologist 2016, 21, 1326–1336. [Google Scholar] [CrossRef] [PubMed]
- Duan, W.; Shen, X.; Lei, J.; Xu, Q.; Yu, Y.; Li, R.; Wu, E.; Ma, Q. Hyperglycemia, a neglected factor during cancer progression. Biomed. Res. Int. 2014, 2014, 461917. [Google Scholar] [CrossRef] [PubMed]
- Derr, R.L.; Ye, X.; Islas, M.U.; Desideri, S.; Saudek, C.D.; Grossman, S.A. Association between hyperglycemia and survival in patients with newly diagnosed glioblastoma. J. Clin. Oncol. 2009, 27, 1082–1086. [Google Scholar] [CrossRef] [PubMed]
- Xue, F.; Lin, L.L.; Dehdashti, F.; Miller, T.R.; Siegel, B.A.; Grigsby, P.W. F-18 fluorodeoxyglucose uptake in primary cervical cancer as an indicator of prognosis after radiation therapy. Gynecol. Oncol. 2006, 101, 147–151. [Google Scholar] [CrossRef]
- Novikova, L.; Smirnova, I.V.; Rawal, S.; Dotson, A.L.; Benedict, S.H.; Stehno-Bittel, L. Variations in rodent models of type 1 diabetes: Islet morphology. J. Diabetes Res. 2013, 2013, 965832. [Google Scholar] [CrossRef]
- Pietuch, A.; Brückner, B.R.; Janshoff, A. Membrane tension homeostasis of epithelial cells through surface area regulation in response to osmotic stress. Biochim. Biophys. Acta 2013, 1833, 712–722. [Google Scholar] [CrossRef]
- Cohen, B.E. Membrane Thickness as a Key Factor Contributing to the Activation of Osmosensors and Essential Ras Signaling Pathways. Front. Cell Dev. Biol. 2018, 6, 76. [Google Scholar] [CrossRef]
- Taubenberger, A.V.; Baum, B.; Matthews, H.K. The Mechanics of Mitotic Cell Rounding. Front. Cell Dev. Biol. 2020, 8, 687. [Google Scholar] [CrossRef]
- Stewart, M.P.; Helenius, J.; Toyoda, Y.; Ramanathan, S.P.; Muller, D.J.; Hyman, A.A. Hydrostatic pressure and the actomyosin cortex drive mitotic cell rounding. Nature 2011, 469, 226–230. [Google Scholar] [CrossRef]
- Fischer-Friedrich, E.; Hyman, A.A.; Jülicher, F.; Müller, D.J.; Helenius, J. Quantification of surface tension and internal pressure generated by single mitotic cells. Sci. Rep. 2014, 4, 6213. [Google Scholar] [CrossRef]
- Toyoda, Y.; Cattin, C.J.; Stewart, M.P.; Poser, I.; Theis, M.; Kurzchalia, T.V.; Buchholz, F.; Hyman, A.A.; Müller, D.J. Genome-scale single-cell mechanical phenotyping reveals disease-related genes involved in mitotic rounding. Nat. Commun. 2017, 8, 1266. [Google Scholar] [CrossRef]
- Madonna, R.; Giovannelli, G.; Confalone, P.; Renna, F.V.; Geng, Y.J.; De Caterina, R. High glucose-induced hyperosmolarity contributes to COX-2 expression and angiogenesis: Implications for diabetic retinopathy. Cardiovasc. Diabetol. 2016, 15, 18. [Google Scholar] [CrossRef]
- Russo, V.C.; Higgins, S.; Werther, G.A.; Cameron, F.J. Effects of fluctuating glucose levels on neuronal cells in vitro. Neurochem. Res. 2012, 37, 1768–1782. [Google Scholar] [CrossRef]
- John, S.; Bacoside, A. Induces Tumor Cell Death in Human Glioblastoma Cell Lines through Catastrophic Macropinocytosis. Front. Mol. Neurosci. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Ao, Q.; Su, W.; Guo, S.; Cai, L.; Huang, L. SENP1 desensitizes hypoxic ovarian cancer cells to cisplatin by up-regulating HIF-1α. Sci. Rep. 2015, 5, 16396. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. Available online: https://www.nature.com/articles/nmeth.2019 (accessed on 2 February 2023). [CrossRef] [PubMed]
- Wu, J.; Lei, H.; Zhang, J.; Chen, X.; Tang, C.; Wang, W.; Xu, H.; Xiao, W.; Gu, W.; Wu, Y. Momordin Ic, a new natural SENP1 inhibitor, inhibits prostate cancer cell proliferation. Oncotarget 2016, 7, 58995–59005. [Google Scholar] [CrossRef]
- Wang, D.; Bao, B. Gallic Acid Impedes Non-Small Cell Lung Cancer Progression via Suppression of EGFR-Dependent CARM1-PELP1 Complex. Drug Des. Dev. Ther. 2020, 14, 1583–1592. [Google Scholar] [CrossRef]
- Piccinini, F.; Tesei, A.; Arienti, C.; Bevilacqua, A. Cancer multicellular spheroids: Volume assessment from a single 2D projection. Comput. Methods Programs Biomed. 2015, 118, 95–106. [Google Scholar] [CrossRef]
- Piccinini, F.; Tesei, A.; Bevilacqua, A. Single-image based methods used for non-invasive volume estimation of cancer spheroids: A practical assessing approach based on entry-level equipment. Comput. Methods Programs Biomed. 2016, 135, 51–60. [Google Scholar] [CrossRef]
- Luzhna, L.; Kathiria, P.; Kovalchuk, O. Micronuclei in genotoxicity assessment: From genetics to epigenetics and beyond. Front. Genet. 2013, 4, 131. [Google Scholar] [CrossRef] [PubMed]
- Eifler, K.; Vertegaal, A.C.O. SUMOylation-Mediated Regulation of Cell Cycle Progression and Cancer. Trends Biochem. Sci. 2015, 40, 779–793. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, I.A.; D’Souza, R.C.; Chang, J.G.; Mann, M.; Vertegaal, A.C. System-wide identification of wild-type SUMO-2 conjugation sites. Nat. Commun. 2015, 6, 7289. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, I.A.; Lyon, D.; Young, C.; Jensen, L.J.; Vertegaal, A.C.; Nielsen, M.L. Site-specific mapping of the human SUMO proteome reveals co-modification with phosphorylation. Nat. Struct. Mol. Biol. 2017, 24, 325–336. [Google Scholar] [CrossRef]
- Prigent, C.; Dimitrov, S. Phosphorylation of serine 10 in histone H3, what for? J. Cell Sci. 2003, 116 (Pt 18), 3677–3685. [Google Scholar] [CrossRef]
- Li, D.W.; Yang, Q.; Chen, J.T.; Zhou, H.; Liu, R.M.; Huang, X.T. Dynamic distribution of Ser-10 phosphorylated histone H3 in cytoplasm of MCF-7 and CHO cells during mitosis. Cell Res. 2005, 15, 120–126. [Google Scholar] [CrossRef]
- Kim, J.I.; Choi, K.U.; Lee, I.S.; Choi, Y.J.; Kim, W.T.; Shin, D.H.; Kim, K.; Lee, J.H.; Kim, J.Y.; Sol, M.Y. Expression of hypoxic markers and their prognostic significance in soft tissue sarcoma. Oncol. Lett. 2015, 9, 1699–1706. [Google Scholar] [CrossRef]
- Burri, P.; Djonov, V.; Aebersold, D.M.; Lindel, K.; Studer, U.; Altermatt, H.J.; Mazzucchelli, L.; Greiner, R.H.; Gruber, G. Significant correlation of hypoxia-inducible factor-1alpha with treatment outcome in cervical cancer treated with radical radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2003, 56, 494–501. [Google Scholar] [CrossRef]
- Peng, Z.; Mizianty, M.J.; Xue, B.; Kurgan, L.; Uversky, V.N. More than just tails: Intrinsic disorder in histone proteins. Mol. Biosyst. 2012, 8, 1886–1901. [Google Scholar] [CrossRef]
- Lara-Ureña, N.; Jafari, V.; García-Domínguez, M. Cancer-Associated Dysregulation of Sumo Regulators: Proteases and Ligases. Int. J. Mol. Sci. 2022, 23, 8012. [Google Scholar] [CrossRef]
- Hoppe-Seyler, K.; Bossler, F.; Lohrey, C.; Bulkescher, J.; Rösl, F.; Jansen, L.; Mayer, A.; Vaupel, P.; Dürst, M.; Hoppe-Seyler, F. Induction of dormancy in hypoxic human papillomavirus-positive cancer cells. Proc. Natl. Acad. Sci. USA 2017, 114, E990–E998. [Google Scholar] [CrossRef] [PubMed]
- Butturini, E.; Carcereri de Prati, A.; Boriero, D.; Mariotto, S. Tumor Dormancy and Interplay with Hypoxic Tumor Microenvironment. Int. J. Mol. Sci. 2019, 20, 4305. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Sung, K.; Beebe, D.; Huang, W.; Shapiro, D.; Miyamoto, S.; Abel, E.J. The SUMO protease SENP1 promotes aggressive behaviors of high HIF2α expressing renal cell carcinoma cells. Oncogenesis 2022, 11, 65. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Hu, J.; Cui, Y.; Liang, S.; Gao, X.; Zhang, J.; Jia, W. Knockdown of SENP1 inhibits HIF-1α SUMOylation and suppresses oncogenic CCNE1 in Wilms tumor. Mol. Ther. Oncolytics 2021, 23, 355–366. [Google Scholar] [CrossRef]
- Zhang, Q.S.; Zhang, M.; Huang, X.J.; Liu, X.J.; Li, W.P. Downregulation of SENP1 inhibits cell proliferation, migration and promotes apoptosis in human glioma cells. Oncol. Lett. 2016, 12, 217–221. [Google Scholar] [CrossRef]
- Li, Y.; De Bolòs, A.; Amador, V.; Reverter, D. Structural Basis for the SUMO2 Isoform Specificity of SENP7. J. Mol. Biol. 2022, 434, 167875. [Google Scholar] [CrossRef]
- Martin, E.W.; Holehouse, A.S. Intrinsically disordered protein regions and phase separation: Sequence determinants of assembly or lack thereof. Emerg. Top. Life Sci. 2020, 4, 307–329. [Google Scholar] [CrossRef]
- Gâtel, P.; Brockly, F.; Reynes, C.; Pastore, M.; Hicheri, Y.; Cartron, G.; Piechaczyk, M.; Bossis, G. Ubiquitin and SUMO conjugation as biomarkers of acute myeloid leukemias response to chemotherapies. Life Sci. Alliance 2020, 3, e201900577. [Google Scholar] [CrossRef]
- Chang, H.M.; Yeh, E.T.H. SUMO: From Bench to Bedside. Physiol. Rev. 2020, 100, 1599–1619. [Google Scholar] [CrossRef]
- Jia, S.; Shen, M.; Zhang, F.; Xie, J. Recent Advances in Momordica charantia: Functional Components and Biological Activities. Int. J. Mol. Sci. 2017, 18, 2555. [Google Scholar] [CrossRef] [PubMed]
- Bortolotti, M.; Mercatelli, D.; Polito, L. Momordica charantia, a Nutraceutical Approach for Inflammatory Related Diseases. Front. Pharmacol. 2019, 10, 486. [Google Scholar] [CrossRef]
- Bai, J.; Zhang, Y.; Tang, C.; Hou, Y.; Ai, X.; Chen, X.; Zhang, Y.; Wang, X.; Meng, X. Gallic acid: Pharmacological activities and molecular mechanisms involved in inflammation-related diseases. Biomed. Pharmacother. 2021, 133, 110985. [Google Scholar] [CrossRef] [PubMed]
- Tuli, H.S.; Mistry, H.; Kaur, G.; Aggarwal, D.; Garg, V.K.; Mittal, S.; Yerer, M.B.; Sak, K.; Khan, M.A. Gallic Acid: A Dietary Polyphenol that Exhibits Anti-neoplastic Activities by Modulating Multiple Oncogenic Targets. Anticancer Agents Med. Chem. 2022, 22, 499–514. [Google Scholar] [CrossRef] [PubMed]
- Kahkeshani, N.; Farzaei, F.; Fotouhi, M.; Alavi, S.S.; Bahramsoltani, R.; Naseri, R.; Momtaz, S.; Abbasabadi, Z.; Rahimi, R.; Farzaei, M.H.; et al. Pharmacological effects of gallic acid in health and diseases: A mechanistic review. Iran J. Basic Med. Sci. 2019, 22, 225–237. [Google Scholar] [CrossRef]
- Aborehab, N.M.; Osama, N. Effect of Gallic acid in potentiating chemotherapeutic effect of Paclitaxel in HeLa cervical cancer cells. Cancer Cell Int. 2019, 19, 154. [Google Scholar] [CrossRef]
- Cui, X.; Harada, S.; Shen, D.; Siegal, G.P.; Wei, S. The Utility of Phosphohistone H3 in Breast Cancer Grading. Appl. Immunohistochem. Mol. Morphol. 2015, 23, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Hans, F.; Dimitrov, S. Histone H3 phosphorylation and cell division. Oncogene 2001, 20, 3021–3027. [Google Scholar] [CrossRef]
- Burgess, A.; Rasouli, M.; Rogers, S. Stressing mitosis to death. Front. Oncol. 2014, 4, 140. [Google Scholar] [CrossRef]
- Xia, W.; Spector, S.; Hardy, L.; Zhao, S.; Saluk, A.; Alemane, L.; Spector, N.L. Tumor selective G2/M cell cycle arrest and apoptosis of epithelial and hematological malignancies by BBL22, a benzazepine. Proc. Natl. Acad. Sci. USA 2000, 97, 7494–7499. [Google Scholar] [CrossRef]
- Cui, C.P.; Wong, C.C.; Kai, A.K.; Ho, D.W.; Lau, E.Y.; Tsui, Y.M.; Chan, L.K.; Cheung, T.T.; Chok, K.S.; Chan, A.C.Y.; et al. SENP1 promotes hypoxia-induced cancer stemness by HIF-1α deSUMOylation and SENP1/HIF-1α positive feedback loop. Gut 2017, 66, 2149–2159. [Google Scholar] [CrossRef]
- Visconti, R.; Della Monica, R.; Grieco, D. Cell cycle checkpoint in cancer: A therapeutically targetable double-edged sword. J. Exp. Clin. Cancer Res. 2016, 35, 153. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Gong, J.; Huang, W.; Lu, F.; Dong, H. The Effect of Momordica charantia in the Treatment of Diabetes Mellitus: A Review. Evid. Based Complement. Alternat. Med. 2021, 2021, 3796265. [Google Scholar] [CrossRef]
- Peter, E.L.; Deyno, S.; Mtewa, A.; Kasali, F.M.; Nagendrappa, P.B.; Sesaazi, D.; Tolo, C.U.; Ogwang, P.E. Safety and efficacy of Momordica charantia Linnaeus in pre-diabetes and type 2 diabetes mellitus patients: A systematic review and meta-analysis protocol. Syst. Rev. 2018, 7, 192. [Google Scholar] [CrossRef]
- Rauniyar, G.P.; Sinha, R.; Chapagain, K.; Maskey, R.; Pandey, D.R. Effects of Momordica Charantia (Karela/bitterguord) in Type 2 Diabetic Patients Taking Allopathic Drugs: A pilot study. Kathmandu Univ. Med. J 2021, 19, 143–147. [Google Scholar] [CrossRef]
- Kumar, V.; Aneesh, K.A.; Kshemada, K.; Ajith, K.G.S.; Binil, R.S.S.; Deora, N.; Sanjay, G.; Jaleel, A.; Muraleedharan, T.S.; Anandan, E.M.; et al. Amalaki rasayana, a traditional Indian drug enhances cardiac mitochondrial and contractile functions and improves cardiac function in rats with hypertrophy. Sci. Rep. 2017, 7, 8588. [Google Scholar] [CrossRef] [PubMed]
- Shengule, S.A.; Mishra, S.; Joshi, K.; Apte, K.; Patil, D.; Kale, P.; Shah, T.; Deshpande, M.; Puranik, A. Anti-hyperglycemic and anti-hyperlipidaemic effect of Arjunarishta in high-fat fed animals. J. Ayurveda Integr. Med. 2018, 9, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, P.; Patel, R.K. Quantification of Gallic acid and Ellagic acid in Arjunarishta by validated HPTLC densitometry. Int. J. Pharm. Sci. Res. 2012, 3, 2215. [Google Scholar] [CrossRef]















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
G., G.K.; Shinde, P.L.; John, S.; C., S.K.; Mishra, R. Understanding the Combined Effects of High Glucose Induced Hyper-Osmotic Stress and Oxygen Tension in the Progression of Tumourigenesis: From Mechanism to Anti-Cancer Therapeutics. Cells 2023, 12, 825. https://doi.org/10.3390/cells12060825
G. GK, Shinde PL, John S, C. SK, Mishra R. Understanding the Combined Effects of High Glucose Induced Hyper-Osmotic Stress and Oxygen Tension in the Progression of Tumourigenesis: From Mechanism to Anti-Cancer Therapeutics. Cells. 2023; 12(6):825. https://doi.org/10.3390/cells12060825
Chicago/Turabian StyleG., Gayathri K., Puja Laxmanrao Shinde, Sebastian John, Sivakumar K. C., and Rashmi Mishra. 2023. "Understanding the Combined Effects of High Glucose Induced Hyper-Osmotic Stress and Oxygen Tension in the Progression of Tumourigenesis: From Mechanism to Anti-Cancer Therapeutics" Cells 12, no. 6: 825. https://doi.org/10.3390/cells12060825