
Tubule-Derived Follistatin Is Increased in the Urine of Rats with Renal Ischemia and Reflects the Severity of Acute Tubular Damage
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Protocols
2.2. Measurement of Renal Function
2.3. Immunohistochemical Analysis
2.4. Histological Examination
2.5. ELISA
2.6. In Situ Hybridization
2.7. Real-Time PCR
2.8. Statistical Analysis
3. Results
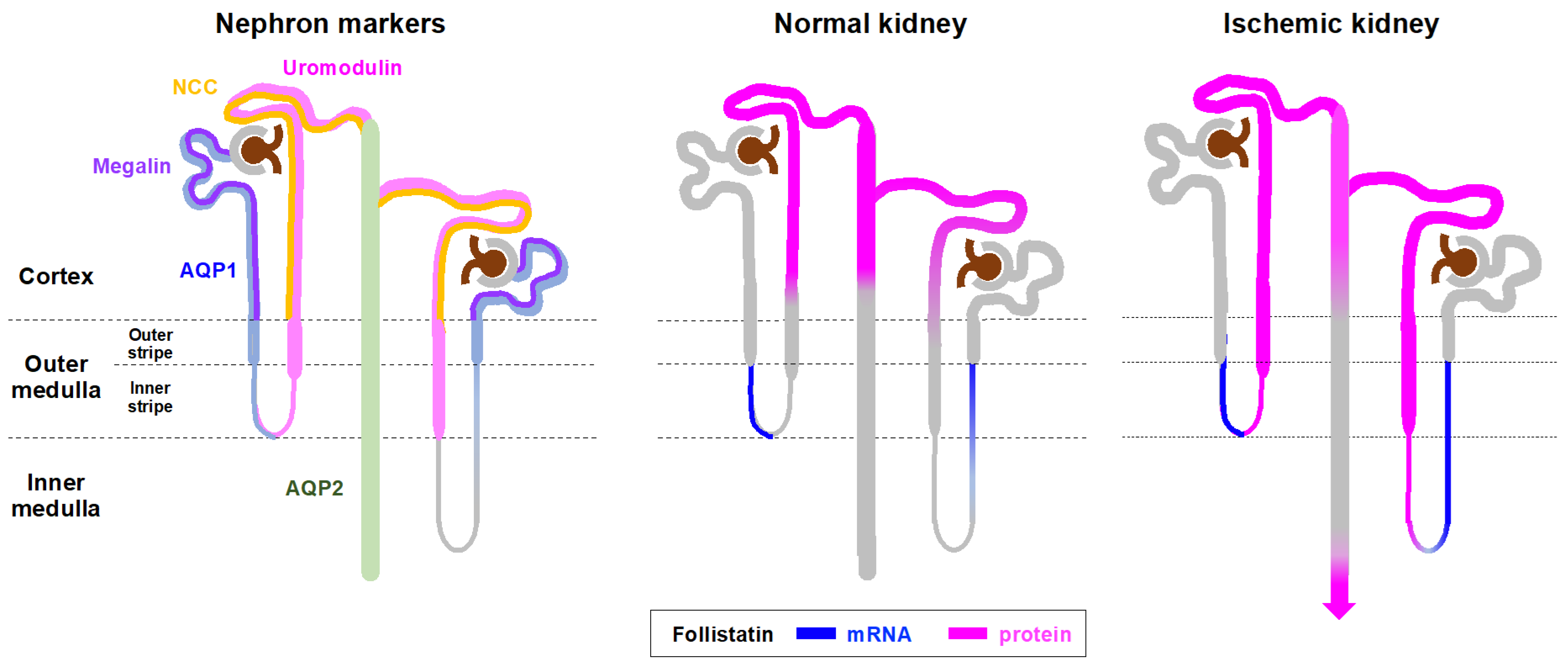
3.1. Localization of Follistatin in Normal and Ischemic Rat Kidneys
3.2. Expression and Localization of Follistatin mRNA in Normal and Ischemic Rat Kidneys
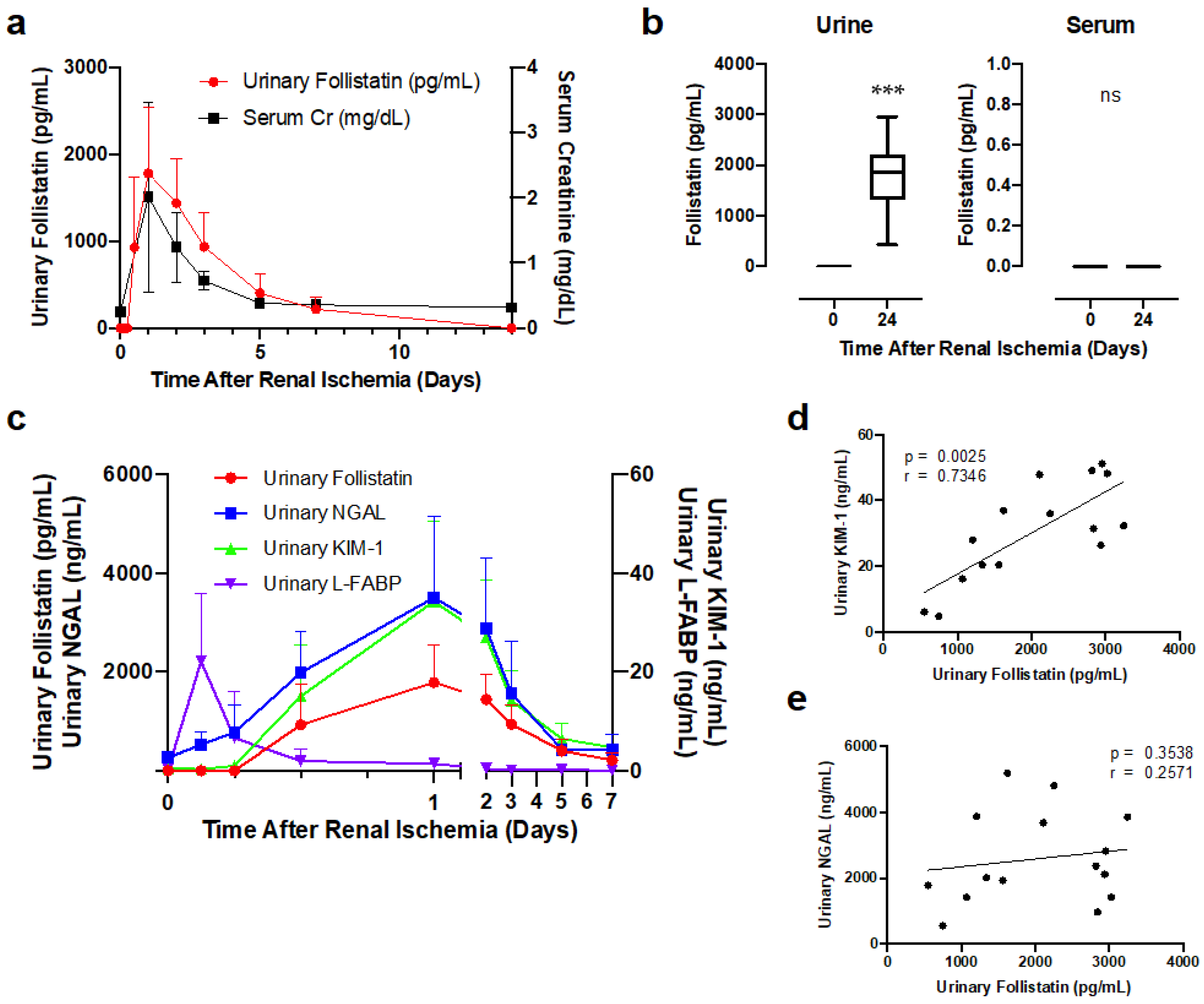
3.3. Urinary Follistatin Level in Rats with Renal Ischemia
3.4. Correlation of Urinary Follistatin with Severity of Acute Tubular Damage in Ischemic Rats
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marino, F.E.; Risbridger, G.; Gold, E. The therapeutic potential of blocking the activin signalling pathway. Cytokine Growth Factor Rev. 2013, 24, 477–484. [Google Scholar] [CrossRef]
- Phillips, D.J.; de Kretser, D.M.; Hedger, M.P. Activin and related proteins in inflammation: Not just interested bystanders. Cytokine Growth Factor Rev. 2009, 20, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Nakasatomi, M.; Takei, Y.; Ikeuchi, H.; Sakairi, T.; Kaneko, Y.; Hiromura, K.; Nojima, Y.; Maeshima, A. Identification of Urinary Activin A as a Novel Biomarker Reflecting the Severity of Acute Kidney Injury. Sci. Rep. 2018, 8, 5176. [Google Scholar] [CrossRef] [Green Version]
- Maeshima, A.; Zhang, Y.Q.; Nojima, Y.; Naruse, T.; Kojima, I. Involvement of the activin-follistatin system in tubular regeneration after renal ischemia in rats. J. Am. Soc. Nephrol. 2001, 12, 1685–1695. [Google Scholar] [CrossRef]
- Maeshima, A.; Maeshima, K.; Nojima, Y.; Kojima, I. Involvement of Pax-2 in the action of activin A on tubular cell regeneration. J. Am. Soc. Nephrol. 2002, 13, 2850–2859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeshima, A.; Nojima, Y.; Kojima, I. Activin A: An autocrine regulator of cell growth and differentiation in renal proximal tubular cells. Kidney Int. 2002, 62, 446–454. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, S.; Maeshima, A.; Kojima, I.; Nojima, Y. Activin A is a potent activator of renal interstitial fibroblasts. J. Am. Soc. Nephrol. 2004, 15, 91–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeshima, A.; Mishima, K.; Yamashita, S.; Nakasatomi, M.; Miya, M.; Sakurai, N.; Sakairi, T.; Ikeuchi, H.; Hiromura, K.; Hasegawa, Y.; et al. Follistatin, an activin antagonist, ameliorates renal interstitial fibrosis in a rat model of unilateral ureteral obstruction. BioMed. Res. Int. 2014, 2014, 376191. [Google Scholar] [CrossRef] [Green Version]
- Maeshima, A.; Miya, M.; Mishima, K.; Yamashita, S.; Kojima, I.; Nojima, Y. Activin A: Autocrine regulator of kidney development and repair. Endocr. J. 2008, 55, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Maeshima, A.; Nojima, Y.; Kojima, I. The role of the activin-follistatin system in the developmental and regeneration processes of the kidney. Cytokine Growth Factor Rev. 2001, 12, 289–298. [Google Scholar] [CrossRef]
- Nakamura, T.; Takio, K.; Eto, Y.; Shibai, H.; Titani, K.; Sugino, H. Activin-binding protein from rat ovary is follistatin. Science 1990, 247, 836–838. [Google Scholar] [CrossRef] [PubMed]
- Robertson, D.M.; Klein, R.; de Vos, F.L.; McLachlan, R.I.; Wettenhall, R.E.; Hearn, M.T.; Burger, H.G.; de Kretser, D.M. The isolation of polypeptides with FSH suppressing activity from bovine follicular fluid which are structurally different to inhibin. Biochem. Biophys. Res. Commun. 1987, 149, 744–749. [Google Scholar] [CrossRef]
- Ueno, N.; Ling, N.; Ying, S.Y.; Esch, F.; Shimasaki, S.; Guillemin, R. Isolation and partial characterization of follistatin: A single-chain Mr 35,000 monomeric protein that inhibits the release of follicle-stimulating hormone. Proc. Natl. Acad. Sci. USA 1987, 84, 8282–8286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Kretser, D.M.; O’Hehir, R.E.; Hardy, C.L.; Hedger, M.P. The roles of activin A and its binding protein, follistatin, in inflammation and tissue repair. Mol. Cell. Endocrinol. 2012, 359, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Kanamoto, M.; Shimada, M.; Morine, Y.; Yoshizumi, T.; Imura, S.; Ikegami, T.; Mori, H.; Arakawa, Y. Beneficial effects of follistatin in hepatic ischemia-reperfusion injuries in rats. Dig. Dis. Sci. 2011, 56, 1075–1081. [Google Scholar] [CrossRef]
- Chen, Y.; Rothnie, C.; Spring, D.; Verrier, E.; Venardos, K.; Kaye, D.; Phillips, D.J.; Hedger, M.P.; Smith, J.A. Regulation and actions of activin A and follistatin in myocardial ischaemia-reperfusion injury. Cytokine 2014, 69, 255–262. [Google Scholar] [CrossRef]
- Koken, E.; Oyar, E.O.; Uyanikgil, Y.; Pazarlar, B.A.; Bilister, C.; Aksun, S.; Yigitturk, G.; Koken, E.C. Exogenous follistatin administration ameliorates cisplatin-induced acute kidney injury through anti-inflammation and anti-apoptosis effects. Bratisl. Lek. Listy 2020, 121, 143–150. [Google Scholar] [CrossRef] [Green Version]
- Kogawa, K.; Ogawa, K.; Hayashi, Y.; Nakamura, T.; Titani, K.; Sugino, H. Immunohistochemical localization of follistatin in rat tissues. Endocrinol. Jpn. 1991, 38, 383–391. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Sugino, K.; Titani, K.; Sugino, H. Follistatin, an activin-binding protein, associates with heparan sulfate chains of proteoglycans on follicular granulosa cells. J. Biol. Chem. 1991, 266, 19432–19437. [Google Scholar] [CrossRef]
- Watanabe, N.; Kawashima, H.; Li, Y.F.; Miyasaka, M. Identification and characterization of ligands for L-selectin in the kidney. III. Characterization of L-selectin reactive heparan sulfate proteoglycans. J. Biochem. 1999, 125, 826–831. [Google Scholar] [CrossRef]
- Borges, F.T.; Michelacci, Y.M.; Aguiar, J.A.; Dalboni, M.A.; Garofalo, A.S.; Schor, N. Characterization of glycosaminoglycans in tubular epithelial cells: Calcium oxalate and oxalate ions effects. Kidney Int. 2005, 68, 1630–1642. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, O.; Nakamura, T.; Shoji, H.; Shimasaki, S.; Hayashi, Y.; Sugino, H. A novel role of follistatin, an activin-binding protein, in the inhibition of activin action in rat pituitary cells. Endocytotic degradation of activin and its acceleration by follistatin associated with cell-surface heparan sulfate. J. Biol. Chem. 1997, 272, 13835–13842. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, E.P.; Overdier, K.H.; Sun, X.; Lin, L.; Liu, X.; Yang, Y.; Ammons, L.A.; Hiller, T.D.; Suflita, M.A.; Yu, Y.; et al. Urinary Glycosaminoglycans Predict Outcomes in Septic Shock and Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2016, 194, 439–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Liu, K.; Han, B.; Xu, Z.; Gao, X. The emerging role of follistatin under stresses and its implications in diseases. Gene 2018, 639, 111–116. [Google Scholar] [CrossRef]
- de Groot, E.; Veltmaat, J.; Caricasole, A.; Defize, L.; van den Eijnden-van Raaij, A. Cloning and analysis of the mouse follistatin promoter. Mol. Biol. Rep. 2000, 27, 129–139. [Google Scholar] [CrossRef]
- Lin, C.; Zhao, X.; Sun, D.; Zhang, L.; Fang, W.; Zhu, T.; Wang, Q.; Liu, B.; Wei, S.; Chen, G.; et al. Transcriptional activation of follistatin by Nrf2 protects pulmonary epithelial cells against silica nanoparticle-induced oxidative stress. Sci. Rep. 2016, 6, 21133. [Google Scholar] [CrossRef] [Green Version]
- Han, X.H.; Jin, Y.R.; Tan, L.; Kosciuk, T.; Lee, J.S.; Yoon, J.K. Regulation of the follistatin gene by RSPO-LGR4 signaling via activation of the WNT/beta-catenin pathway in skeletal myogenesis. Mol. Cell. Biol. 2014, 34, 752–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winters, S.J.; Ghooray, D.; Fujii, Y.; Moore, J.P., Jr.; Nevitt, J.R.; Kakar, S.S. Transcriptional regulation of follistatin expression by GnRH in mouse gonadotroph cell lines: Evidence for a role for cAMP signaling. Mol. Cell. Endocrinol. 2007, 271, 45–54. [Google Scholar] [CrossRef]
- Mutiara, S.; Kanasaki, H.; Oride, A.; Purwana, I.N.; Shimasaki, S.; Yamamoto, H.; Miyazaki, K. Follistatin gene expression by gonadotropin-releasing hormone: A role for cyclic AMP and mitogen-activated protein kinase signaling pathways in clonal gonadotroph LbetaT2 cells. Mol. Cell. Endocrinol. 2009, 307, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Dong, H.; Lin, C.; Sheng, J.; Zhang, F.; Su, J.; Xu, Z. Reduction of AUF1-mediated follistatin mRNA decay during glucose starvation protects cells from apoptosis. Nucleic. Acids Res. 2014, 42, 10720–10730. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Wei, S.; Lai, K.; Sheng, J.; Su, J.; Zhu, J.; Dong, H.; Hu, H.; Xu, Z. Nucleolar follistatin promotes cancer cell survival under glucose-deprived conditions through inhibiting cellular rRNA synthesis. J. Biol. Chem. 2010, 285, 36857–36864. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.Q.; Kanzaki, M.; Shibata, H.; Kojima, I. Regulation of the expression of follistatin in rat hepatocytes. Biochim. Biophys. Acta 1997, 1354, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Ow, C.P.; Abdelkader, A.; Hilliard, L.M.; Phillips, J.K.; Evans, R.G. Determinants of renal tissue hypoxia in a rat model of polycystic kidney disease. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 307, R1207–R1215. [Google Scholar] [CrossRef]
- Fahling, M.; Mathia, S.; Paliege, A.; Koesters, R.; Mrowka, R.; Peters, H.; Persson, P.B.; Neumayer, H.H.; Bachmann, S.; Rosenberger, C. Tubular von Hippel-Lindau knockout protects against rhabdomyolysis-induced AKI. J. Am. Soc. Nephrol. 2013, 24, 1806–1819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronco, C.; Bellomo, R.; Kellum, J.A. Acute kidney injury. Lancet 2019, 394, 1949–1964. [Google Scholar] [CrossRef] [PubMed]
- See, E.J.; Jayasinghe, K.; Glassford, N.; Bailey, M.; Johnson, D.W.; Polkinghorne, K.R.; Toussaint, N.D.; Bellomo, R. Long-term risk of adverse outcomes after acute kidney injury: A systematic review and meta-analysis of cohort studies using consensus definitions of exposure. Kidney Int. 2019, 95, 160–172. [Google Scholar] [CrossRef]
- Kellum, J.A.; Romagnani, P.; Ashuntantang, G.; Ronco, C.; Zarbock, A.; Anders, H.J. Acute kidney injury. Nat. Rev. Dis. Primers 2021, 7, 52. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagayama, I.; Takayanagi, K.; Hasegawa, H.; Maeshima, A. Tubule-Derived Follistatin Is Increased in the Urine of Rats with Renal Ischemia and Reflects the Severity of Acute Tubular Damage. Cells 2023, 12, 801. https://doi.org/10.3390/cells12050801
Nagayama I, Takayanagi K, Hasegawa H, Maeshima A. Tubule-Derived Follistatin Is Increased in the Urine of Rats with Renal Ischemia and Reflects the Severity of Acute Tubular Damage. Cells. 2023; 12(5):801. https://doi.org/10.3390/cells12050801
Chicago/Turabian StyleNagayama, Izumi, Kaori Takayanagi, Hajime Hasegawa, and Akito Maeshima. 2023. "Tubule-Derived Follistatin Is Increased in the Urine of Rats with Renal Ischemia and Reflects the Severity of Acute Tubular Damage" Cells 12, no. 5: 801. https://doi.org/10.3390/cells12050801