

Analysis of Wild Type and Variant B Cystatin C Interactome in Retinal Pigment Epithelium Cells Reveals Variant B Interacting Mitochondrial Proteins

Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Plasmids
2.3. Expression of Halo-Tagged Cystatin C
2.4. Pull down Interaction Assay
2.5. SDS-PAGE and Silver Stain
2.6. nanoLC-ESI-MS/MS Analysis
2.7. Data Analysis and Protein Identification
2.8. Subcellular Fractionation
2.9. Immunoblotting
2.10. Mitochondrial ROS Production Measurement
2.11. Measurement of Mitochondrial Membrane Potential (Δψm) Using Mitotracker Red FM
3. Results
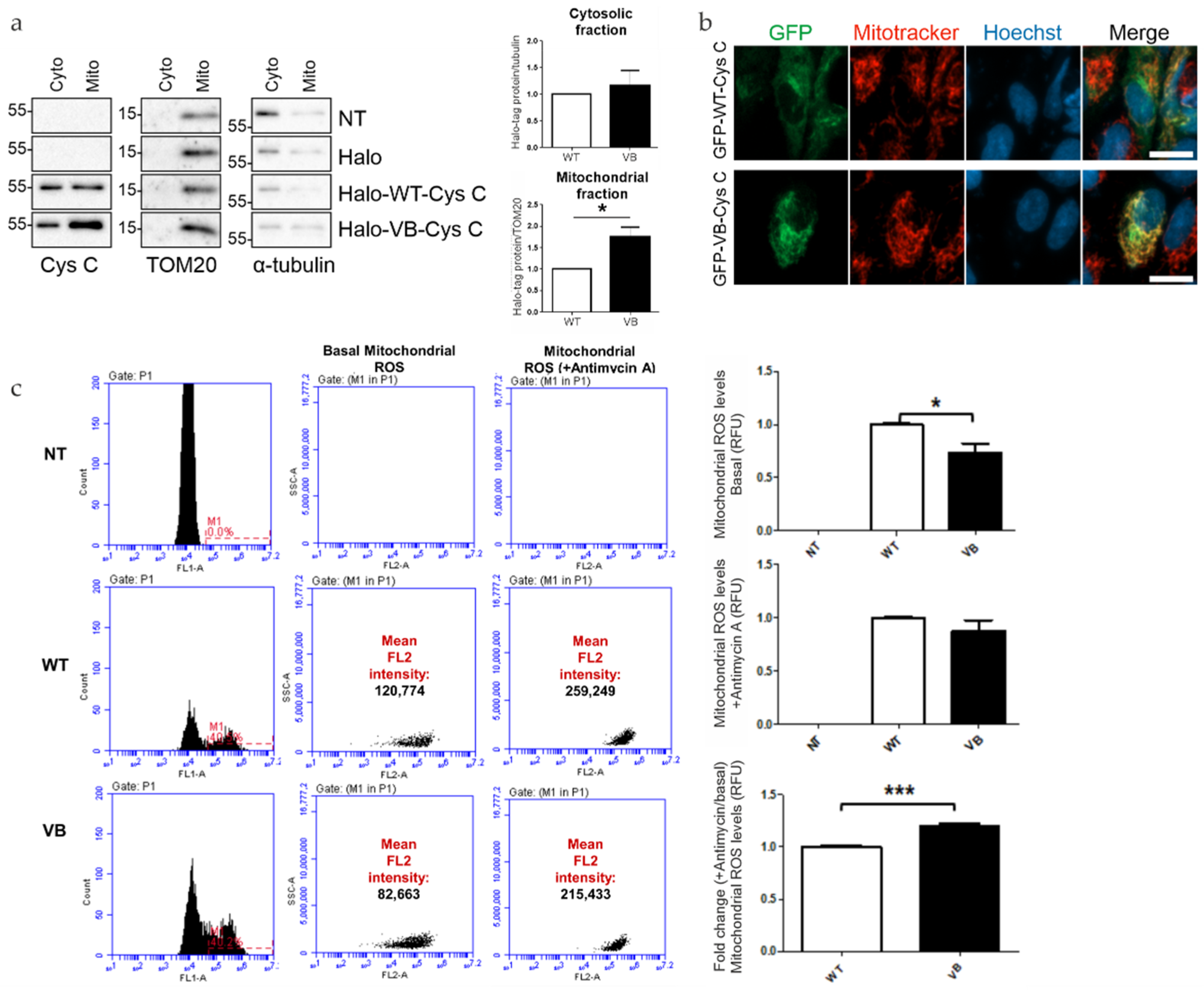
3.1. Expression of Halo-Tagged Proteins
3.2. Interactome Analysis Reveals Specific Protein Interactions of Variant B Cystatin C with Mitochondrial Proteins
3.3. Expression of Variant B Cystatin C Leads to Alterations of RPE Mitochondrial Function
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pennington, K.L.; DeAngelis, M.M. Epidemiology of age-related macular degeneration (AMD): Associations with cardiovascular disease phenotypes and lipid factors. Eye Vision 2016, 3, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambati, J.; Fowler, B.J. Mechanisms of age-related macular degeneration. Neuron 2012, 75, 26–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinnunen, K.; Petrovski, G.; Moe, M.C.; Berta, A.; Kaarniranta, K. Molecular mechanisms of retinal pigment epithelium damage and development of age-related macular degeneration. Acta Ophthalmol. 2012, 90, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, L.G.; Fariss, R.N.; Stambolian, D.; Abecasis, G.R.; Curcio, C.A.; Swaroop, A. Age-related macular degeneration: Genetics and biology coming together. Annu. Rev. Genomics Hum. Genet. 2014, 15, 151–171. [Google Scholar] [CrossRef] [Green Version]
- Fritsche, L.G.; Igl, W.; Bailey, J.N.; Grassmann, F.; Sengupta, S.; Bragg-Gresham, J.L.; Burdon, K.P.; Hebbring, S.J.; Wen, C.; Gorski, M.; et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet. 2016, 48, 134–143. [Google Scholar] [CrossRef] [Green Version]
- Wistow, G.; Bernstein, S.L.; Wyatt, M.K.; Fariss, R.N.; Behal, A.; Touchman, J.W.; Bouffard, G.; Smith, D.; Peterson, K. Expressed sequence tag analysis of human RPE/choroid for the NEIBank Project: Over 6000 non-redundant transcripts, novel genes and splice variants. Mol. Vis. 2022, 8, 205–220. [Google Scholar]
- Paraoan, L.; Grierson, I.; Maden, B.E.H. Analysis of expressed sequence tags of retinal pigment epithelium: Cystatin C is an abundant transcript. Int. J. Biochem. Cell B 2000, 32, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Paraoan, L.; White, M.R.H.; Spiller, D.G.; Grierson, I.; Maden, B.E.H. Precursor cystatin C in cultured retinal pigment epithelium cells: Evidence for processing through the secretory pathway. Mol. Membr. Biol. 2001, 18, 229–236. [Google Scholar] [PubMed]
- Kay, P.; Yang, Y.C.; Paraoan, L. Directional protein secretion by the retinal pigment epithelium: Roles in retinal health and the development of age-related macular degeneration. J. Cell Mol. Med. 2013, 17, 833–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paraoan, L.; Sharif, U.; Carlsson, E.; Supharattanasitthi, W.; Mahmud, N.M.; Kamalden, T.A.; Hiscott, P.; Jackson, M.; Grierson, I. Secretory proteostasis of the retinal pigmented epithelium: Impairment links to age-related macular degeneration. Prog. Retin. Eye Res. 2020, 79, 100859. [Google Scholar] [CrossRef]
- Butler, J.M.; Sharif, U.; Ali, M.; McKibbin, M.; Thompson, J.P.; Gale, R.; Yang, Y.C.; Inglehearn, C.; Paraoan, L. A missense variant in CST3 exerts a recessive effect on susceptibility to age-related macular degeneration resembling its association with Alzheimer’s disease. Hum. Genet. 2015, 134, 705–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zurdel, J.; Finckh, U.; Menzer, G.; Nitsch, R.M.; Richard, G. CST3 genotype associated with exudative age related macular degeneration. Br. J. Ophthalmol. 2002, 86, 214–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paraoan, L.; Hiscott, P.; Gosden, C.; Grierson, I. Cystatin C in macular and neuronal degenerations: Implications for mechanism(s) of age-related macular degeneration. Vision Res. 2010, 50, 737–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertram, L.; McQueen, M.B.; Mullin, K.; Blacker, D.; Tanzi, R.E. Systematic meta-analyses of Alzheimer disease genetic association studies: The AlzGene database. Nat. Genet. 2007, 39, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Finckh, U.; von der Kammer, H.; Velden, J.; Michel, T.; Andresen, B.; Deng, A.; Zhang, J.; Muller-Thomsen, T.; Zuchowski, K.; Menzer, G.; et al. Genetic association of a cystatin C gene polymorphism with late-onset Alzheimer disease. Arch. Neurol. 2000, 57, 1579–1583. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Zhao, H.; Lu, X.; Kong, Y.; Jin, H. Meta-analysis of the cystatin C(CST3) gene G73A polymorphism and susceptibility to Alzheimer’s disease. Int. J. Neurosci. 2012, 122, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Benussi, L.; Ghidoni, R.; Steinhoff, T.; Alberici, A.; Villa, A.; Mazzoli, F.; Nicosia, F.; Barbiero, L.; Broglio, L.; Feudatari, E.; et al. Alzheimer disease-associated cystatin C variant undergoes impaired secretion. Neurobiol. Dis. 2003, 13, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Paraoan, L.; Ratnayaka, A.; Spiller, D.G.; Hiscott, P.; White, M.R.; Grierson, I. Unexpected intracellular localization of the AMD-associated cystatin C variant. Traffic 2004, 5, 884–895. [Google Scholar] [CrossRef]
- Paraoan, L.; Grierson, I. Focus on molecules: Cystatin C. Exp. Eye Res. 2007, 84, 1019–1020. [Google Scholar] [CrossRef] [PubMed]
- Paraoan, L.; Grierson, I.; Maden, B.E. Fate of cystatin C lacking the leader sequence in RPE cells. Exp. Eye Res. 2003, 76, 753–756. [Google Scholar] [CrossRef]
- Supharattanasitthi, W.; Carlsson, E.; Sharif, U.; Paraoan, L. CRISPR/Cas9-mediated one step bi-allelic change of genomic DNA in iPSCs and human RPE cells in vitro with dual antibiotic selection. Sci. Rep. 2019, 9, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlsson, E.; Supharattanasitthi, W.; Jackson, M.; Paraoan, L. Increased Rate of Retinal Pigment Epithelial Cell Migration and Pro-Angiogenic Potential Ensuing From Reduced Cystatin C Expression. Investig. Ophthalmol. Vis. Sci. 2020, 61, 9. [Google Scholar] [CrossRef] [Green Version]
- Ratnayaka, A.; Paraoan, L.; Spiller, D.G.; Hiscott, P.; Nelson, G.; White, M.R.; Grierson, I. A dual Golgi- and mitochondria-localised Ala25Ser precursor cystatin C: An additional tool for characterising intracellular mis-localisation leading to increased AMD susceptibility. Exp. Eye Res. 2007, 84, 1135–1139. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Fisher, C.R.; Ferrington, D.A. Perspective on AMD Pathobiology: A Bioenergetic Crisis in the RPE. Investig. Ophthalmol. Vis. Sci. 2018, 59, AMD41–AMD47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terluk, M.R.; Kapphahn, R.J.; Soukup, L.M.; Gong, H.; Gallardo, C.; Montezuma, S.R.; Ferrington, D.A. Investigating mitochondria as a target for treating age-related macular degeneration. J. Neurosci. 2015, 35, 7304–7311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordgaard, C.L.; Berg, K.M.; Kapphahn, R.J.; Reilly, C.; Feng, X.; Olsen, T.W.; Ferrington, D.A. Proteomics of the retinal pigment epithelium reveals altered protein expression at progressive stages of age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2006, 47, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.M.; Supharattanasitthi, W.; Yang, Y.C.; Paraoan, L. RNA-seq analysis of ageing human retinal pigment epithelium: Unexpected up-regulation of visual cycle gene transcription. J. Cell Mol. Med. 2021, 25, 5572–5585. [Google Scholar] [CrossRef] [PubMed]
- Dhirachaikulpanich, D.; Lagger, C.; Chatsirisupachai, K.; de Magalhaes, J.P.; Paraoan, L. Intercellular communication analysis of the human retinal pigment epithelial and choroidal cells predicts pathways associated with aging, cellular senescence and age-related macular degeneration. Front. Aging Neurosci. 2022, 14, 1016293. [Google Scholar] [CrossRef] [PubMed]
- Dhirachaikulpanich, D.; Li, X.; Porter, L.F.; Paraoan, L. Integrated Microarray and RNAseq Transcriptomic Analysis of Retinal Pigment Epithelium/Choroid in Age-Related Macular Degeneration. Front. Cell Dev. Biol. 2020, 8, 808. [Google Scholar] [CrossRef] [PubMed]
- Golestaneh, N.; Chu, Y.; Xiao, Y.Y.; Stoleru, G.L.; Theos, A.C. Dysfunctional autophagy in RPE, a contributing factor in age-related macular degeneration. Cell Death Dis. 2017, 8, e2537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apta-Smith, M.J.; Hernandez-Fernaud, J.R.; Bowman, A.J. Evidence for the nuclear import of histones H3.1 and H4 as monomers. EMBO J. 2018, 37, e98714. [Google Scholar] [CrossRef] [PubMed]
- Malki, I.; Liepina, I.; Kogelnik, N.; Watmuff, H.; Robinson, S.; Lightfoot, A.; Gonchar, O.; Bottrill, A.; Fry, A.M.; Dominguez, C. Cdk1-mediated threonine phosphorylation of Sam68 modulates its RNA binding, alternative splicing activity and cellular functions. Nucleic Acids Res. 2022, 50, 13045–13062. [Google Scholar] [CrossRef] [PubMed]
- Narvaez, A.J.; Ber, S.; Crooks, A.; Emery, A.; Hardwick, B.; Almeida, E.G.; Huggins, D.J.; Perera, D.; Roberts-Thomson, M.; Azzarelli, R.; et al. Modulating Protein-Protein Interactions of the Mitotic Polo-like Kinases to Target Mutant KRAS. Cell Chem. Biol. 2017, 24, 1017–1028.e1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larance, M.; Ramm, G.; Stockli, J.; van Dam, E.M.; Winata, S.; Wasinger, V.; Simpson, F.; Graham, M.; Junutula, J.R.; Guilhaus, M.; et al. Characterization of the role of the Rab GTPase-activating protein AS160 in insulin-regulated GLUT4 trafficking. J. Biol. Chem. 2005, 280, 37803–37813. [Google Scholar] [CrossRef] [Green Version]
- Kandror, K.V.; Yu, L.; Pilch, P.F. The major protein of GLUT4-containing vesicles, gp160, has aminopeptidase activity. J. Biol. Chem. 1994, 269, 30777–30780. [Google Scholar] [CrossRef] [PubMed]
- Keller, S.R.; Scott, H.M.; Mastick, C.C.; Aebersold, R.; Lienhard, G.E. Cloning and characterization of a novel insulin-regulated membrane aminopeptidase from Glut4 vesicles. J. Biol. Chem. 1995, 270, 23612–23618. [Google Scholar] [CrossRef] [Green Version]
- Repalli, J. Translocator protein (TSPO) role in aging and Alzheimer’s disease. Curr. Aging Sci. 2014, 7, 168–175. [Google Scholar] [CrossRef] [Green Version]
- Tournier, B.B.; Tsartsalis, S.; Rigaud, D.; Fossey, C.; Cailly, T.; Fabis, F.; Pham, T.; Gregoire, M.C.; Kovari, E.; Moulin-Sallanon, M.; et al. TSPO and amyloid deposits in sub-regions of the hippocampus in the 3xTgAD mouse model of Alzheimer’s disease. Neurobiol. Dis. 2019, 121, 95–105. [Google Scholar] [CrossRef] [Green Version]
- Christensen, A.; Pike, C.J. TSPO ligand PK11195 improves Alzheimer-related outcomes in aged female 3xTg-AD mice. Neurosci. Lett. 2018, 683, 7–12. [Google Scholar] [CrossRef]
- Sant’Anna, R.; Navarro, S.; Ventura, S.; Paraoan, L.; Foguel, D. Amyloid properties of the leader peptide of variant B cystatin C: Implications for Alzheimer and macular degeneration. FEBS Lett. 2016, 590, 644–654. [Google Scholar] [CrossRef] [Green Version]
- Mitter, S.K.; Song, C.; Qi, X.; Mao, H.; Rao, H.; Akin, D.; Lewin, A.; Grant, M.; Dunn, W., Jr.; Ding, J.; et al. Dysregulated autophagy in the RPE is associated with increased susceptibility to oxidative stress and AMD. Autophagy 2014, 10, 1989–2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, S.; Cano, M.; Ebrahimi, K.; Wang, L.; Handa, J.T. The impact of oxidative stress and inflammation on RPE degeneration in non-neovascular AMD. Prog. Retin. Eye Res. 2017, 60, 201–218. [Google Scholar] [CrossRef] [PubMed]
- Casellas, P.; Galiegue, S.; Basile, A.S. Peripheral benzodiazepine receptors and mitochondrial function. Neurochem. Int. 2022, 40, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Gatliff, J.; East, D.; Crosby, J.; Abeti, R.; Harvey, R.; Craigen, W.; Parker, P.; Campanella, M. TSPO interacts with VDAC1 and triggers a ROS-mediated inhibition of mitochondrial quality control. Autophagy 2014, 10, 2279–2296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gavish, M.; Bachman, I.; Shoukrun, R.; Katz, Y.; Veenman, L.; Weisinger, G.; Weizman, A. Enigma of the peripheral benzodiazepine receptor. Pharmacol. Rev. 1999, 51, 629–650. [Google Scholar]
- Papadopoulos, V.; Lecanu, L. Translocator protein (18 kDa) TSPO: An emerging therapeutic target in neurotrauma. Exp. Neurol. 2009, 219, 53–57. [Google Scholar] [CrossRef] [Green Version]
- Papadopoulos, V.; Lecanu, L.; Brown, R.C.; Han, Z.; Yao, Z.X. Peripheral-type benzodiazepine receptor in neurosteroid biosynthesis, neuropathology and neurological disorders. Neuroscience 2006, 138, 749–756. [Google Scholar] [CrossRef]
- Veenman, L.; Papadopoulos, V.; Gavish, M. Channel-like functions of the 18-kDa translocator protein (TSPO): Regulation of apoptosis and steroidogenesis as part of the host-defense response. Curr. Pharm. Des. 2007, 13, 2385–2405. [Google Scholar] [CrossRef] [PubMed]
- Bhatia-Kissova, I.; Camougrand, N. Mitophagy: A process that adapts to the cell physiology. Int. J. Biochem. Cell Biol. 2013, 45, 30–33. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Smigelskaite, J.; Doblander, C.; Janakiraman, M.; Hermann, M.; Wurm, M.; Scheidl, S.F.; Sucher, R.; Deutschmann, A.; Troppmair, J. Survival signaling by C-RAF: Mitochondrial reactive oxygen species and Ca2+ are critical targets. Mol. Cell Biol. 2008, 28, 2304–2313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Kim, S.H.; Kim, K.Y.; Park, S.G.; Yu, S.N.; Kim, Y.W.; Nam, H.W.; An, H.H.; Kim, Y.W.; Ahn, S.C. Mitochondrial ROS activates ERK/autophagy pathway as a protected mechanism against deoxypodophyllotoxin-induced apoptosis. Oncotarget 2017, 8, 111581–111596. [Google Scholar] [CrossRef] [Green Version]
- Kaarniranta, K.; Pawlowska, E.; Szczepanska, J.; Jablkowska, A.; Blasiak, J. Role of Mitochondrial DNA Damage in ROS-Mediated Pathogenesis of Age-Related Macular Degeneration (AMD). Int. J. Mol. Sci. 2019, 20, 2374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulua, A.C.; Simon, A.; Maddipati, R.; Pelletier, M.; Park, H.; Kim, K.Y.; Sack, M.N.; Kastner, D.L.; Siegel, R.M. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J. Exp. Med. 2011, 208, 519–533. [Google Scholar] [CrossRef] [PubMed]
- Kay, P.; Yang, Y.C.; Hiscott, P.; Gray, D.; Maminishkis, A.; Paraoan, L. Age-related changes of cystatin C expression and polarized secretion by retinal pigment epithelium: Potential age-related macular degeneration links. Investig. Ophthalmol. Vis. Sci. 2014, 55, 926–934. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Hypoxia-inducible factor 1: Regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochim. Biophys. Acta 2011, 1813, 1263–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.J.; Middleton, R.J.; Kam, W.W.; Chin, D.Y.; Hatty, C.R.; Chan, R.H.; Banati, R.B. Functional gains in energy and cell metabolism after TSPO gene insertion. Cell Cycle 2017, 16, 436–447. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Yu, S.; Wang, C.Y.; Wang, Y.; Liu, H.X.; Cui, Y.; Zhang, L.D. Advanced glycation end products induce oxidative stress and mitochondrial dysfunction in SH-SY5Y cells. In Vitro Cell Dev. Biol. Anim. 2015, 51, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chang, Y.; Ye, N.; Chen, Y.; Zhang, N.; Sun, Y. Advanced glycation end productsinduced mitochondrial energy metabolism dysfunction alters proliferation of human umbilical vein endothelial cells. Mol. Med. Rep. 2017, 15, 2673–2680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharif, U.; Mahmud, N.M.; Kay, P.; Yang, Y.C.; Harding, S.P.; Grierson, I.; Kamalden, T.A.; Jackson, M.J.; Paraoan, L. Advanced glycation end products-related modulation of cathepsin L and NF-kappaB signalling effectors in retinal pigment epithelium lead to augmented response to TNFalpha. J. Cell Mol. Med. 2019, 23, 405–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteras, N.; Rohrer, J.D.; Hardy, J.; Wray, S.; Abramov, A.Y. Mitochondrial hyperpolarization in iPSC-derived neurons from patients of FTDP-17 with 10+16 MAPT mutation leads to oxidative stress and neurodegeneration. Redox Biol. 2017, 12, 410–422. [Google Scholar] [CrossRef] [PubMed]
- Suski, J.; Lebiedzinska, M.; Bonora, M.; Pinton, P.; Duszynski, J.; Wieckowski, M.R. Relation Between Mitochondrial Membrane Potential and ROS Formation. Methods Mol. Biol. 2018, 1782, 357–381. [Google Scholar] [PubMed]
- Kang, H.T.; Lee, H.I.; Hwang, E.S. Nicotinamide extends replicative lifespan of human cells. Aging Cell 2006, 5, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.Y.; Kang, H.T.; Hwang, E.S. Nicotinamide-induced mitophagy: Event mediated by high NAD+/NADH ratio and SIRT1 protein activation. J. Biol. Chem. 2012, 287, 19304–19314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, S.B.; Jang, S.Y.; Kang, H.T.; Wei, B.; Jeoun, U.W.; Yoon, G.S.; Hwang, E.S. Modulation of Mitochondrial Membrane Potential and ROS Generation by Nicotinamide in a Manner Independent of SIRT1 and Mitophagy. Mol. Cells 2017, 40, 503–514. [Google Scholar] [PubMed] [Green Version]
- Szabadkai, G.; Duchen, M.R. Mitochondria: The hub of cellular Ca2+ signaling. Physiology (Bethesda) 2008, 23, 84–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Supnet, C.; Bezprozvanny, I. Neuronal calcium signaling, mitochondrial dysfunction, and Alzheimer’s disease. J. Alzheimers Dis. 2010, 20 (Suppl. 2), S487–S498. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Identified Protein | Gene Name | Uniprot ID | p Value (-LOG10) | Difference (LOG2) | Unique Peptides | Unique Sequence Coverage (%) | Molecular Weight (kDa) |
|---|---|---|---|---|---|---|---|
| Cathepsin C | CTSC | P53634 | 3.128994029 | 11.26671028 | 15 | 39.1 | 51.853 |
| Cathepsin L | CTSL | P07711 | 3.885627129 | 8.967143377 | 6 | 25.2 | 37.564 |
| Cathepsin K | CTSK | P43235 | 3.642980692 | 8.719130198 | 10 | 50.5 | 36.966 |
| Cathepsin H | CTSH | P09668 | 3.443870854 | 8.583513896 | 9 | 31.6 | 36.269 |
| Cystatin B | CSTB | P04080 | 3.834472823 | 8.368869781 | 5 | 77.6 | 11.139 |
| Ubiquitin carboxyl-terminal hydrolase 24 | USP24 | Q9UPU5 | 4.302084411 | 6.91021347 | 2 | 0.8 | 294.36 |
| Cathepsin B | CTSB | P07858 | 5.215647829 | 6.847566605 | 18 | 50.7 | 37.821 |
| Cathepsin O | CTSO | P43234 | 5.722981088 | 5.925343196 | 6 | 18.4 | 35.957 |
| Leucyl-cystinyl aminopeptidase | LNPEP | Q9UIQ6 | 2.964108501 | 5.812737147 | 26 | 25.6 | 117.35 |
| Protein zer-1 homolog | ZER1 | Q7Z7L7 | 6.50224812 | 5.582649867 | 19 | 26.1 | 88.169 |
| Baculoviral IAP repeat-containing protein 6 | BIRC6 | Q9NR09 | 3.072196881 | 5.047833761 | 21 | 6.5 | 530.25 |
| Cathepsin F | CTSF | Q9UBX1 | 3.643260716 | 4.351509094 | 5 | 15.3 | 53.365 |
| Transcription elongation factor B polypeptide 1 | TCEB1 | Q15369 | 2.590215898 | 4.142413457 | 5 | 70.8 | 9.9601 |
| Emopamil-Binding Protein | EBP | Q15125 | 3.145526514 | 3.908337275 | 3 | 16.5 | 26.352 |
| Pre-mRNA-processing factor 40 homolog A | PRPF40A | O75400 | 4.823116829 | 3.851610184 | 8 | 11.3 | 108.8 |
| ADP-ribosylation factor-like protein 8B | ARL8B | Q9NVJ2 | 3.917623907 | 3.847599665 | 3 | 13.4 | 21.539 |
| Cathepsin V | CTSV | O60911 | 3.596574475 | 3.820123037 | 3 | 14.1 | 37.329 |
| Reticulocalbin-2 | RCN2 | Q14257 | 2.957438588 | 2.969583511 | 8 | 35.6 | 36.876 |
| RNA-binding protein 25 | RBM25 | P49756 | 2.47628457 | 2.762470245 | 9 | 15.1 | 100.18 |
| Solute Carrier Family 25 Member 12 | SLC25A12 | O75746 | 4.617153607 | 2.696769714 | 10 | 18 | 74.761 |
| Identified Protein | Gene Name | Uniprot ID | p Value (-LOG10) | Difference (LOG2) | Unique Peptides | Unique Sequence Coverage (%) | Molecular Weight (kDa) |
|---|---|---|---|---|---|---|---|
| Cathepsin C | CTSC | P53634 | 3.542433544 | 11.42620722 | 15 | 39.1 | 51.853 |
| Cathepsin L | CTSL | P07711 | 4.944991294 | 9.40882047 | 6 | 25.2 | 37.564 |
| Cystatin B | CSTB | P04080 | 3.90733015 | 8.848660151 | 5 | 77.6 | 11.139 |
| Cathepsin K | CTSK | P43235 | 3.778308165 | 8.52559344 | 10 | 50.5 | 36.966 |
| Cathepsin H | CTSH | P09668 | 3.620166418 | 7.520583471 | 9 | 31.6 | 36.269 |
| Cathepsin B | CTSB | P07858 | 4.369058919 | 6.979846319 | 18 | 50.7 | 37.821 |
| Cathepsin O | CTSO | P43234 | 7.050883497 | 6.113189697 | 6 | 18.4 | 35.957 |
| Protein zer-1 homolog | ZER1 | Q7Z7L7 | 5.61326774 | 4.726226807 | 19 | 26.1 | 88.169 |
| Translocator protein | TSPO | P30536 | 3.089099339 | 4.6859773 | 3 | 36.4 | 11.896 |
| ADP-ribosylation factor-like protein 8B | ARL8B | Q9NVJ2 | 4.124792469 | 4.229539871 | 3 | 13.4 | 21.539 |
| Baculoviral IAP repeat-containing protein 6 | BIRC6 | Q9NR09 | 2.727875293 | 4.118724187 | 21 | 6.5 | 530.25 |
| Emopamil-Binding Protein | EBP | Q15125 | 3.565297847 | 3.988709768 | 3 | 16.5 | 26.352 |
| Tubulin beta-8 chain | TUBB8 | Q3ZCM7 | 2.606538895 | 3.97833252 | 2 | 7.4 | 49.775 |
| Cathepsin F | CTSF | Q9UBX1 | 3.123290332 | 3.924125671 | 5 | 15.3 | 53.365 |
| Cathepsin V | CTSV | O60911 | 3.457522376 | 3.883289973 | 3 | 14.1 | 37.329 |
| Pre-mRNA-processing factor 40 homolog A | PRPF40A | O75400 | 3.826395659 | 3.662123362 | 8 | 11.3 | 108.8 |
| Leucyl-cystinyl aminopeptidase | LNPEP | Q9UIQ6 | 2.430111396 | 3.348843892 | 26 | 25.6 | 117.35 |
| Prosaposin | PSAP | P07602 | 2.525074435 | 2.975476583 | 14 | 28.3 | 58.44 |
| Ras-related protein Rab-6C | RAB6C | Q9H0N0 | 2.182205955 | 2.939130147 | 2 | 12.2 | 28.242 |
| DnaJ homolog subfamily C member 5 | DNAJC5 | Q9H3Z4 | 4.486646762 | 2.830143611 | 2 | 24.2 | 22.149 |
| RNA-binding protein 25 | RBM25 | P49756 | 2.382926143 | 2.787614187 | 9 | 15.1 | 100.18 |
| Transcription elongation factor B polypeptide 1 | TCEB1 | Q15369 | 2.059233321 | 2.78107961 | 5 | 70.8 | 9.9601 |
| 60S acidic ribosomal protein P1 | RPLP1 | P05386 | 2.349200506 | 2.666696548 | 2 | 51.8 | 11.514 |
| Tubulin alpha-1C chain | TUBA1C | Q9BQE3 | 2.518650635 | 2.564816793 | 4 | 13.1 | 57.73 |
| Cytochrome b5 type B | CYB5B | O43169 | 3.694849566 | 2.431503296 | 2 | 18 | 16.694 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carlsson, E.; Sharif, U.; Supharattanasitthi, W.; Paraoan, L. Analysis of Wild Type and Variant B Cystatin C Interactome in Retinal Pigment Epithelium Cells Reveals Variant B Interacting Mitochondrial Proteins. Cells 2023, 12, 713. https://doi.org/10.3390/cells12050713
Carlsson E, Sharif U, Supharattanasitthi W, Paraoan L. Analysis of Wild Type and Variant B Cystatin C Interactome in Retinal Pigment Epithelium Cells Reveals Variant B Interacting Mitochondrial Proteins. Cells. 2023; 12(5):713. https://doi.org/10.3390/cells12050713
Chicago/Turabian StyleCarlsson, Emil, Umar Sharif, Wasu Supharattanasitthi, and Luminita Paraoan. 2023. "Analysis of Wild Type and Variant B Cystatin C Interactome in Retinal Pigment Epithelium Cells Reveals Variant B Interacting Mitochondrial Proteins" Cells 12, no. 5: 713. https://doi.org/10.3390/cells12050713