
Study of the Bcl-2 Interactome by BiFC Reveals Differences in the Activation Mechanism of Bax and Bak

{kind=link}
{kind=link}
{kind=link}
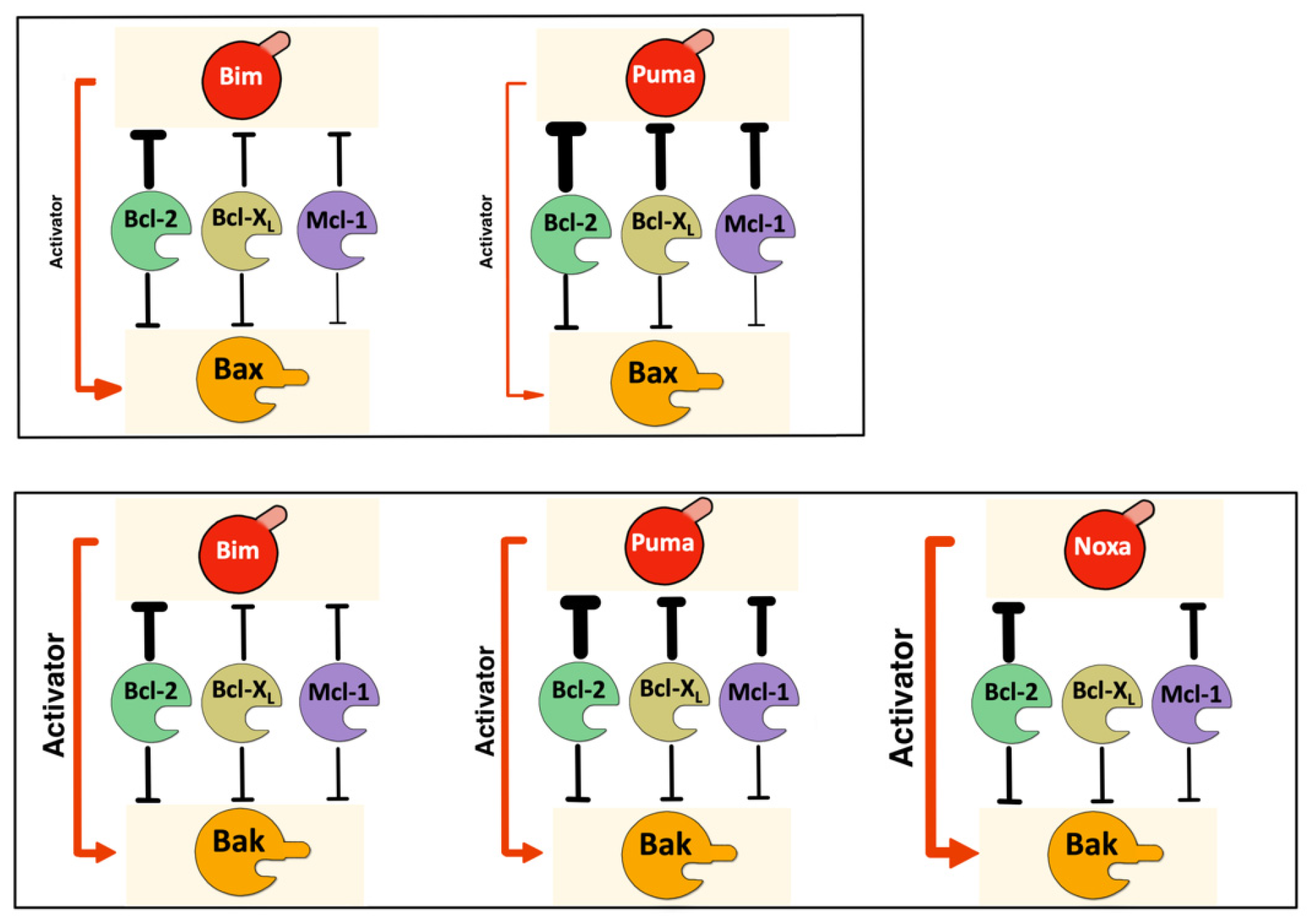{kind=link}
{kind=link}
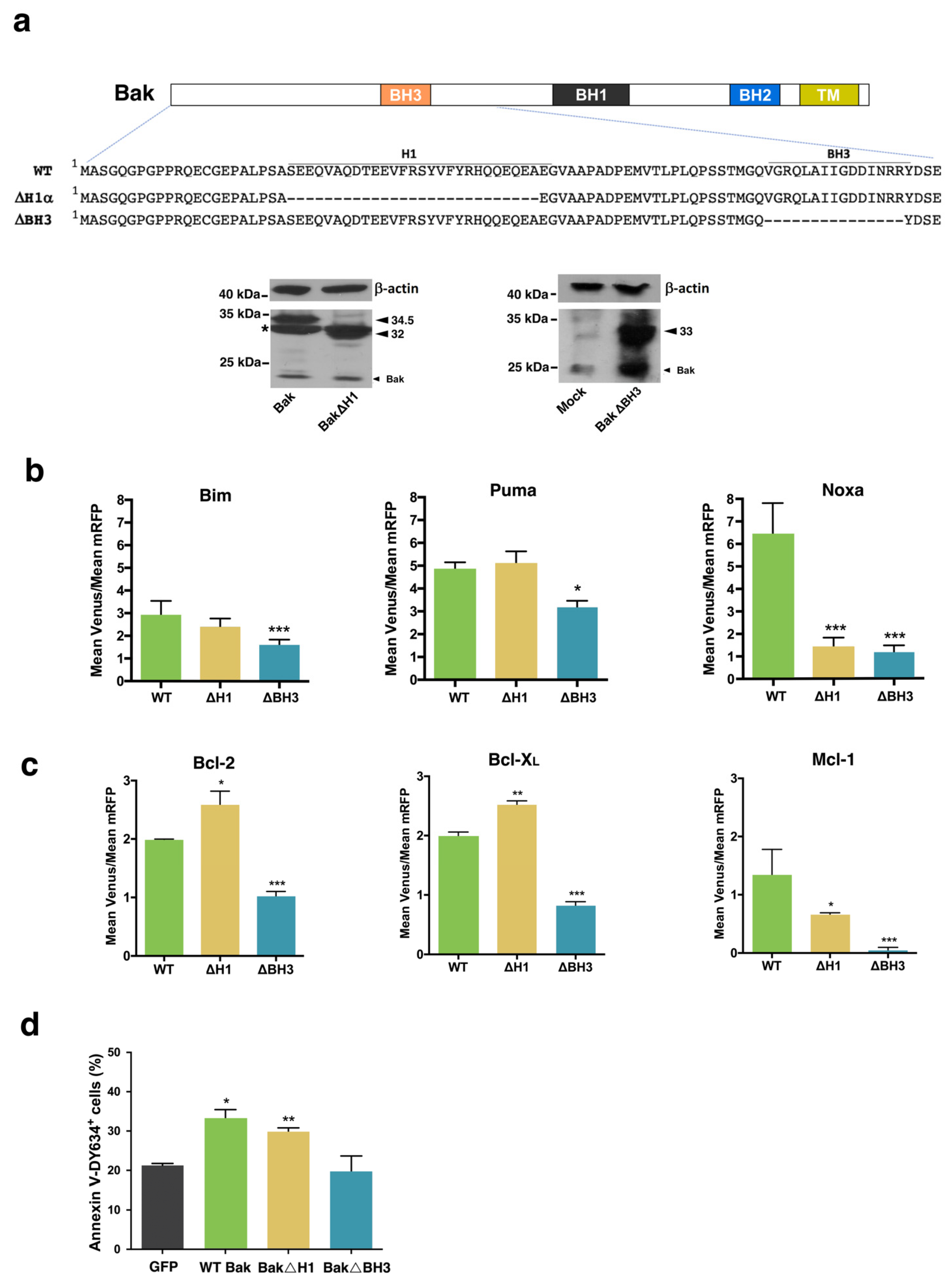{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Construction of pBiFC and pBabe Vectors
2.2. Cell Lines
2.3. BiFC Assays
2.4. Cell Death Analysis
2.5. CRISPR/Cas9
2.6. Western Blot Analysis
2.7. Fluorescence Microscopy
2.8. Statistical Analysis
3. Results
3.1. The Interactome of the Bcl-2 Family in Living Cells
3.2. Involvement of the α1 Helix and the BH3 Domain in the Interaction of Bax and Bak with BH3-Only and Antiapoptotic Proteins
3.3. Involvement of the BH3 Domain and the α1–α6 Interface in Dimerization of Bax and Bak
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wei, M.C.; Zong, W.-X.; Cheng, E.H.-Y.; Lindsten, T.; Panoutsakopoulou, V.; Ross, A.J.; Roth, K.A.; MacGregor, G.R.; Thompson, C.B.; Korsmeyer, S.J. Proapoptotic BAX and BAK: A requisite gateway to mitochondrial dysfunction and death. Science 2001, 292, 727–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yalniz, F.F.; Wierda, W.G. Targeting BCL2 in Chronic Lymphocytic Leukemia and Other Hematologic Malignancies. Drugs 2019, 79, 1287–1304. [Google Scholar] [CrossRef] [PubMed]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Willis, S.N.; Fletcher, J.I.; Kaufmann, T.; van Delft, M.F.; Chen, L.; Czabotar, P.E.; Ierino, H.; Lee, E.F.; Fairlie, W.D.; Bouillet, P.; et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science 2007, 315, 856–859. [Google Scholar] [CrossRef] [Green Version]
- Kuwana, T.; Bouchier-Hayes, L.; Chipuk, J.E.; Bonzon, C.; Sullivan, B.A.; Green, D.R.; Newmeyer, D.D. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol. Cell 2005, 17, 525–535. [Google Scholar] [CrossRef]
- Dai, H.; Pang, Y.P.; Ramirez-Alvarado, M.; Kaufmann, S.H. Evaluation of the BH3-only protein Puma as a direct Bak activator. J. Biol. Chem. 2014, 289, 89–99. [Google Scholar] [CrossRef] [Green Version]
- Dai, H.; Smith, A.; Meng, X.W.; Schneider, P.A.; Pang, Y.P.; Kaufmann, S.H. Transient binding of an activator BH3 domain to the Bak BH3-binding groove initiates Bak oligomerization. J. Cell Biol. 2011, 194, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Merino, D.; Giam, M.; Hughes, P.D.; Siggs, O.M.; Heger, K.; O’Reilly, L.A.; Adams, J.M.; Strasser, A.; Lee, E.F.; Fairlie, W.D.; et al. The role of BH3-only protein Bim extends beyond inhibiting Bcl-2-like prosurvival proteins. J. Cell Biol. 2009, 186, 355–362. [Google Scholar] [CrossRef] [Green Version]
- Gallenne, T.; Gautier, F.; Oliver, L.; Hervouet, E.; Noel, B.; Hickman, J.A.; Geneste, O.; Cartron, P.F.; Vallette, F.M.; Manon, S.; et al. Bax activation by the BH3-only protein Puma promotes cell dependence on antiapoptotic Bcl-2 family members. J. Cell Biol. 2009, 185, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Gavathiotis, E.; Suzuki, M.; Davis, M.L.; Pitter, K.; Bird, G.H.; Katz, S.G.; Tu, H.C.; Kim, H.; Cheng, E.H.; Tjandra, N.; et al. BAX activation is initiated at a novel interaction site. Nature 2008, 455, 1076–1081. [Google Scholar] [CrossRef] [Green Version]
- Vela, L.; Gonzalo, O.; Naval, J.; Marzo, I. Direct Interaction of Bax and Bak Proteins with Bcl-2 Homology Domain 3 (BH3)-only Proteins in Living Cells Revealed by Fluorescence Complementation. J. Biol. Chem. 2013, 288, 4935–4946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leber, B.; Lin, J.; Andrews, D.W. Embedded together: The life and death consequences of interaction of the Bcl-2 family with membranes. Apoptosis 2007, 12, 897–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robin, A.Y.; Krishna Kumar, K.; Westphal, D.; Wardak, A.Z.; Thompson, G.V.; Dewson, G.; Colman, P.M.; Czabotar, P.E. Crystal structure of Bax bound to the BH3 peptide of Bim identifies important contacts for interaction. Cell Death Dis. 2015, 6, e1809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cartron, P.F.; Gallenne, T.; Bougras, G.; Gautier, F.; Manero, F.; Vusio, P.; Meflah, K.; Vallette, F.M.; Juin, P. The first alpha helix of Bax plays a necessary role in its ligand-induced activation by the BH3-only proteins Bid and PUMA. Mol. Cell 2004, 16, 807–818. [Google Scholar] [CrossRef]
- Edwards, A.L.; Gavathiotis, E.; LaBelle, J.L.; Braun, C.R.; Opoku-Nsiah, K.A.; Bird, G.H.; Walensky, L.D. Multimodal interaction with BCL-2 family proteins underlies the proapoptotic activity of PUMA BH3. Chem. Biol. 2013, 20, 888–902. [Google Scholar] [CrossRef] [Green Version]
- Gavathiotis, E.; Reyna, D.E.; Davis, M.L.; Bird, G.H.; Walensky, L.D. BH3-triggered structural reorganization drives the activation of proapoptotic BAX. Mol. Cell 2010, 40, 481–492. [Google Scholar] [CrossRef] [Green Version]
- Nechushtan, A.; Smith, C.L.; Hsu, Y.T.; Youle, R.J. Conformation of the Bax C-terminus regulates subcellular location and cell death. EMBO J. 1999, 18, 2330–2341. [Google Scholar] [CrossRef] [Green Version]
- Renault, T.T.; Manon, S. Bax: Addressed to kill. Biochimie 2011, 93, 1379–1391. [Google Scholar] [CrossRef] [PubMed]
- Westphal, D.; Dewson, G.; Czabotar, P.E.; Kluck, R.M. Molecular biology of Bax and Bak activation and action. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2011, 1813, 521–531. [Google Scholar] [CrossRef] [Green Version]
- Dewson, G.; Kratina, T.; Czabotar, P.; Day, C.L.; Adams, J.M.; Kluck, R.M. Bak activation for apoptosis involves oligomerization of dimers via their alpha6 helices. Mol. Cell 2009, 36, 696–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewson, G.; Ma, S.; Frederick, P.; Hockings, C.; Tan, I.; Kratina, T.; Kluck, R.M. Bax dimerizes via a symmetric BH3:groove interface during apoptosis. Cell Death Differ. 2012, 19, 661–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aluvila, S.; Mandal, T.; Hustedt, E.; Fajer, P.; Choe, J.Y.; Oh, K.J. Organization of the mitochondrial apoptotic BAK pore: Oligomerization of the BAK homodimers. J. Biol. Chem. 2014, 289, 2537–2551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, S.; Bell, F.; Westphal, D.; Anwari, K.; Gulbis, J.; Smith, B.J.; Dewson, G.; Kluck, R.M. Bak apoptotic pores involve a flexible C-terminal region and juxtaposition of the C-terminal transmembrane domains. Cell Death Differ. 2015, 22, 1665–1675. [Google Scholar] [CrossRef] [Green Version]
- Gahl, R.F.; He, Y.; Yu, S.; Tjandra, N. Conformational rearrangements in the pro-apoptotic protein, Bax, as it inserts into mitochondria: A cellular death switch. J. Biol. Chem. 2014, 289, 32871–32882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uren, R.T.; O’Hely, M.; Iyer, S.; Bartolo, R.; Shi, M.X.; Brouwer, J.M.; Alsop, A.E.; Dewson, G.; Kluck, R.M. Disordered clusters of Bak dimers rupture mitochondria during apoptosis. eLife 2017, 6, e19944. [Google Scholar] [CrossRef]
- Kodama, Y.; Hu, C.D. Bimolecular fluorescence complementation (BiFC): A 5-year update and future perspectives. BioTechniques 2012, 53, 285–298. [Google Scholar] [CrossRef]
- Logue, S.E.; Elgendy, M.; Martin, S.J. Expression, purification and use of recombinant annexin V for the detection of apoptotic cells. Nat. Protoc. 2009, 4, 1383–1395. [Google Scholar] [CrossRef] [PubMed]
- Grinberg, A.V.; Hu, C.D.; Kerppola, T.K. Visualization of Myc/Max/Mad family dimers and the competition for dimerization in living cells. Mol. Cell Biol. 2004, 24, 4294–4308. [Google Scholar] [CrossRef] [Green Version]
- Kerppola, T.K. Design and Implementation of Bimolecular Fluorescence Complementation (BiFC) Assays for the Visualization of Protein Interactions in Living Cells. Nat. Protoc. 2006, 1, 1278–1286. [Google Scholar] [CrossRef] [Green Version]
- Morell, M.; Espargaro, A.; Aviles, F.X.; Ventura, S. Study and selection of in vivo protein interactions by coupling bimolecular fluorescence complementation and flow cytometry. Nat. Protoc. 2008, 3, 22–33. [Google Scholar] [CrossRef]
- Andreu-Fernandez, V.; Sancho, M.; Genoves, A.; Lucendo, E.; Todt, F.; Lauterwasser, J.; Funk, K.; Jahreis, G.; Perez-Paya, E.; Mingarro, I.; et al. Bax transmembrane domain interacts with prosurvival Bcl-2 proteins in biological membranes. Proc. Natl. Acad. Sci. USA 2017, 114, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.D.; Kerppola, T.K. Simultaneous visualization of multiple protein interactions in living cells using multicolor fluorescence complementation analysis. Nat. Biotechnol. 2003, 21, 539–545. [Google Scholar] [CrossRef] [Green Version]
- Cartron, P.-F.; Oliver, L.; Juin, P.; Meflah, K.; Vallette, F.M. The p18 Truncated Form of Bax Behaves Like a Bcl-2 Homology Domain 3-only Protein. J. Biol. Chem. 2004, 279, 11503–11512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotschy, A.; Szlavik, Z.; Murray, J.; Davidson, J.; Maragno, A.L.; Le Toumelin-Braizat, G.; Chanrion, M.; Kelly, G.L.; Gong, J.N.; Moujalled, D.M.; et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 2016, 538, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.J.; Tait, S.W.G. Targeting BCL-2 regulated apoptosis in cancer. Open Biol. 2018, 8, 180002. [Google Scholar] [CrossRef] [Green Version]
- Llambi, F.; Moldoveanu, T.; Tait, S.W.; Bouchier-Hayes, L.; Temirov, J.; McCormick, L.L.; Dillon, C.P.; Green, D.R. A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol. Cell 2011, 44, 517–531. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Tu, H.C.; Ren, D.; Takeuchi, O.; Jeffers, J.R.; Zambetti, G.P.; Hsieh, J.J.; Cheng, E.H. Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol. Cell 2009, 36, 487–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, Y.P.; Dai, H.; Smith, A.; Meng, X.W.; Schneider, P.A.; Kaufmann, S.H. Bak Conformational Changes Induced by Ligand Binding: Insight into BH3 Domain Binding and Bak Homo-Oligomerization. Sci. Rep. 2012, 2, 257. [Google Scholar] [CrossRef] [Green Version]
- Du, H.; Wolf, J.; Schafer, B.; Moldoveanu, T.; Chipuk, J.E.; Kuwana, T. BH3 domains other than Bim and Bid can directly activate Bax/Bak. J. Biol. Chem. 2011, 286, 491–501. [Google Scholar] [CrossRef] [Green Version]
- Bleicken, S.; Hantusch, A.; Das, K.K.; Frickey, T.; Garcia-Saez, A.J. Quantitative interactome of a membrane Bcl-2 network identifies a hierarchy of complexes for apoptosis regulation. Nat. Commun. 2017, 8, 73. [Google Scholar] [CrossRef]
- Yee, K.S.; Vousden, K.H. Contribution of membrane localization to the apoptotic activity of PUMA. Apoptosis 2008, 13, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Matulis, S.M.; Gupta, V.A.; Nooka, A.K.; Hollen, H.V.; Kaufman, J.L.; Lonial, S.; Boise, L.H. Dexamethasone treatment promotes Bcl-2 dependence in multiple myeloma resulting in sensitivity to venetoclax. Leukemia 2016, 30, 1086–1093. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.J.; Dai, H.; Correia, C.; Takahashi, R.; Lee, S.H.; Schmitz, I.; Kaufmann, S.H. Noxa/Bcl-2 protein interactions contribute to bortezomib resistance in human lymphoid cells. J. Biol. Chem. 2011, 286, 17682–17692. [Google Scholar] [CrossRef] [Green Version]
- Willis, S.N.; Chen, L.; Dewson, G.; Wei, A.; Naik, E.; Fletcher, J.I.; Adams, J.M.; Huang, D.C. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005, 19, 1294–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, H.; Meng, X.W.; Lee, S.H.; Schneider, P.A.; Kaufmann, S.H. Context-dependent Bcl-2/Bak interactions regulate lymphoid cell apoptosis. J. Biol. Chem. 2009, 284, 18311–18322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leshchiner, E.S.; Braun, C.R.; Bird, G.H.; Walensky, L.D. Direct activation of full-length proapoptotic BAK. Proc. Natl. Acad. Sci. USA 2013, 110, E986–E995. [Google Scholar] [CrossRef] [Green Version]
- Barclay, L.A.; Wales, T.E.; Garner, T.P.; Wachter, F.; Lee, S.; Guerra, R.M.; Stewart, M.L.; Braun, C.R.; Bird, G.H.; Gavathiotis, E.; et al. Inhibition of Pro-apoptotic BAX by a noncanonical interaction mechanism. Mol. Cell 2015, 57, 873–886. [Google Scholar] [CrossRef] [Green Version]
- Ding, J.; Zhang, Z.; Roberts, G.J.; Falcone, M.; Miao, Y.; Shao, Y.; Zhang, X.C.; Andrews, D.W.; Lin, J. Bcl-2 and Bax interact via the BH1-3 groove-BH3 motif interface and a novel interface involving the BH4 motif. J. Biol. Chem. 2010, 285, 28749–28763. [Google Scholar] [CrossRef] [Green Version]
- Ding, J.; Mooers, B.H.; Zhang, Z.; Kale, J.; Falcone, D.; McNichol, J.; Huang, B.; Zhang, X.C.; Xing, C.; Andrews, D.W.; et al. After embedding in membranes antiapoptotic Bcl-XL protein binds both Bcl-2 homology region 3 and helix 1 of proapoptotic Bax protein to inhibit apoptotic mitochondrial permeabilization. J. Biol. Chem. 2014, 289, 11873–11896. [Google Scholar] [CrossRef] [Green Version]
- Czabotar, P.E.; Lee, E.F.; Thompson, G.V.; Wardak, A.Z.; Fairlie, W.D.; Colman, P.M. Mutation to Bax beyond the BH3 domain disrupts interactions with pro-survival proteins and promotes apoptosis. J. Biol. Chem. 2011, 286, 7123–7131. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Gehring, K. Heterodimerization of BAK and MCL-1 activated by detergent micelles. J. Biol. Chem. 2010, 285, 41202–41210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bleicken, S.; Classen, M.; Padmavathi, P.V.L.; Ishikawa, T.; Zeth, K.; Steinhoff, H.-J.; Bordignon, E. Molecular Details of Bax Activation, Oligomerization, and Membrane Insertion. J. Biol. Chem. 2010, 285, 6636–6647. [Google Scholar] [CrossRef] [Green Version]
- Brouwer, J.M.; Westphal, D.; Dewson, G.; Robin, A.Y.; Uren, R.T.; Bartolo, R.; Thompson, G.V.; Colman, P.M.; Kluck, R.M.; Czabotar, P.E. Bak core and latch domains separate during activation, and freed core domains form symmetric homodimers. Mol. Cell 2014, 55, 938–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czabotar, P.E.; Westphal, D.; Dewson, G.; Ma, S.; Hockings, C.; Fairlie, W.D.; Lee, E.F.; Yao, S.; Robin, A.Y.; Smith, B.J.; et al. Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell 2013, 152, 519–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewson, G.; Kratina, T.; Sim, H.W.; Puthalakath, H.; Adams, J.M.; Colman, P.M.; Kluck, R.M. To trigger apoptosis, Bak exposes its BH3 domain and homodimerizes via BH3:groove interactions. Mol. Cell 2008, 30, 369–380. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhu, W.; Lapolla, S.M.; Miao, Y.; Shao, Y.; Falcone, M.; Boreham, D.; McFarlane, N.; Ding, J.; Johnson, A.E.; et al. Bax forms an oligomer via separate, yet interdependent, surfaces. J. Biol. Chem. 2010, 285, 17614–17627. [Google Scholar] [CrossRef] [Green Version]
- Tan, C.; Dlugosz, P.J.; Peng, J.; Zhang, Z.; Lapolla, S.M.; Plafker, S.M.; Andrews, D.W.; Lin, J. Auto-activation of the apoptosis protein Bax increases mitochondrial membrane permeability and is inhibited by Bcl-2. J. Biol. Chem. 2006, 281, 14764–14775. [Google Scholar] [CrossRef] [Green Version]
- Dai, H.; Ding, H.; Meng, X.W.; Peterson, K.L.; Schneider, P.A.; Karp, J.E.; Kaufmann, S.H. Constitutive BAK activation as a determinant of drug sensitivity in malignant lymphohematopoietic cells. Genes Dev. 2015, 29, 2140–2152. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Subramaniam, S.; Kale, J.; Liao, C.; Huang, B.; Brahmbhatt, H.; Condon, S.G.; Lapolla, S.M.; Hays, F.A.; Ding, J.; et al. BH3-in-groove dimerization initiates and helix 9 dimerization expands Bax pore assembly in membranes. EMBO J. 2016, 35, 208–236. [Google Scholar] [CrossRef] [Green Version]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonzalo, Ó.; Benedi, A.; Vela, L.; Anel, A.; Naval, J.; Marzo, I. Study of the Bcl-2 Interactome by BiFC Reveals Differences in the Activation Mechanism of Bax and Bak. Cells 2023, 12, 800. https://doi.org/10.3390/cells12050800
Gonzalo Ó, Benedi A, Vela L, Anel A, Naval J, Marzo I. Study of the Bcl-2 Interactome by BiFC Reveals Differences in the Activation Mechanism of Bax and Bak. Cells. 2023; 12(5):800. https://doi.org/10.3390/cells12050800
Chicago/Turabian StyleGonzalo, Óscar, Andrea Benedi, Laura Vela, Alberto Anel, Javier Naval, and Isabel Marzo. 2023. "Study of the Bcl-2 Interactome by BiFC Reveals Differences in the Activation Mechanism of Bax and Bak" Cells 12, no. 5: 800. https://doi.org/10.3390/cells12050800