1. Introduction
The natural product nemorosone was isolated in 1996 through the extraction of floral resins from
Clusia rosea, an evergreen tropical plant with several medicinal applications [
1]. Its chemical structure was unraveled in 2001 as a type A polyisoprenylated benzophenone [
2]. The high interest in nemorosone mainly resides in its cytotoxic anti-cancer activity, as shown in a broad spectrum of different human cancer models such as leukemia, colorectal, pancreatic, hepatic, and breast cancer [
3,
4,
5,
6,
7,
8,
9,
10,
11]. In most cases, apoptosis was reported as its mode of cytotoxicity along with the arrest of cell cycle progression. Recently, its antimetastatic potential through the modulation of molecules related to the epithelial-mesenchymal transition (EMT) was also described [
11]. Furthermore, several research works have reported antimutagenic activity without a genotoxic effect, selectivity toward cancer cells, and the capacity to circumvent multidrug-resistance mechanisms [
4,
5,
12,
13,
14]. Altogether, these previous studies show that nemorosone can be considered as a lead compound for the development of novel antiproliferative drugs for cancer therapy.
On the other hand, we found that nemorosone disrupts the mitochondrial bioenergetic status by acting as a potent protonophoric mitochondrial uncoupler [
6,
9]. Therefore, despite the well-established capacity of nemorosone to induce apoptosis [
4,
5], other modes of regulated cell death could be induced depending on the cell type. Since the conceptualization of ferroptosis in 2012, several compounds previously described as inducers of apoptosis, such as cisplatin, sorafenib, and withaferin A, have been found to elicit ferroptosis [
15,
16,
17]. Moreover, the induction of ferroptosis has also been identified in a myriad of natural products [
18].
Ferroptosis is a form of regulated necrosis mediated by iron-catalyzed excessive lipid peroxidation [
19,
20], often referred to as biological rust of lipid membranes [
21]. Ferroptosis can be induced by inactivating glutathione peroxidase 4 (GPX4), which detoxifies lipid hydroperoxides. GPX4 can be inactivated through direct targeting and inhibition, by class II and III inducers, or alternatively by indirect mechanisms through the depletion of intracellular GSH, an essential co-factor of GPX4, by class I inducers [
22]. Furthermore, ferroptosis can be activated by the increase in the cellular labile iron pool (LIP), which is the intracellular non-protein bound redox-active iron, or iron oxidation mediated by class IV inducers [
21].
Recently, Gao et al. showed that mitochondria play a central role in cysteine-deprivation-induced and erastin-induced ferroptosis (Class I FINs), but not in the case of the ferroptosis induced by GPX4 inhibition (Class II FINs). Mechanistically, cysteine deprivation leads to a transient hyperpolarization of the mitochondrial membrane potential and lipid peroxide production [
23]. On the other hand, cell death induced by mitochondrial uncoupling is accompanied by depolarization of the mitochondrial membrane potential, reduced ATP levels, increased ROS, and diminished antioxidant defense by decreasing GSH levels [
24]. The latter may resemble a similar cellular effect observed in erastin-treated cells. Therefore, we hypothesized and examined whether mitochondrial uncoupling by nemorosone could initiate a ferroptotic response.
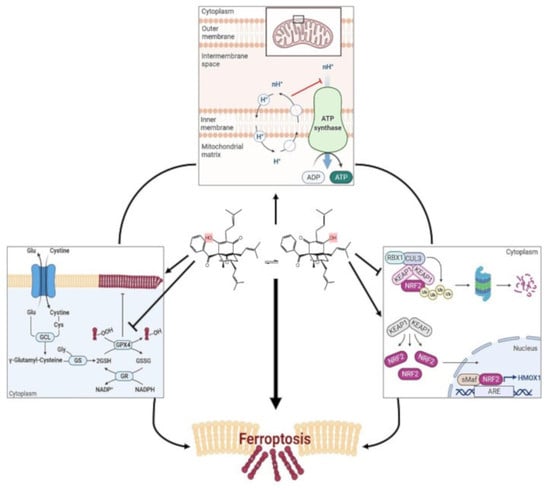
Using different cell lines, we found that nemorosone triggers ferroptosis, as detected by lipid peroxidation. We elucidated that nemorosone-induced ferroptosis involves a double-edged mechanism. Nemorosone decreases the glutathione levels by blocking the cystine/glutamate antiporter and induces lipid peroxidation as an early event. At later time points, nemorosone administration results in activation of the KEAP1–NRF2–HMOX1 axis, causing an increase in the intracellular labile Fe2+ pool and consequent reactive oxygen species (ROS) production. Methylnemorosone, a structural variant of nemorosone that has lost the capacity to uncouple mitochondrial respiration, does not trigger cell death anymore. The classical mitochondrial uncoupler carbonyl cyanide 3-chlorophenylhydrazone (CCCP) has a similar biological effect as nemorosone, in inducing ferroptosis. In summary, our results demonstrate that nemorosone as well as other mitochondrial uncouplers drive intrinsic ferroptosis. The present findings could open new perspectives for a better insight into ferroptosis initiated by mitochondrial dysfunction and for the development of novel ferroptosis inducers for cancer treatment.
2. Materials and Methods
2.1. Antibodies and Reagents
The following antibodies were used in this study: β-tubulin (Abcam, Cambridge, UK, ab21058), GPX4 (Abcam, ab41787), HMOX1 (Enzo Life Sciences, Farmingdale, NY, USA, ADI-SPA-896-F), NRF2 (Abcam, ab62352), and KEAP1 (Proteintech, Rosemont, IL, USA, 10503-2-AP). The following chemicals were used: SytoxGreen (Thermo Fisher Scientific, Waltham, MA, USA, S7020): 1.7 μM, BODIPY 581/591 C11 probe (Invitrogen, Waltham, MA, USA, D-3861): 2 μM, SytoxBlue (Thermo Fisher Scientific, S11348): 1.25 μM, DRAQ7 (BioStatus, Loughborough, UK, DR71000): 0.3 μM, TMRE: tetramethylrhodamine ethyl ester (Thermo Fisher Scientific, T669): 200 nM, FeRhoNox-1 (Goryo Chemical, Sapporo, Hokkaido, Japan, GC901): 10 μM, MitoSOX Red (Thermo Fisher Scientific, M36008): 5 μM, erastin (Selleckchem, Houston, TX, USA, S7242): 10 and 20 μM, CCCP: carbonyl cyanide 3-chlorophenylhydrazone (Sigma, St. Louis, MO, USA, Cat#C2759), Nec-1s (Calbiochem, San Diego, CA, USA, 480065): 10 μM, Fer1 (Xcess Biosciences, Chicago, IL, USA, 053224): 1 μM, DFO (Sigma-Aldrich, St. Louis, MI, USA, D-9533): 50 μM, and CPX (Sigma-Aldrich, C0415): 5 μM. Z-VAD-FMK (Bachem, Bubendorf, Switzerland, N-1510), a caspase peptide inhibitor, was used at a concentration of 10 μM, while DEVD-AMC (Pepta Nova, Sandhausen, Germany, 3171-V), a fluorogenic substrate for caspase-3, was used at 20 μM. The mitochondrial complexes inhibitors rotenone (Sigma, Cat#R8875), antimycin A (Sigma, Cat#A8674), and oligomycin (Sigma, Cat#75351) were used at 0.5 and 10 μM, 0.5 and 50 μM, and 1.5 μM, respectively. Hemin (Sigma-Aldrich, H9039) was used at 5 and 10 μM, zinc protoporphyrin: ZnPP (Enzo Life Sciences, ALX-430-049-M025) was used at 1 μM, and ferrous ammonium sulfate: Fe(NH4)2(SO4)2·6H2O (Sigma-Aldrich, FX0245) was used at 1 mM. Trimethylsilyldiazomethane (Sigma-Aldrich, 362832): a 2.0 M solution in hexanes was used. Toluene, methanol, n-hexane, and diethyl ether were purchased from Chem-Lab NV (Zedelgem, Belgium, HPLC grade).
2.2. Nemorosone Isolation and Characterization. Synthesis and Characterization of Methylated Derivative
TLC was performed using precoated silica gel plates (Macherey-Nagel SIL G-25 UV254). Chemical shifts for the 1H NMR and 13C NMR spectra, recorded on a Bruker Avance 400 spectrometer, were reported in parts per million with reference to the residual solvent signal (CDCl3: 7.26 ppm; CD3OD: 3.30 ppm and 49.00 ppm). Coupling constants (J) are expressed in hertz. Electrospray mass spectra were recorded by means of an Agilent 1100 series single quadrupole MS detector type VL, with APCI and API-ES sources, and provided with a Phenomenex Luna C18 (2) column (5 µm 250 mm × 4.60 mm). An Agilent 1100 series connected to a 6220A TOF-MS detector, equipped with an APCI-ESI multi-mode source, was used to conduct high resolution mass spectrometry (HRMS). A Perkin-Elmer 1000 FT-IR infrared spectrometer (HATR) was utilized to record the infrared spectra. A Perkin Elmer 241 polarimeter was used to measure optical rotation.
Nemorosone was isolated, as previously described [
2], from the floral resin of
Clusia rosea. Concisely, an EtOH-H
2O solution was used to crystallize nemorosone from the resin of the flowers of this plant species.
Figure S1A shows the structure of nemorosone: C
33H
42O
4, a mixture of tautomers (1
S,5
R,7
R)-5-benzoyl-4-hydroxy-6,6-dimethyl-1,3,7-tris(3-methylbut-2-en-1-yl)bicyclo [3.3.1]non-3-ene-2,9-dione and (1
S,5
S,7
R)-1-benzoyl-4-hydroxy-8,8-dimethyl-3,5,7-tris(3-methylbut-2-en-1-yl)bicyclo[3.3.1]non-3-ene-2,9-dione.
Purity of isolated nemorosone was >99%, as determined via reversed-phase HPLC, with detection at 214/254 nm (retention time: 6.9 min; see
Figure S4). Eluting conditions: eluent A/eluent B (50/50) for 30 s, followed by gradient elution (A/B from 50/50 to 0/100) over 6 min (eluent A: 0.1% HCOOH in water; eluent B: acetonitrile) on a Phenomenex Luna C18 (2) column (5 µm 250 mm × 4.60 mm). Analytical data are in agreement with the literature and identical to recorded data of commercially available nemorosone (purchased from Cayman Chemical, Ann Arbor, MI, USA, 24256). Rf 0.22 in hexane/EtOAc 8/2 [Lit: ≈0.21 [
25]].
1H NMR (CD
3OD, 400 MHz): 7.53 (br d,
J = 7.7 Hz, 2H), 7.43 (app tt,
J = 7.4 Hz/1.2 Hz, 1H), 7.22–7.28 (m, 2H), 4.96–5.11 (m, 3H), 3.13 (dd,
J = 15.0 Hz/7.8 Hz, 1H, A-part of ABX-system), 3.07 (dd,
J = 14.8 Hz/7.4 Hz, 1H, B-part of ABX-system), 2.54 (dd,
J = 14.7 Hz/7.8 Hz, 1H, A-part of ABX-system), 2.47 (dd,
J = 14.5 Hz/7.3 Hz, 1H, B-part of ABX-system), 2.09–2.19 (m, 1H), 2.01 (br dd,
J = 13.1 Hz/2.7 Hz, 1H), 1.65–1.85 (m, 2H), 1.68 (s, 3H), 1.64 (app s, 12H), 1.58 (s, 3H), 1.39–1.50 (m, 1H), 1.33 (s, 3H), 1.10 (s, 3H) ppm.
13C NMR (CD
3OD, 100 MHz): 209.26 (C), 194.88 (C), 138.23 (C), 135.25 (C), 134.24 (C), 133.66 (C), 133.12 (CH), 129.61 (CH), 128.78 (CH), 123.99 (CH), 122.12 (CH), 121.18 (C), 120.79 (CH), 44.64 (CH), 30.40 (CH
2), 28.19 (CH
2), 26.30 (CH
3), 26.02 (CH
3), 24.35 (CH
3), 22.32 (CH
2), 18.30 (CH
3), 18.11 (CH
3), 17.97 (CH
3), 16.20 (CH
3) ppm. [a]
D20 +99° (c 0.095, MeOH) [Lit: −98.3° (ent-nemorosone, c 1.19, MeOH) [
26]]. IR (HATR): 3534 (m), 3418 (m), 2966 (m), 2916 (m), 2882 (m), 1711 (s), 1699 (s), 1582 (vs), 1446 (m), 1391 (m), 1368 (vs), 1317 (m), 1262 (m), 1242 (m), 1215 (s), 1197 (m), 1185 (m), 1172 (m), 1157 (m), 1128 (m), 1101 (m), 1063 (m), 1032 (w), 1019 (m), 1003 (w), 959 (w), 936 (w), 922 (w), 897 (w), 839 (s), 799 (m), 772 (w), 751 (m), 730 (w), 690 (m), 666 (m) cm
−1. HRMS (ESI, positive mode): calculated for C
33H
43O
4+ [M+H
+]: 503.3156, found: 503.3149. Data on isolated natural nemorosone: [
2]; data on synthetic ent-nemorosone: [
26]; data on synthetic racemic nemorosone: [
25]. Copies of
1H and
13C (APT) spectra can be found in
Supplementary Materials (
Figures S5 and S6, respectively).
For the synthesis of
O-methylated nemorosone, to a solution of nemorosone (200 mg, 0.398 mmol, 1 eq) in toluene/methanol (10 mL, 4/1), trimethylsilyldiazomethane (1.2 mL, 2.0 M in hexanes, 2.4 mmol, 6 eq) was added dropwise. After stirring the reaction mixture (a pale yellow solution) at room temperature for 30 min, silica was added to quench the excess of reagent. The resulting suspension was filtered and, under reduced pressure, the filtrate was concentrated. The
1H NMR spectrum of the crude mixture of both formed isomers was consistent with findings in the literature data on pure individual compounds [
25]; integration of diagnostic methyl ester signals showed an isomer ratio of 78/22 (see
Figure S7). The residue was partially purified using flash chromatography (gradient elution: hexane/ether 99/1–93/7), affording pure major isomer (48 mg, 0.093 mmol, 23% yield) and a mixture of major and minor isomers (153 mg, 0.296 mmol, 74% yield, ratio major/minor: 72.4/27.6, as determined via RP-HPLC/MS, integration of peaks at 214 nm, retention times 6.3 min and 6.5 min; see
Figure S8). Eluting conditions: eluent A/eluent B (100/0) during 30 s, followed by gradient elution (A/B from 100/0 to 0/100) over 6 min (eluent A: 5 mM NH
4OAc in water; eluent B: acetonitrile) on a Phenomenex Luna C18 (2) column (5 µm 250 mm × 4.60 mm). Major isomer: Rf 0.25 in hexane/ether 9/1. HRMS (ESI, positive mode): calculated for C
34H
45O
4+ [M + H
+]: 517.3312, found: 517.3335. Minor isomer: Rf 0.17 in hexane/ether 9/1. HRMS (ESI, positive mode): calculated for C
34H
45O
4+ [M + H
+]: 517.3312, found: 517.3330.
2.3. Conditions for Cell Culture
DMEM medium supplemented with 10% (
v/
v) fetal calf serum (FCS), sodium pyruvate (1 mM), l-glutamine (1 mM), and non-essential amino acids (1 mM) was used to cultivate U87MG and U373MG human glioblastoma cells and HT22 cells (non-tumorigenic mouse hippocampal neuronal cell line); while IMR-32 (human neuroblastoma cell line) and HT1080 human fibrosarcoma cells were cultured in RPMI 1640 and EMEM medium, respectively, both supplemented in the same way as DMEM medium. Each cell line was obtained from ATCC. Every 3–4 days, cells cultures were split using a trypsin/EDTA solution and maintained at 37 °C in a humid 5% CO
2 environment. It is important to highlight that these cancer cell lines were chosen both for their clinical relevance (they are representative cell lines of tumors refractory to conventional therapy) and for their reported sensitivity to the induction of non-apoptotic regulated cell death [
17,
19,
27,
28]. In addition, nemorosone had not been tested in any of these cell lines.
2.4. Analysis of Cell Death and Caspase-3 Activity
Using the FLUOstar Omega fluorescence plate reader (BMG Labtech GmbH), cell death and caspase-3 activity were measured as previously reported [
29,
30]. Briefly, cells were seeded in a 96-well plate, and all experiments were carried out in triplicate. The following day, after being preincubated with the selected inhibitors, cells were treated with stimuli at desired concentrations in the presence of SytoxGreen and DEVD-AMC. At 1 h intervals, the fluorescence intensity of both fluorescent probes was measured. Percent cell death was calculated using Triton X-100 (0.05%) as a reference for 100% cell death. Following this same procedure, live cell images of seeded cells were obtained using a Zeiss LSM780 confocal microscope. The ImageJ program was used to merge the images. To analyze the induction of cell death, SytoxBlue staining was also used in conjunction with flow cytometry (BD LSR-Fortessa, BD Biosciences, Franklin Lakes, NJ, USA).
2.5. Lipid ROS Analysis
Lipid ROS generation was determined by a previously described methodology [
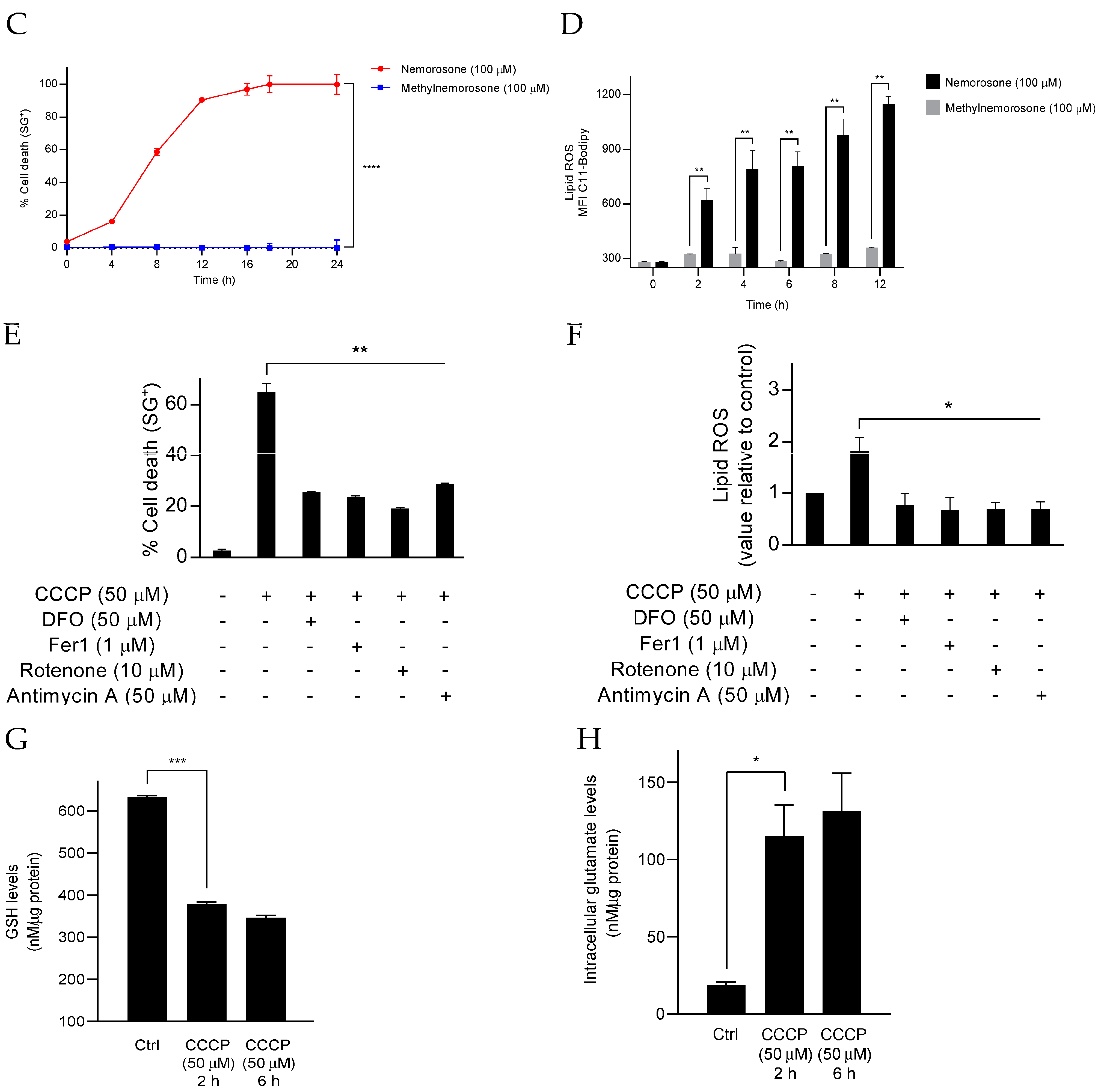
30]. In short, in a 6-well plate, HT1080 (300,000 cells/well) and IMR-32 (500,000 cells/well) cells were seeded. Cells were stimulated the following day and harvested. Fluorescent probes, C11-BODIPY and DRAQ7, were added to the wells 10 min prior to each time point, and lipid ROS accumulation was measured by flow cytometry (BD LSRFortessa, BD Biosciences). B530 (C11-BODIPY) and R780 (DRAQ7) channels were used to measure fluorescence. Only non-permeable live cell fluorescence was evaluated. Per condition, a minimum of 10,000 cells were examined.
2.6. Mitochondrial ROS Analysis
Mitochondrial ROS generation was determined using MitoSOX Red. In brief, HT1080 cells (300,000 cells/well) were seeded in a 6-well plate and incubated overnight. Afterward, cells were exposed to the test compounds according to the instructions of the experiment. After being washed with pre-warmed HBSS (Thermo Fisher Scientific, 14025076), cells were then incubated with fresh medium containing MitoSox Red for 15 min at 37 °C. Subsequently, cells were washed with HBSS and collected in PBS (Thermo Fisher Scientific, 10010023) containing SytoxBlue for measurement using BD FACSVerse (BD Biosciences). B586 (MitoSOX Red) and V448 (SytoxBlue) channels were used to measure fluorescence. Only non-permeable live cell fluorescence was evaluated. Per condition, a minimum of 10,000 cells were examined.
2.7. Determination of Cellular Labile Fe2+ Pool
FeRhoNox-1 dye was used to measure iron levels as previously described [
17]. HT1080 cells (300,000 cells/well) were seeded in a 6-well plate and incubated overnight. Cells were harvested the next day and centrifuged at 300×
g for 5 min. Then, cells were centrifuged at 300×
g for 5 min after being washed with PBS buffer. The collected cells were stained with FeRhoNox-1 in PBS and kept in a CO
2 incubator for 30 min. Following HBSS washing, cell culture was dissolved in 300 μL of HBSS containing SytoxBlue, and examined using BD LSRFortessa (BD Biosciences). Y585 (FeRhoNox-1) and V450 (SytoxBlue) channels were used to measure fluorescence. Only non-permeable live cell fluorescence was evaluated. Per condition, a minimum of 10,000 cells were examined.
2.8. Measurement of Mitochondrial Membrane Potential
In a 6-well plate, HT1080 (300,000 cells/well) and IMR-32 (500,000 cells/well) cells were seeded. The following day, after cells had received the indicated treatment, TMRE (200 nM) was added, and the mixture was incubated for 30 min. The cells were washed with PBS to remove extra TMRE before being collected for analysis with the BD LSRFortessa (BD Biosciences). B575 (TMRE) and V450 (SytoxBlue) channels were used to measure fluorescence. Only non-permeable live cell fluorescence was evaluated. Per condition, a minimum of 10,000 cells were examined.
2.9. Measurement of GSH Levels
As previously reported [
17], glutathione levels were determined using QuantiChrom Glutathione Assay Kit (BioAssay Systems, Hayward, CA, USA, DIGT-250). Concisely, 1,000,000 cells per condition (HT1080 or IMR-32 cells) were seeded in a 6-well plate. The next day, cells were treated as indicated in each experiment. After that, cells were gathered, transferred to a new tube, and centrifuged at 425×
g for 5 min at 4 °C. After being resuspended in 300 μL of PBS, each cell pellet was lysed using ultrasound. Each lysate was centrifuged at 14,000 rpm for 10 min at 4 °C. The cleared lysate was then used to calculate the amount of GSH present in each sample following the kit descriptions.
2.10. Measurement of Intracellular Glutamate Levels
Intracellular glutamate levels were measured using Amplex® Red Glutamic Acid/Glutamate Oxidase Assay Kit (Thermo Fisher Scientific, A12221). HT1080 cells (400,000 cells/well) were seeded in a 6-well plate and incubated overnight. The following day, cells were treated according to the conditions described in each experiment and collected by centrifugation at 300× g for 5 min. After removing the supernatant, the pellet was resuspended in PBS buffer. Afterward, each sample was centrifuged again at 300× g for 5 min, and the pellet was resuspended in 100 μL of Tris HCl buffer (0.1 M, pH = 7.5). Then, cells were lysed by sonication, and each sample was diluted (2×) with Tris HCl buffer. Next, 50 μL of the diluted samples were transferred into separate wells of a microplate (OptiPlate 96-well plate), and the amount of intracellular glutamate was calculated following the kit descriptions.
2.11. Measurement of ATP Levels
ATP levels were determined using CellTiter-Glo 2.0 Assay Kit (Promega, Madison, WI, USA, Cat# G9242/3) based on the firefly luciferin–luciferase assay system. Briefly, HT1080 cells (400,000 cells/well) were seeded in a 96-well plate in the absence (control) or presence of nemorosone, CCCP, or oligomycin, in line with the conditions described in the experiment legend. The measurement was performed in accordance with the instructions of the kit.
2.12. Measurement of Mitochondrial Respiration in Intact Cells
Intact HT1080 or IMR-32 cells were added to a 2 mL chamber at a concentration of 1,000,000 cells/mL. Oxygen consumption was measured at 37 °C using a high-resolution respirometer (Oxygraph-2k Oroboros Instruments, Innsbruck, Austria). Oxygen flow per cell (pmol·s
−1·mL
−1) was recorded continuously using DatLab software 6 (Oroboros Instruments). After approximately 10 min of monitoring oxygen consumption, corresponding sequential injections of selected compounds and inhibitors were performed as indicated by the phosphorylation control protocol [
31].
2.13. OCR and ECAR Measurement
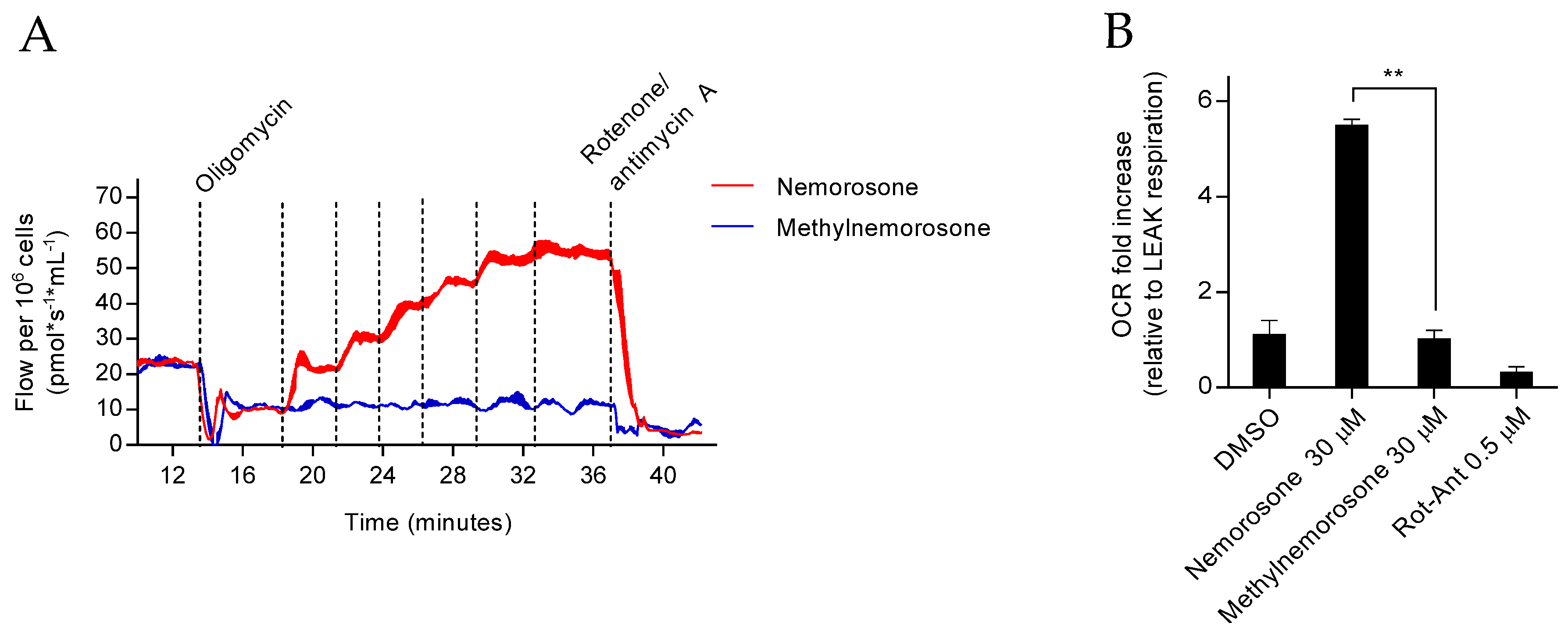
The Seahorse XFe96 Analyzer (Agilent) was used to measure the oxygen consumption rate (OCR) and extracellular acidification rate (ECAR). HT1080 cells (200,000 cells/well) were seeded into 96-well plates and incubated for 24 h. Before the assay, the culture medium was changed to a similar medium without phenol red and with 25 mM glucose, 1 mM sodium pyruvate, and 1 mM glutamine, and the cells were equilibrated for 30 min at 37 °C. During the assay, the compounds of interest were added, and the OCR and ECAR values were measured at intervals of approximately 6 min.
2.14. RNA Sequencing and Data Analysis
An RNA 6000 nano chip (Agilent Technologies, Santa Clara, CA, USA) as well as an RNA labchip (Caliper GX-Perkin Elmer) were used to assess the total RNA quality of gall and control samples. Concentrations were determined using a Quant-it Ribogreen RNA assay (Life Technologies). Then, 265 ng of RNA were employed for the library prep through the QuantSeq 3′ mRNA libr prep FWD kit (Lexogen). Library prep was carried out in accordance with the recommendations of the manufacturer. In brief, first-strand cDNA synthesis was performed, followed by an RNA removal step. Then, second-strand synthesis was performed with the use of UMIs, after which the cDNA was purified using beads (Lexogen). In addition to being purified with beads, the cDNA was used for 13 cycles of enrichment PCR. Using a high sensitivity DNA chip from Agilent Technologies, the quality was examined. To enable equimolar library pooling, a qPCR assay was used to quantify the libraries in accordance with the Illumina protocol. Finally, sequencing was carried out on a Nextseq500 using 20% Phix spike-in (single-end reads, 76 cycles).
Through the use of FastQC (version 0.11.9), the quality of the reads was confirmed [
32]. The following parameters were used to trim reads with Trimmomatic (version 0.39): ILLUMINACLIP:<TruSeq3-SE adapter file>:3:30:10, SLIDINGWINDOW:5:20, MINLEN:20 [
33]. STAR (version 2.7.8a) was used for mapping with the subsequent parameters: readFilesCommand zcat, outFilterMultimapNmax 1 and outSAMtype BAM SortedByCoordinate using the GRCh38.106 genome build [
34].
These R packages were utilized to create a count table: GenomicFeatures (version 1.44.2), to convert the GRCh38.106 GTF file into a Granges object, and GenomicAlignments (version 1.28.0), for the summarizeOverlaps function to create the count table [
35]. The counting options were as follows: mode = ‘Union’, singleEnd = TRUE, and ignore.strand = FALSE. To find differentially expressed genes, DESeq2 (version 1.32.0) was used with a Benjamini–Hochberg FDR cutoff of 0.05 [
36]. Lists of differentially expressed genes were used for downstream analysis using Ingenuity pathway analysis (Qiagen). An R (v 4.1.3) environment was used for the analysis.
2.15. RNA Isolation and Analysis by RT-qPCR
Total RNA from treated HT1080 cells was extracted according to the NucleoSpin® RNA Plus protocol (fifth revision, corresponding to January 2021) prepared by MACHEREY-NAGEL GmbH & Co. KG (Düren, Germany). For DNA synthesis, a C1000 Touch® thermocycler (Bio-Rad) was used. The qPCR analysis was performed under the following conditions: 95 °C for denaturation, 60 °C for hybridization, and 70 °C for elongation. qbase+ software (Biogazelle) was used to calculate the expression levels of mRNA (HMOX1: Bio-Rad, Hercules, CA, USA, qHsaCIP0033307) from the structural genes (housekeeping genes), HMBS (Bio-Rad, qHsaCID0038839) and RPL3 (Bio-Rad, qHsaCED0038656), which were used as internal references.
2.16. Protein Extraction and Western Blot Analysis
At designated times, test compound-treated HT1080 cells were harvested and subjected to two washes with cold PBS solution. A cell lysis buffer (Cell Signaling Technology, Danvers, MA, USA) was used to extract the total cytosolic proteins, and their concentrations were determined by the Bradford method. In the wells of the 10% SDS-PAGE gel, 25 µg of protein were loaded along with the molecular weight marker. After performing the run (1 h, 100 V), the transfer of the proteins from the gel to nitrocellulose membranes was carried out. Subsequently, the membranes were blocked with 5% skim milk powder prepared in TBST saline (0.05% Tween 20). The membranes were incubated for 24 h at 4 °C with each primary antibody of interest (except in the case of β-tubulin, with which they were incubated for 1 h). Peroxidase-labeled secondary antibodies (PerkinElmer Life Sciences) were used to detect immunoreactive proteins.
2.17. Statistical Analysis
Unpaired Student’s t-test was carried out, using GraphPad Prism version 9.2.0 (GraphPad Software, San Diego, CA, USA), to calculate p values (* p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001; see figure legends for more information), with the exception of Figure 3C, where a two-way ANOVA test was employed. Unless otherwise stated, data are displayed as the mean ± SD of three separate experiments.
4. Discussion
In the current work, nemorosone, a phytochemical isolated from the floral resin of the
C. rosea plant, was identified to induce ferroptosis in fibrosarcoma and neuroblastoma cells. In 20 years of nemorosone anticancer-effect research, this is the first report that expands the potential of nemorosone by showing its capacity to induce another mechanism of cell death than apoptosis [
4,
11].
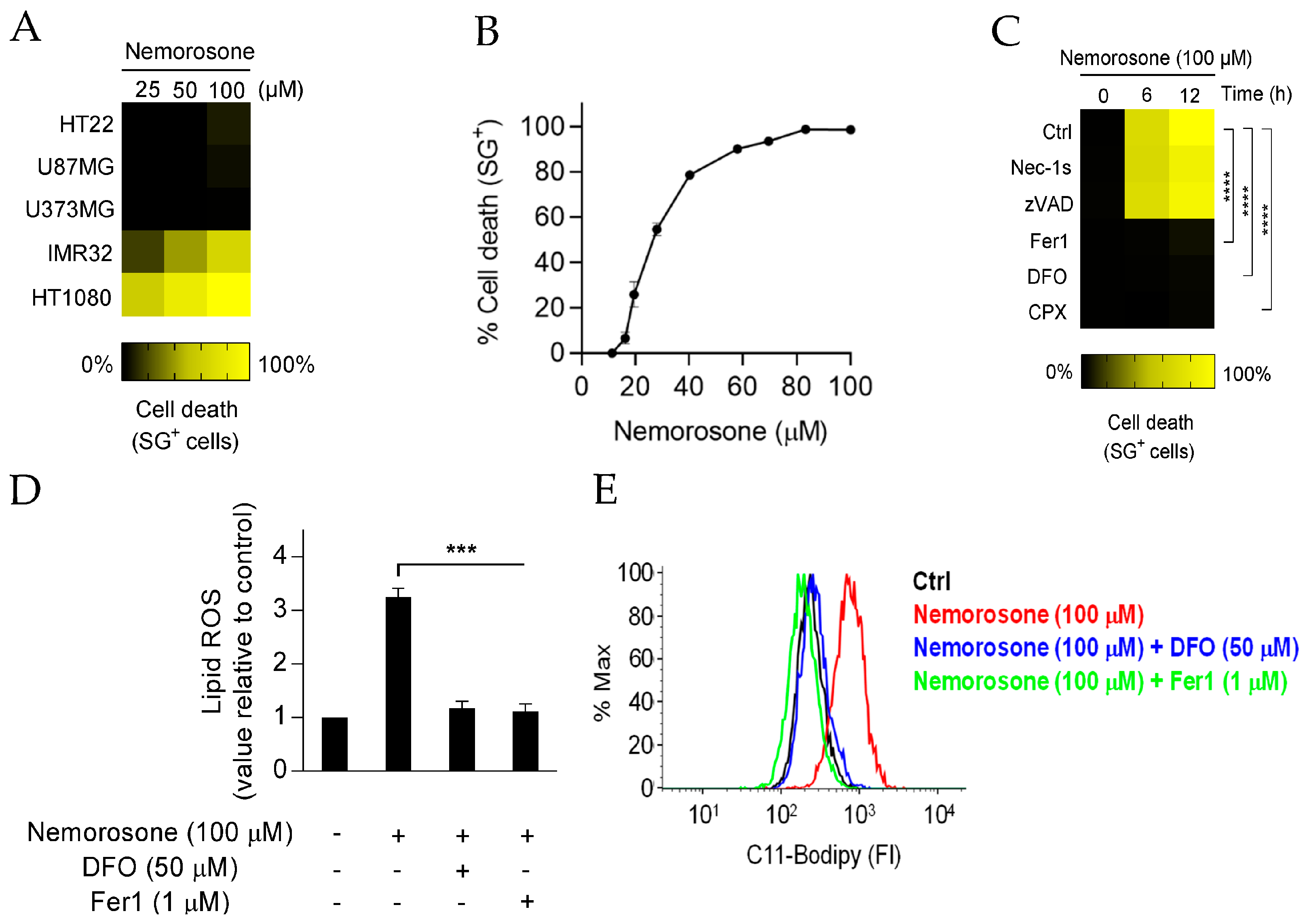
Notably, while nemorosone was cytotoxic against the neuroblastoma and fibrosarcoma cell lines, it did not show any effect on glioblastoma U87MG and U373MG cells. In general, the sensitivity to ferroptosis depends on the different endogenous mechanisms that protect cells against the lipid peroxidation that drives ferroptosis [
46]. Recently, ferroptosis suppressor protein 1 (FSP1) was shown to play an essential role in the resistance of U373MG cells to erastin-induced System xc inhibition, while some unidentified mechanisms that support GPX4 function, independent of System xc activity, were also observed [
47]. In line with this, a higher methionine uptake has been reported in gliomas than in normal astrocytes, which positively correlated with tumor viability and aggressiveness and indicated a greater reliance on transsulfuration, a metabolic pathway that connects methionine with glutathione biosynthesis independent of the System xc antiporter [
48,
49]. These reports allow us to explain a priori the resistance of glioblastoma cell lines to nemorosone-induced ferroptosis, although new experimental results are required to corroborate the aforementioned hypotheses. Conversely, based on the literature data, HT1080 and IMR-32 cells appear to be sensitive to the ferroptosis induced by decreased GSH levels through System xc inhibition and the increased labile iron pool (LIP) [
17,
19]. In this investigation, it was found that nemorosone induces ferroptosis by modulating these two parameters (
Section 3.2 and
Section 3.4).
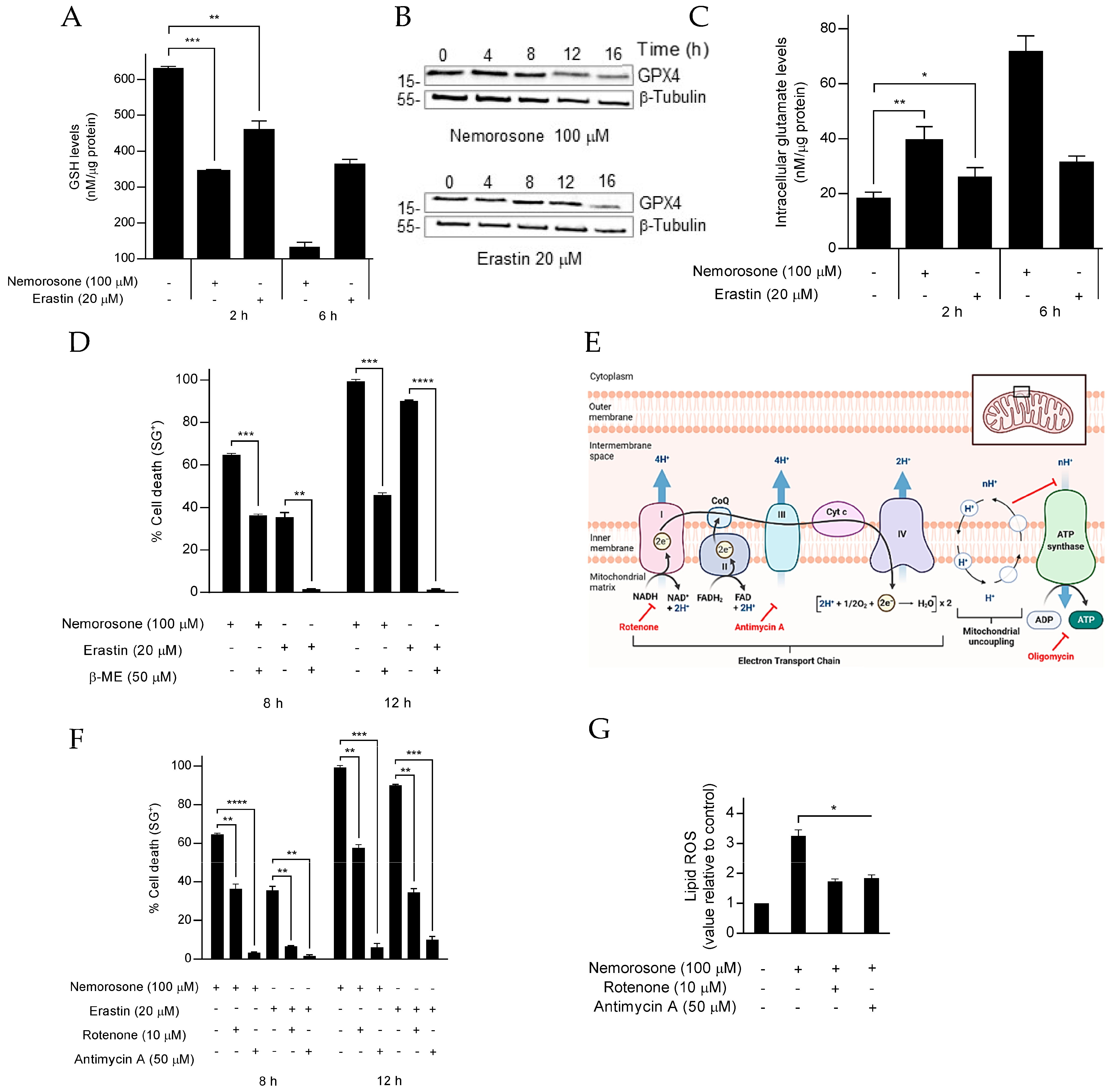
On one hand, nemorosone-induced ferroptosis in HT1080 and IMR-32 cells involves the drop in the glutathione levels that can be associated with a blockade of the System xc cystine/glutamate antiporter or SLC7A11, which resembles the canonical ferroptosis-inducing pathway triggered by a class I FIN, such as erastin, also known as cysteine-deprivation-induced ferroptosis [
19,
23]. Inhibition of cystine import, which is required for GSH synthesis, results in depletion of intracellular GSH levels [
16,
33,
37], an important cofactor for selenium-dependent GPX4. Therefore, GSH depletion by nemorosone could indirectly inactivate GPX4, leading to production of lipid ROS, which in turn results in lipid peroxidation and ferroptotic cell death [
19,
42]. It should be mentioned that more direct experimental approaches, such as the [
14C]-cystine uptake assay [
19], are required to confirm nemorosone-induced blockade of the System xc antiporter. Furthermore, inhibition of cystine entry is not necessarily the only pathway by which nemorosone could be lowering GSH levels. In fact, the decrease in the levels of NADPH, an electron-donor agent, relevant in the reduction of oxidized substrates, was reported as a common phenotypic effect for different structurally divergent uncoupling compounds [
24]. It has been suggested that the dissipation of the mitochondrial membrane potential renders nicotinamide nucleotide transhydrogenase (NNT) incapable of maintaining the reduced NADPH state, which in turn can affect GSH regeneration via glutathione reductase (GR) [
50,
51]. In effect, the abundance of NADPH functions as a biomarker that is inversely correlated with the sensitivity of cells to the inducers of ferroptosis [
52]. This important experimental issue should also be studied in future research.
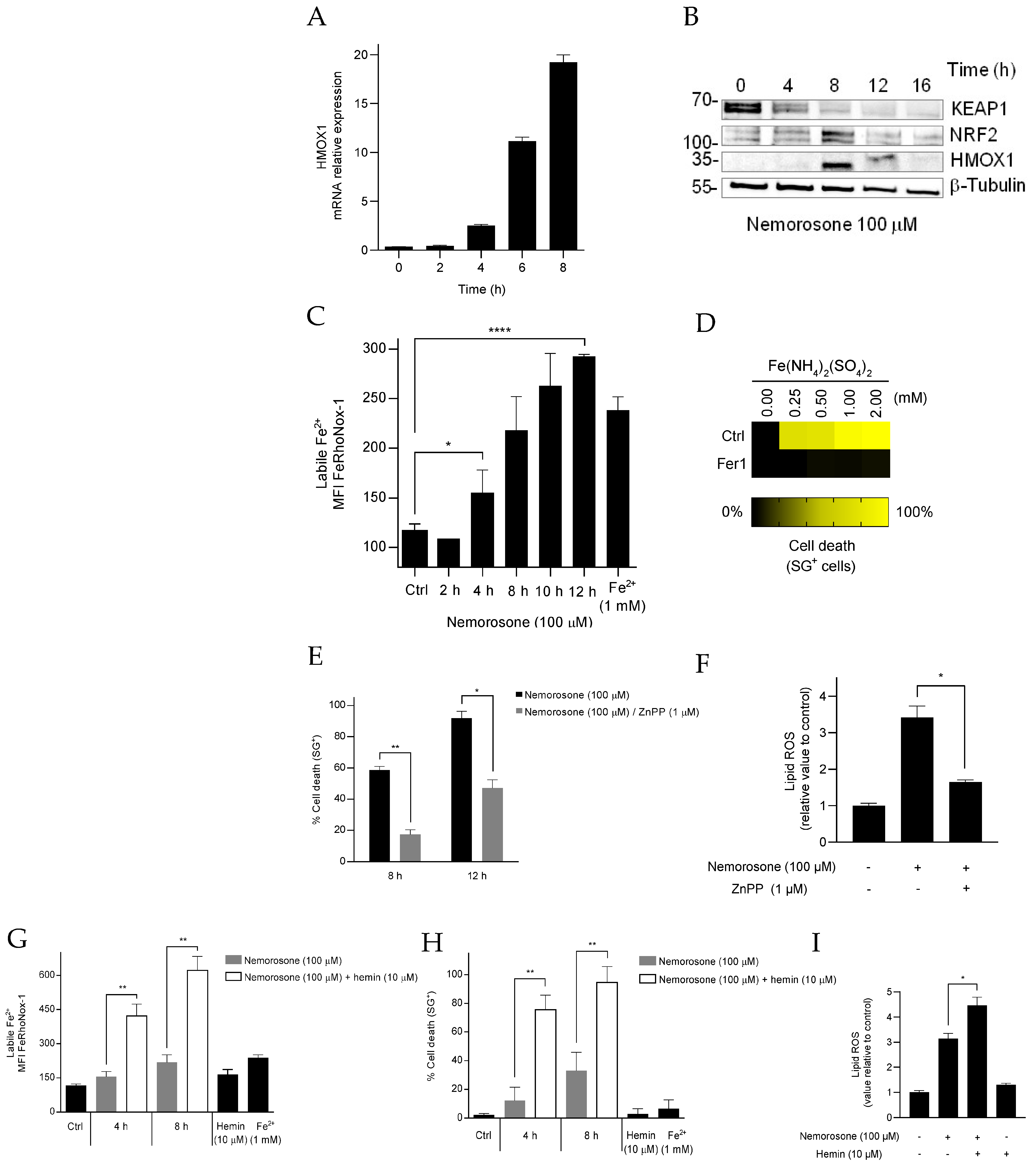
On the other hand, nemorosone induces a non-canonical mechanism of ferroptosis by increasing the LIP, in response to the excessive activation of heme oxygenase-1 by targeting KEAP1 and NRF2, which is sufficient to trigger toxic lipid peroxidation. This result is comparable with the effect of withaferin A (WA), a natural FIN isolated from
Withania somnifera roots, which at a medium dose induces ferroptosis through a massive upregulation of HMOX1 [
17]. Likewise, Tagitinin C, another natural compound, induces ferroptosis in colorectal cancer cells through the PERK–NRF2–HMOX1 signaling pathway, and again the significant overexpression of HMOX1 led to the increase in the LIP, which promoted lipid peroxidation and ferroptosis [
53]. As can be seen, these results with nemorosone add to a still small, but apparently growing, list of natural compounds that modulate the NRF2–HMOX1 axis to induce ferroptotic cell death, which could be related to some natural protection mechanism of plants (not yet reported) against pathogenic microorganisms. Similarly, HMOX1 was also shown as an essential enzyme that is involved in iron supplementation and lipid peroxidation in erastin-induced ferroptosis [
54].
By giving cancerous cells antioxidant and cytoprotective effects and by removing toxic intracellular heme, the inducible intracellular enzyme HMOX1 was also shown to play a role in cancer progression [
55]. This is in line with the fact that HMOX1 is elevated in various human malignancies such as, for example, fibrosarcoma tumors and HT1080 cells [
56,
57]. In consequence, HMOX1 inhibition was explored to reduce tumor growth [
43,
44,
46]. Nevertheless, based on the current data, a massive activation of HMOX1 is important to kill HT1080 cells, strongly suggesting the efficacy of an opposite strategy: making tumor cells sensitive to the induction of ferroptosis via the therapeutic overactivation of HMOX1. At the same time, the active role of HMOX1 in tumor cells constitutes a significant difference compared to the healthy tissue and is, consequently, a way by which compounds such as nemorosone could induce a selective ferroptosis mechanism in cancer cells such as fibrosarcoma.
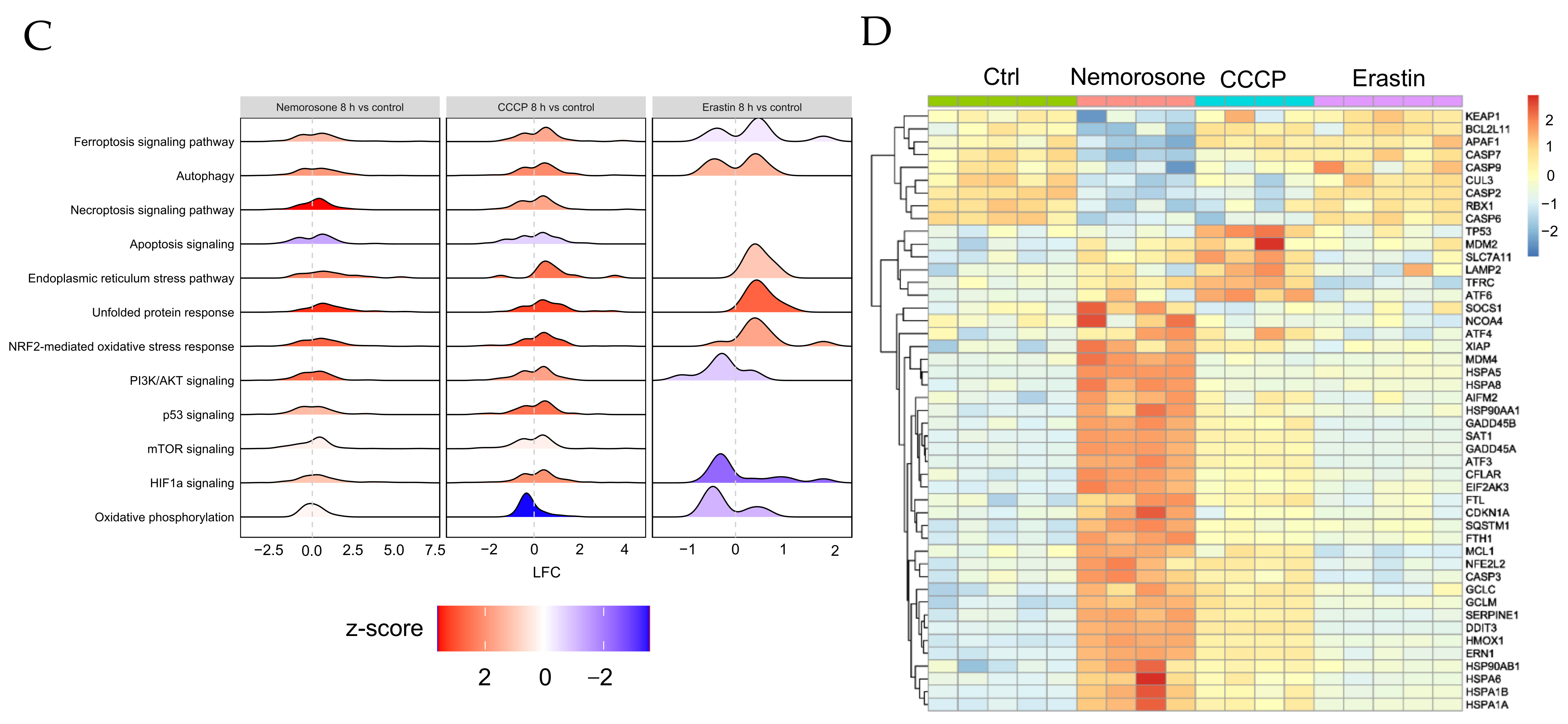
However, nemorosone-induced activation of the NRF2-mediated oxidative stress response pathway (
Figure 4C), with the consequent modulation of NRF2 target genes (
Table S1), shows that nemorosone activated the NRF2 pathway as an antioxidant and antiferroptotic response. It is the disproportionate upregulation of HMOX1 compared to other genes that results in a pro-ferroptotic effect. It is notable that
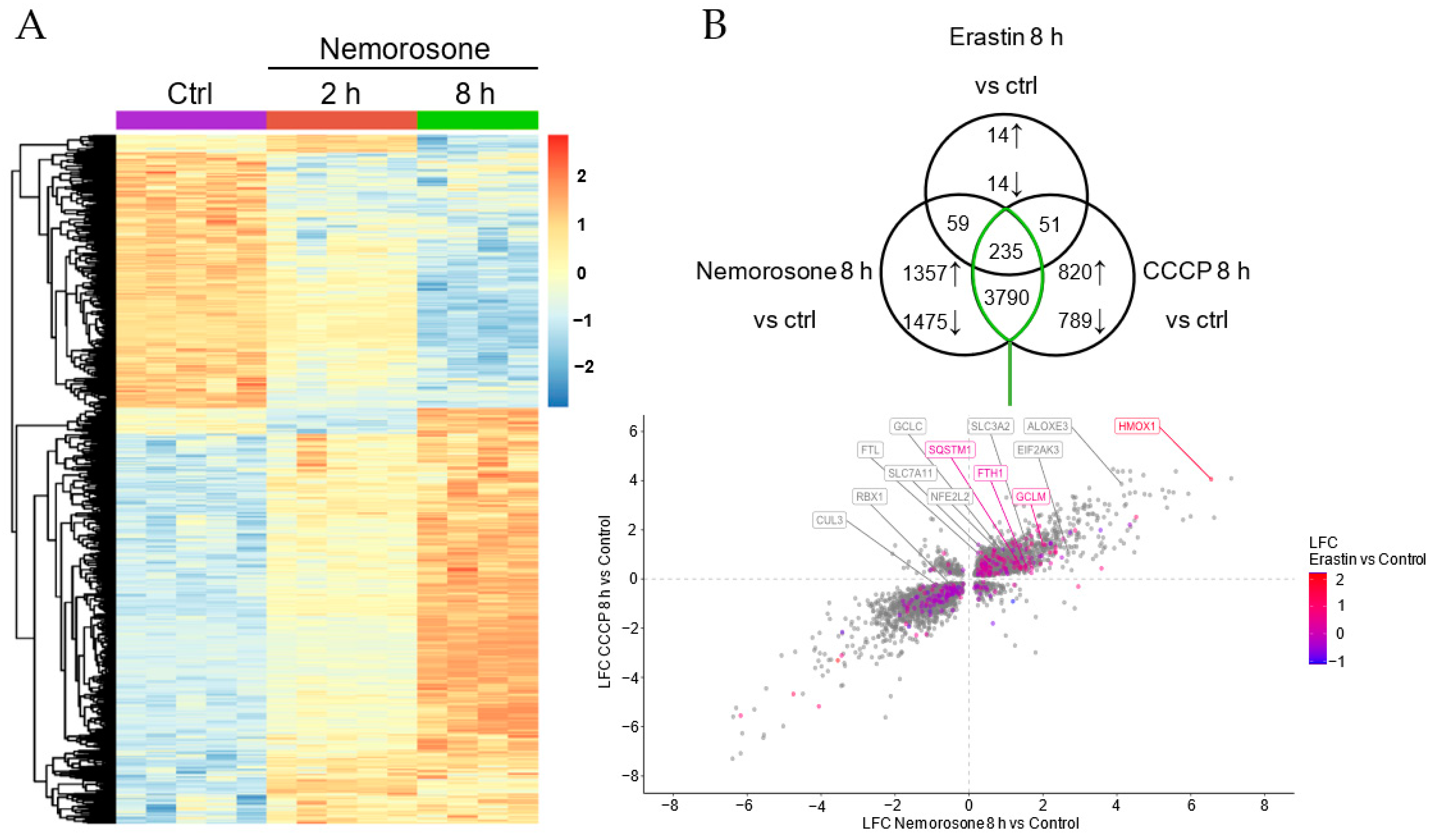
HMOX1 is the gene most upregulated by nemorosone: more than 90 times compared to the control, while
FTH1 is upregulated less than 3 times (
Figure 4B and
Table S1). That is, nemorosone generates an expression of
HMOX1 30 times higher than the induced expression of
FTH1, which, similar to what was reported for WA, suggests the induction of ferroptosis by raising the LIP in a context of insufficient ferritin buffering capacity [
17]. In other words, the effect of nemorosone reveals a hormetic response associated with the NRF2–HMOX1 axis: a protective effect after moderate activation (classical and most common reports) vs. a cytotoxic effect after excessive activation (reported for some naturally occurring ferroptosis-inducing compounds).
It remains to be answered why nemorosone and other natural compounds generate such an overactivation of heme oxygenase-1. First of all, it must be taken into account that activation of HMOX1 by pathways other than NRF2 cannot be excluded. Several classes of stress-responsive transcription factors that activate
HMOX1 gene have also been identified, such as members of the heat-shock factor (HSF), nuclear factor-κB (NF-κB), and activator protein-1 (AP-1) families [
58]. On the other hand, nemorosone was identified as a natural activator of the p300 histone acetyltransferase that enhanced histone acetylation in cells [
59]. At the same time, p300-mediated NRF2 acetylation was shown to be essential for the maximal binding of NRF2 to specific ARE (antioxidant response element)-containing promoters [
60]. Moreover, p300 was recently reported to compete with KEAP1 for the regulation of NRF2, enhancing the protein level of NRF2 and allowing NRF2 to translocate to the nucleus to upregulate the transcription of target genes [
61]. This possible nemorosone-induced epigenetic regulation of the KEAP1–NRF2–HMOX1 axis could also explain the capacity of nemorosone to induce non-canonical ferroptosis through excessive activation of HMOX1. Consistently, we observed that nemorosone also induces the downregulation of
KEAP1,
CUL3, and
RBX1, while it upregulates
SQSTM1 and
EIF2AK3 (PERK) (
Figure 4B,D), all of which suggests the activation of the SQSTM1–KEAP1–NRF2–HMOX1 and PERK–NRF2–HMOX1 pathways as part of
HMOX1 overactivation-mediated cytotoxicity. It is important to highlight that the regulation of the expression at gene level of the KEAP1–CUL3–RBX1–NRF2 complex does not exclude the possibility of regulation at the protein level by a direct binding between nemorosone and KEAP1, as was reported in the aforementioned case of withaferin A [
17]. All these factors need to be addressed in future experimental activities.
The time relation existing between the two ferroptosis mechanisms triggered by nemorosone is also noteworthy: the drop of GSH, resulting in a lipid peroxidation increase, appears from 2 h (an early event), while HMOX1 overexpression, with its consequent increase in the intracellular labile Fe
2+ levels, only begins at 6 to 8 h (a later event). Moreover, before the execution of the later event, there is already cell death induction in some cells. However, the cell death level is accelerated and enhanced at the time points in which HMOX1 expression can be associated with labile Fe
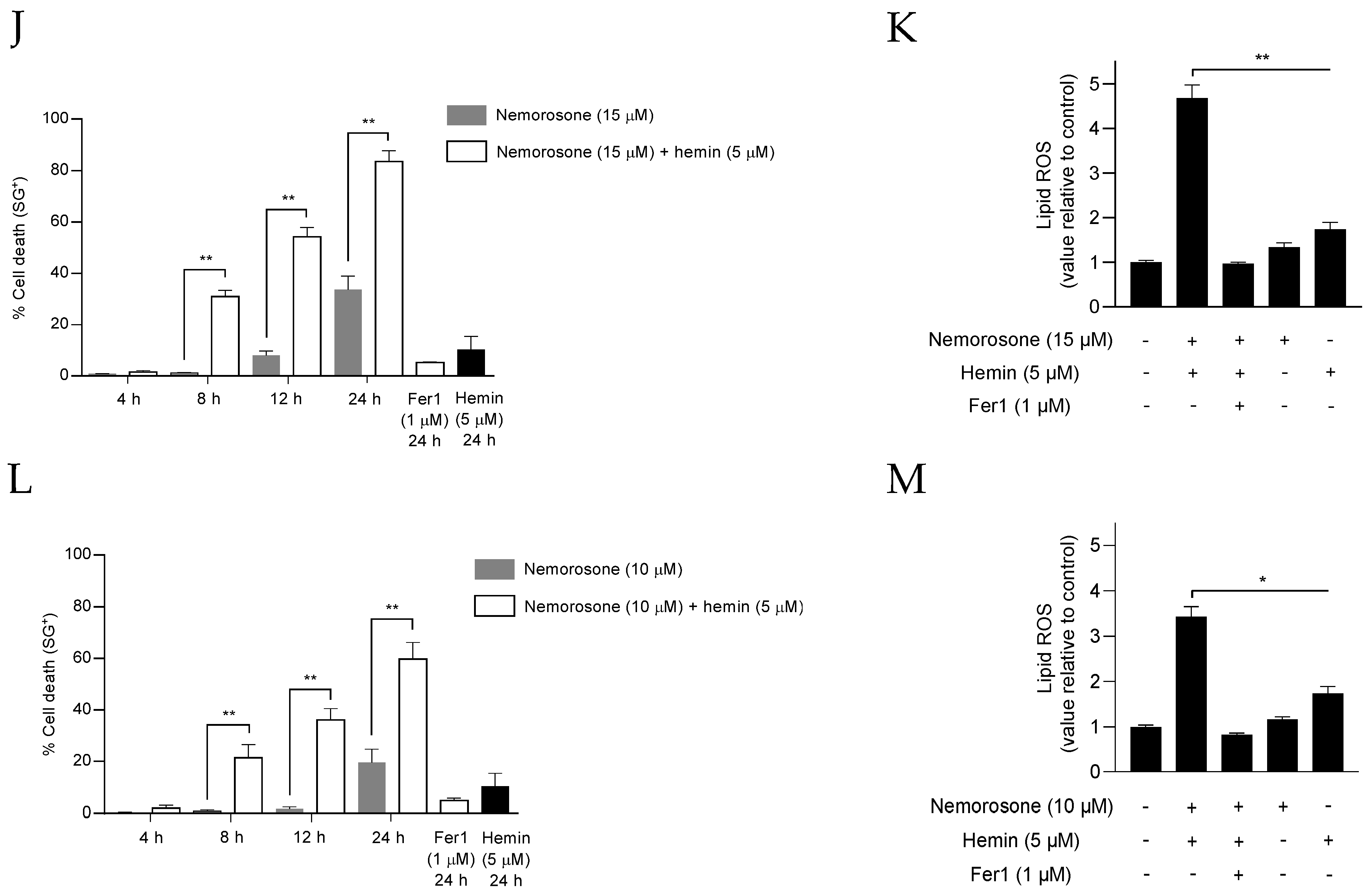
2+ and an additional lipid peroxidation increase. It is unclear whether this difference in cell death by early lipid peroxidation due to blockage of cystine import and by the later event represent two distinct responding populations, in which cell resistance to cell death during the early lipid peroxidation wave receives a second ferroptotic hit due to HMOX1 upregulation, hemin degradation, and the increase in the labile Fe
2+ pool. Importantly, high sensitization values were achieved by combining nemorosone and the substrate of HMOX1 hemin, which confirms the cytotoxic role of nemorosone-induced HMOX1 activation and points out a possible therapeutic approach to be experimentally tested in in vivo experiments. The aforementioned results allow for the conclusion that nemorosone exerts an erastin-like ferroptosis (intrinsic ferroptosis) in fibrosarcoma cells that is characterized by the concurrence of both canonical (decreasing GSH levels) and non-canonical (increasing LIP through HMOX1 upregulation) mechanisms. This may confer more therapeutic efficacy to nemorosone by circumventing the resistance mechanisms of the tumor cells that bypass the System xc blockade or the depletion of GSH levels, an effect suggested by the persistence of the induction of cell death, unlike erastin, in the presence of β-ME (
Figure 2D).
On the other hand, erastin-induced cell death and, in general, cysteine-deprivation-induced (CDI) ferroptosis are exerted by transient mitochondrial membrane potential (MMP) hyperpolarization, in such a way that low concentrations (10 μM) of the mitochondrial uncoupler CCCP can prevent (by the drop of MMP) CDI lipid ROS accumulation and protect against ferroptosis [
23]. However, we confirmed that nemorosone acts as a mitochondrial uncoupler, dissipating (similar to CCCP) the transmembrane proton gradient prior to cell death execution. The possible involvement of mitochondrial uncoupling in ferroptosis induced by nemorosone was approached by using a high concentration of CCCP (50 μM) and methylnemorosone. While CCCP acted similarly to nemorosone regarding ferroptosis induction, methylnemorosone, which cannot exert mitochondrial uncoupling activity anymore, completely lost cytotoxicity. Altogether, this suggests that mitochondrial uncoupling is indeed required for nemorosone to trigger ferroptosis in fibrosarcoma cells. Furthermore,
Figure S3 shows a possible link between HMOX1 over-activation and mitochondrial uncoupling: CCCP, a classic mitochondrial uncoupler, also increases Fe
2+ levels upon
HMOX1 upregulation.
In addition, the obtained results at a high concentration of CCCP and the reported capacity to protect against erastin-induced ferroptosis at a low concentration [
23] show a dual role as uncoupler compounds to induce ferroptosis or protect against it by varying the concentration. The protective mechanism could be an important approach to treat several ferroptosis-associated diseases such as ischemic organ injury, brain damage, and kidney failure [
62,
63], expanding the potential application of mitochondrial uncouplers.
To sum up, here, we connect, for the first time, mitochondrial uncoupling with ferroptotic cell death induction by the use of two closely related agents: proficient (nemorosone) and deficient (methylnemorosone). The cascade of cellular effects leading to ferroptosis induced by the mitochondrial uncoupler compounds in cancer cells is still an unexplored and emerging area of research and therapeutic opportunities.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







