
Oxidative Stress and Lipid Accumulation Augments Cell Death in LDLR-Deficient RPE Cells and Ldlr−/− Mice

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
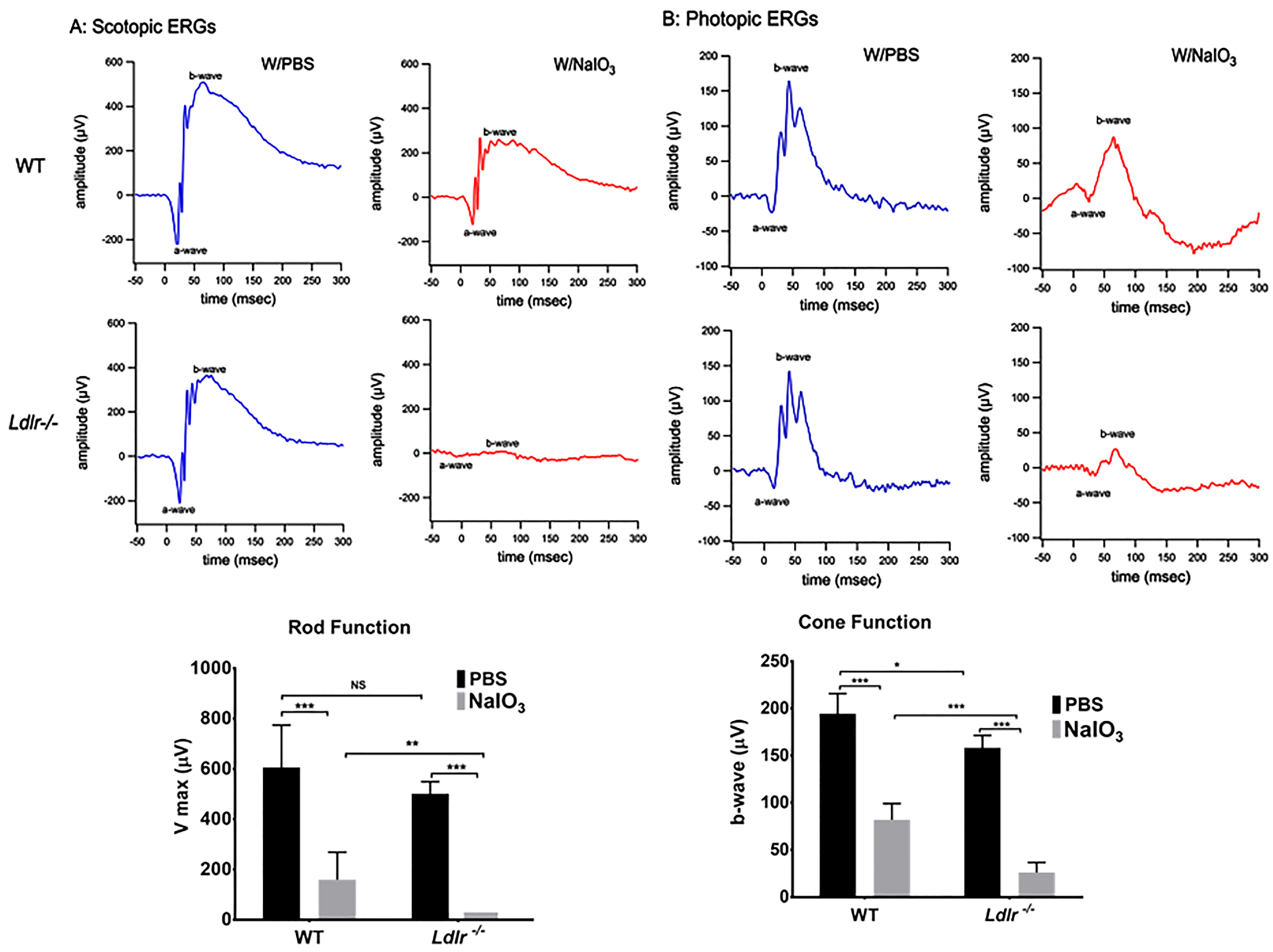{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Retinal Pigment Epithelial Cell Culture
2.2. Localization of LDLR in fhRPE Cells with and without Oxidative Stress
2.3. siRNA-Mediated Knockdown of LDLR and Caspase-3/7 Activation Using IncuCyte Cell Apoptosis Assay
2.4. NaIO3-Induced RPE Degeneration Mouse Model
2.5. Spectral Domain Optical Coherence Tomography (SD-OCT) and Fundus Imaging
2.6. Electrophysiology
2.7. Retinal Histology
2.8. TUNEL Staining
2.9. Immunofluorescence Staining
2.10. Oil Red O Staining
2.11. Data Analysis
3. Results
3.1. LDLR RPE Localization
3.2. LDLR Knockdown Accelerated and Exacerbated tBH-Induced Cell Death
3.3. Deficiency of LDLR Exacerbated Retinal Degeneration In Vivo
3.4. LDLR Deficiency and or Acute NaIO3 Administration Reduced Visual Performance
3.5. Histopathological Changes and Retinal Degeneration in NaIO3 Administered Ldlr −/− Mice
3.6. Oxidative Stress Exacerbates Apoptosis in Ldlr −/− Mouse Retina
3.7. LDLR Deficiency Resulted in Lipid Accumulation in the Outer Retina
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fleckenstein, M.; Keenan, T.D.L.; Guymer, R.H.; Chakravarthy, U.; Schmitz-Valckenberg, S.; Klaver, C.C.; Wong, W.T.; Chew, E.Y. Age-related macular degeneration. Nat. Rev. Dis. Primers 2021, 7, 31. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, e106–e116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Handa, J.T.; Bowes Rickman, C.; Dick, A.D.; Gorin, M.B.; Miller, J.W.; Toth, C.A.; Ueffing, M.; Zarbin, M.; Farrer, L.A. A systems biology approach towards understanding and treating non-neovascular age-related macular degeneration. Nat. Commun. 2019, 10, 3347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curcio, C.A.; Millican, C.L. Basal linear deposit and large drusen are specific for early age-related maculopathy. Arch. Ophthalmol. 1999, 117, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Curcio, C.A.; Millican, C.L.; Bailey, T.; Kruth, H.S. Accumulation of cholesterol with age in human Bruch’s membrane. Invest. Ophthalmol. Vis. Sci. 2001, 42, 265–274. [Google Scholar]
- Curcio, C.A.; Johnson, M.; Rudolf, M.; Huang, J.D. The oil spill in ageing Bruch membrane. Br. J. Ophthalmol. 2011, 95, 1638–1645. [Google Scholar] [CrossRef]
- Wang, L.; Clark, M.E.; Crossman, D.K.; Kojima, K.; Messinger, J.D.; Mobley, J.A.; Curcio, C.A. Abundant lipid and protein components of drusen. PLoS ONE 2010, 5, e10329. [Google Scholar] [CrossRef]
- Chang, C.C.; Huang, C.H.; Chou, Y.C.; Chang, J.Y.; Sun, C.A. Association Between Age-Related Macular Degeneration and Risk of Heart Failure: A Population-Based Nested Case-Control Study. J. Am. Heart Assoc. 2021, 10, e020071. [Google Scholar] [CrossRef]
- Wang, S.B.; Mitchell, P.; Chiha, J.; Liew, G.; Plant, A.J.; Thiagalingam, A.; Burlutsky, G.; Gopinath, B. Severity of coronary artery disease is independently associated with the frequency of early age-related macular degeneration. Br. J. Ophthalmol. 2015, 99, 365–370. [Google Scholar] [CrossRef]
- Fernandez, A.B.; Ballard, K.D.; Wong, T.Y.; Guo, M.; McClelland, R.L.; Burke, G.; Cotch, M.F.; Klein, B.; Allison, M.; Klein, R. Age-related macular degeneration and progression of coronary artery calcium: The Multi-Ethnic Study of Atherosclerosis. PLoS ONE 2018, 13, e0201000. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, H.; Ji, J.; Wang, L.; Lv, W.; He, Y.; Li, X.; Feng, G.; Chen, K. A histological study of atherosclerotic characteristics in age-related macular degeneration. Heliyon 2022, 8, e08973. [Google Scholar] [CrossRef] [PubMed]
- Oppi, S.; Luscher, T.F.; Stein, S. Mouse Models for Atherosclerosis Research-Which Is My Line? Front. Cardiovasc. Med. 2019, 6, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bentzon, J.F.; Falk, E. Atherosclerotic lesions in mouse and man: Is it the same disease? Curr. Opin. Lipidol. 2010, 21, 434–440. [Google Scholar] [CrossRef]
- Sonoda, S.; Sreekumar, P.G.; Kase, S.; Spee, C.; Ryan, S.J.; Kannan, R.; Hinton, D.R. Attainment of polarity promotes growth factor secretion by retinal pigment epithelial cells: Relevance to age-related macular degeneration. Aging 2009, 2, 28–42. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, S.; Spee, C.; Barron, E.; Ryan, S.J.; Kannan, R.; Hinton, D.R. A protocol for the culture and differentiation of highly polarized human retinal pigment epithelial cells. Nat. Protoc. 2009, 4, 662–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sreekumar, P.G.; Ishikawa, K.; Spee, C.; Mehta, H.H.; Wan, J.; Yen, K.; Cohen, P.; Kannan, R.; Hinton, D.R. The Mitochondrial-Derived Peptide Humanin Protects RPE Cells From Oxidative Stress, Senescence, and Mitochondrial Dysfunction. Invest. Ophthalmol. Vis. Sci. 2016, 57, 1238–1253. [Google Scholar] [CrossRef] [PubMed]
- Totsuka, K.; Ueta, T.; Uchida, T.; Roggia, M.F.; Nakagawa, S.; Vavvas, D.G.; Honjo, M.; Aihara, M. Oxidative stress induces ferroptotic cell death in retinal pigment epithelial cells. Exp. Eye Res. 2019, 181, 316–324. [Google Scholar] [CrossRef]
- Sreekumar, P.G.; Li, Z.; Wang, W.; Spee, C.; Hinton, D.R.; Kannan, R.; MacKay, J.A. Intra-vitreal alphaB crystallin fused to elastin-like polypeptide provides neuroprotection in a mouse model of age-related macular degeneration. J. Control. Release 2018, 283, 94–104. [Google Scholar] [CrossRef]
- Shihan, M.H.; Novo, S.G.; Le Marchand, S.J.; Wang, Y.; Duncan, M.K. A simple method for quantitating confocal fluorescent images. Biochem. Biophys. Rep. 2021, 25, 100916. [Google Scholar] [CrossRef]
- Wang, M.; Lau, L.I.; Sreekumar, P.G.; Spee, C.; Hinton, D.R.; Sadda, S.R.; Kannan, R. Characterization and Regulation of Carrier Proteins of Mitochondrial Glutathione Uptake in Human Retinal Pigment Epithelium Cells. Invest. Ophthalmol. Vis. Sci. 2019, 60, 500–516. [Google Scholar] [CrossRef] [Green Version]
- Su, F.; Spee, C.; Araujo, E.; Barron, E.; Wang, M.; Ghione, C.; Hinton, D.R.; Nusinowitz, S.; Kannan, R.; Reddy, S.T.; et al. A Novel HDL-Mimetic Peptide HM-10/10 Protects RPE and Photoreceptors in Murine Models of Retinal Degeneration. Int. J. Mol. Sci. 2019, 20, 4807. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Kannan, R.; Spee, C.; Sreekumar, P.G.; Dou, G.; Hinton, D.R. Protection of retina by alphaB crystallin in sodium iodate induced retinal degeneration. PLoS ONE 2014, 9, e98275. [Google Scholar] [CrossRef]
- Tserentsoodol, N.; Sztein, J.; Campos, M.; Gordiyenko, N.V.; Fariss, R.N.; Lee, J.W.; Fliesler, S.J.; Rodriguez, I.R. Uptake of cholesterol by the retina occurs primarily via a low density lipoprotein receptor-mediated process. Mol. Vis. 2006, 12, 1306–1318. [Google Scholar] [PubMed]
- Wang, S.H.; Huang, Y.; Yuan, Y.; Xia, W.Q.; Wang, P.; Huang, R. LDL receptor knock-out mice show impaired spatial cognition with hippocampal vulnerability to apoptosis and deficits in synapses. Lipids Health Dis. 2014, 13, 175. [Google Scholar] [CrossRef] [Green Version]
- Poreba, M.; Szalek, A.; Kasperkiewicz, P.; Rut, W.; Salvesen, G.S.; Drag, M. Small Molecule Active Site Directed Tools for Studying Human Caspases. Chem. Rev. 2015, 115, 12546–12629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauleikhoff, D.; Barondes, M.; Minassian, D.; Chrisholm, J.; Wessing, A.; Bird, A.C. Drusen and their significance in age related macular degeneration. Fortschr. Ophthalmol. 1990, 87, 429–432. [Google Scholar]
- Curcio, C.A. Antecedents of Soft Drusen, the Specific Deposits of Age-Related Macular Degeneration, in the Biology of Human Macula. Invest. Ophthalmol. Vis. Sci. 2018, 59, AMD182–AMD194. [Google Scholar] [CrossRef] [Green Version]
- Itabe, H.; Yamaguchi, T.; Nimura, S.; Sasabe, N. Perilipins: A diversity of intracellular lipid droplet proteins. Lipids Health Dis. 2017, 16, 83. [Google Scholar] [CrossRef] [Green Version]
- Girard, V.; Jollivet, F.; Knittelfelder, O.; Celle, M.; Arsac, J.N.; Chatelain, G.; Van den Brink, D.M.; Baron, T.; Shevchenko, A.; Kuhnlein, R.P.; et al. Abnormal accumulation of lipid droplets in neurons induces the conversion of alpha-Synuclein to proteolytic resistant forms in a Drosophila model of Parkinson’s disease. PLoS Genet. 2021, 17, e1009921. [Google Scholar] [CrossRef]
- Shimabukuro, M.K.; Langhi, L.G.; Cordeiro, I.; Brito, J.M.; Batista, C.M.; Mattson, M.P.; Mello Coelho, V. Lipid-laden cells differentially distributed in the aging brain are functionally active and correspond to distinct phenotypes. Sci. Rep. 2016, 6, 23795. [Google Scholar] [CrossRef] [Green Version]
- Orban, T.; Palczewska, G.; Palczewski, K. Retinyl ester storage particles (retinosomes) from the retinal pigmented epithelium resemble lipid droplets in other tissues. J. Biol. Chem. 2011, 286, 17248–17258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, D.; Tedeschi, A. The Role of Lipids, Lipid Metabolism and Ectopic Lipid Accumulation in Axon Growth, Regeneration and Repair after CNS Injury and Disease. Cells 2021, 10, 1078. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Brown, M.S. The LDL receptor. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 431–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques, L.R.; Diniz, T.A.; Antunes, B.M.; Rossi, F.E.; Caperuto, E.C.; Lira, F.S.; Goncalves, D.C. Reverse Cholesterol Transport: Molecular Mechanisms and the Non-medical Approach to Enhance HDL Cholesterol. Front. Physiol. 2018, 9, 526. [Google Scholar] [CrossRef] [Green Version]
- Gordiyenko, N.; Campos, M.; Lee, J.W.; Fariss, R.N.; Sztein, J.; Rodriguez, I.R. RPE cells internalize low-density lipoprotein (LDL) and oxidized LDL (oxLDL) in large quantities in vitro and in vivo. Invest. Ophthalmol. Vis. Sci. 2004, 45, 2822–2829. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ma, K.L.; Ruan, X.Z.; Liu, B.C. Dysregulation of the Low-Density Lipoprotein Receptor Pathway Is Involved in Lipid Disorder-Mediated Organ Injury. Int. J. Biol. Sci. 2016, 12, 569–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, K.C.; Lindsey, S.; Stephan, Z.F.; Brecker, D. Retinal pigment epithelium possesses both LDL and scavenger receptor activity. Invest. Ophthalmol. Vis. Sci. 1989, 30, 225–232. [Google Scholar]
- Pfeffer, B.A.; Fliesler, S.J. Reassessing the suitability of ARPE-19 cells as a valid model of native RPE biology. Exp. Eye. Res. 2022, 219, 109046. [Google Scholar] [CrossRef]
- Bhutto, I.A.; Ogura, S.; Baldeosingh, R.; McLeod, D.S.; Lutty, G.A.; Edwards, M.M. An Acute Injury Model for the Phenotypic Characteristics of Geographic Atrophy. Invest. Ophthalmol. Vis. Sci. 2018, 59, AMD143–AMD151. [Google Scholar] [CrossRef] [Green Version]
- Koster, C.; van den Hurk, K.T.; Ten Brink, J.B.; Lewallen, C.F.; Stanzel, B.V.; Bharti, K.; Bergen, A.A. Sodium-Iodate Injection Can Replicate Retinal Degenerative Disease Stages in Pigmented Mice and Rats: Non-Invasive Follow-Up Using OCT and ERG. Int. J. Mol. Sci. 2022, 23, 2918. [Google Scholar] [CrossRef]
- Enzbrenner, A.; Zulliger, R.; Biber, J.; Pousa, A.M.Q.; Schafer, N.; Stucki, C.; Giroud, N.; Berrera, M.; Kortvely, E.; Schmucki, R.; et al. Sodium Iodate-Induced Degeneration Results in Local Complement Changes and Inflammatory Processes in Murine Retina. Int. J. Mol. Sci. 2021, 22, 9218. [Google Scholar] [CrossRef] [PubMed]
- Park, U.C.; Cho, M.S.; Park, J.H.; Kim, S.J.; Ku, S.Y.; Choi, Y.M.; Moon, S.Y.; Yu, H.G. Subretinal transplantation of putative retinal pigment epithelial cells derived from human embryonic stem cells in rat retinal degeneration model. Clin. Exp. Reprod. Med. 2011, 38, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Cui, L.; Qu, Z.; Lu, L.; Wang, F.; Wu, Y.; Zhang, J.; Gao, F.; Tian, H.; Xu, L.; et al. Subretinal transplantation of rat MSCs and erythropoietin gene modified rat MSCs for protecting and rescuing degenerative retina in rats. Curr. Mol. Med. 2013, 13, 1419–1431. [Google Scholar] [CrossRef] [PubMed]
- Carido, M.; Zhu, Y.; Postel, K.; Benkner, B.; Cimalla, P.; Karl, M.O.; Kurth, T.; Paquet-Durand, F.; Koch, E.; Munch, T.A.; et al. Characterization of a mouse model with complete RPE loss and its use for RPE cell transplantation. Invest. Ophthalmol. Vis. Sci. 2014, 55, 5431–5444. [Google Scholar] [CrossRef] [Green Version]
- Petrus-Reurer, S.; Bartuma, H.; Aronsson, M.; Westman, S.; Lanner, F.; Andre, H.; Kvanta, A. Integration of Subretinal Suspension Transplants of Human Embryonic Stem Cell-Derived Retinal Pigment Epithelial Cells in a Large-Eyed Model of Geographic Atrophy. Invest. Ophthalmol. Vis. Sci. 2017, 58, 1314–1322. [Google Scholar] [CrossRef]
- Romero-Vazquez, S.; Llorens, V.; Soler-Boronat, A.; Figueras-Roca, M.; Adan, A.; Molins, B. Interlink between Inflammation and Oxidative Stress in Age-Related Macular Degeneration: Role of Complement Factor H. Biomedicines 2021, 9, 763. [Google Scholar] [CrossRef]
- Kim, S.Y.; Kambhampati, S.P.; Bhutto, I.A.; McLeod, D.S.; Lutty, G.A.; Kannan, R.M. Evolution of oxidative stress, inflammation and neovascularization in the choroid and retina in a subretinal lipid induced age-related macular degeneration model. Exp. Eye. Res. 2021, 203, 108391. [Google Scholar] [CrossRef]
- Datta, S.; Cano, M.; Ebrahimi, K.; Wang, L.; Handa, J.T. The impact of oxidative stress and inflammation on RPE degeneration in non-neovascular AMD. Prog. Retin. Eye Res. 2017, 60, 201–218. [Google Scholar] [CrossRef]
- Kauppinen, A.; Paterno, J.J.; Blasiak, J.; Salminen, A.; Kaarniranta, K. Inflammation and its role in age-related macular degeneration. Cell Mol. Life Sci. 2016, 73, 1765–1786. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Liu, G.; Liu, X.; Wang, Y.; He, F.; Nie, D.; Liu, X.; Liu, X. RNA-seq analysis reveals differentially expressed inflammatory chemokines in a rat retinal degeneration model induced by sodium iodate. J. Int. Med. Res. 2022, 50, 3000605221119376. [Google Scholar] [CrossRef]
- Calvier, L.; Herz, J.; Hansmann, G. Interplay of Low-Density Lipoprotein Receptors, LRPs, and Lipoproteins in Pulmonary Hypertension. JACC Basic Transl. Sci. 2022, 7, 164–180. [Google Scholar] [CrossRef] [PubMed]
- Merkel, M.; Velez-Carrasco, W.; Hudgins, L.C.; Breslow, J.L. Compared with saturated fatty acids, dietary monounsaturated fatty acids and carbohydrates increase atherosclerosis and VLDL cholesterol levels in LDL receptor-deficient, but not apolipoprotein E-deficient, mice. Proc. Natl. Acad. Sci. USA 2001, 98, 13294–13299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bieghs, V.; Van Gorp, P.J.; Wouters, K.; Hendrikx, T.; Gijbels, M.J.; van Bilsen, M.; Bakker, J.; Binder, C.J.; Lutjohann, D.; Staels, B.; et al. LDL receptor knock-out mice are a physiological model particularly vulnerable to study the onset of inflammation in non-alcoholic fatty liver disease. PLoS ONE 2012, 7, e30668. [Google Scholar] [CrossRef]
- Yang, Z.H.; Nill, K.; Takechi-Haraya, Y.; Playford, M.P.; Nguyen, D.; Yu, Z.X.; Pryor, M.; Tang, J.; Rojulpote, K.V.; Mehta, N.N.; et al. Differential Effect of Dietary Supplementation with a Soybean Oil Enriched in Oleic Acid versus Linoleic Acid on Plasma Lipids and Atherosclerosis in LDLR-Deficient Mice. Int. J. Mol. Sci. 2022, 23, 8385. [Google Scholar] [CrossRef] [PubMed]
- Grover, S.P.; Coughlin, T.; Fleifil, S.M.; Posma, J.J.N.; Spronk, H.H.M.; Heitmeier, S.; Owens, A.P., 3rd; Mackman, N. Effect of combining aspirin and rivaroxaban on atherosclerosis in mice. Atherosclerosis 2022, 345, 7–14. [Google Scholar] [CrossRef]
- Komninos, D.; Ramos, L.; van der Heijden, G.W.; Morrison, M.C.; Kleemann, R.; van Herwaarden, A.E.; Kiliaan, A.J.; Arnoldussen, I.A.C. High fat diet-induced obesity prolongs critical stages of the spermatogenic cycle in a Ldlr(−/−).Leiden mouse model. Sci. Rep. 2022, 12, 430. [Google Scholar] [CrossRef] [PubMed]
- Umar, S.; Ruffenach, G.; Moazeni, S.; Vaillancourt, M.; Hong, J.; Cunningham, C.; Cao, N.; Navab, S.; Sarji, S.; Li, M.; et al. Involvement of Low-Density Lipoprotein Receptor in the Pathogenesis of Pulmonary Hypertension. J. Am. Heart Assoc. 2020, 9, e012063. [Google Scholar] [CrossRef] [PubMed]
- Ramachandra Rao, S.; Fliesler, S.J. Cholesterol homeostasis in the vertebrate retina: Biology and pathobiology. J. Lipid Res. 2021, 62, 100057. [Google Scholar] [CrossRef]
- Cao, X.; Guo, Y.; Wang, Y.; Wang, H.; Liu, D.; Gong, Y.; Wang, J.; Chen, X.; Zhang, W. Effects of high-fat diet and Apoe deficiency on retinal structure and function in mice. Sci. Rep. 2020, 10, 18601. [Google Scholar] [CrossRef]
- Ong, J.M.; Zorapapel, N.C.; Aoki, A.M.; Brown, D.J.; Nesburn, A.B.; Rich, K.A.; Kenney, C.M. Impaired electroretinogram (ERG) response in apolipoprotein E-deficient mice. Curr. Eye Res. 2003, 27, 15–24. [Google Scholar] [CrossRef]
- Rudolf, M.; Ivandic, B.; Winkler, J.; Schmidt-Erfurth, U. Accumulation of lipid particles in Bruch’s membrane of LDL receptor knockout mice as a model of age-related macular degeneration. Ophthalmologe 2004, 101, 715–719. [Google Scholar] [CrossRef] [PubMed]
- Dunaief, J.L.; Dentchev, T.; Ying, G.S.; Milam, A.H. The role of apoptosis in age-related macular degeneration. Arch. Ophthalmol. 2002, 120, 1435–1442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, P.; Liew, G.; Gopinath, B.; Wong, T.Y. Age-related macular degeneration. Lancet 2018, 392, 1147–1159. [Google Scholar] [CrossRef] [PubMed]
- Pennington, K.L.; DeAngelis, M.M. Epidemiology of age-related macular degeneration (AMD): Associations with cardiovascular disease phenotypes and lipid factors. Eye Vis. 2016, 3, 34. [Google Scholar] [CrossRef] [Green Version]
- Neale, B.M.; Fagerness, J.; Reynolds, R.; Sobrin, L.; Parker, M.; Raychaudhuri, S.; Tan, P.L.; Oh, E.C.; Merriam, J.E.; Souied, E.; et al. Genome-wide association study of advanced age-related macular degeneration identifies a role of the hepatic lipase gene (LIPC). Proc. Natl. Acad. Sci. USA 2010, 107, 7395–7400. [Google Scholar] [CrossRef] [Green Version]
- Fritsche, L.G.; Chen, W.; Schu, M.; Yaspan, B.L.; Yu, Y.; Thorleifsson, G.; Zack, D.J.; Arakawa, S.; Cipriani, V.; Ripke, S.; et al. Seven new loci associated with age-related macular degeneration. Nat. Genet. 2013, 45, 433–439. [Google Scholar] [CrossRef] [Green Version]
- Fritsche, L.G.; Fariss, R.N.; Stambolian, D.; Abecasis, G.R.; Curcio, C.A.; Swaroop, A. Age-related macular degeneration: Genetics and biology coming together. Annu. Rev. Genom. Hum. Genet. 2014, 15, 151–171. [Google Scholar] [CrossRef] [Green Version]
- Devries-Seimon, T.; Li, Y.; Yao, P.M.; Stone, E.; Wang, Y.; Davis, R.J.; Flavell, R.; Tabas, I. Cholesterol-induced macrophage apoptosis requires ER stress pathways and engagement of the type A scavenger receptor. J. Cell Biol. 2005, 171, 61–73. [Google Scholar] [CrossRef]
- Biswas, L.; Zhou, X.; Dhillon, B.; Graham, A.; Shu, X. Retinal pigment epithelium cholesterol efflux mediated by the 18 kDa translocator protein, TSPO, a potential target for treating age-related macular degeneration. Hum. Mol. Genet. 2017, 26, 4327–4339. [Google Scholar] [CrossRef] [Green Version]
- Ishibashi, S.; Brown, M.S.; Goldstein, J.L.; Gerard, R.D.; Hammer, R.E.; Herz, J. Hypercholesterolemia in low density lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. J. Clin. Invest. 1993, 92, 883–893. [Google Scholar] [CrossRef] [Green Version]
- Boren, J.; Chapman, M.J.; Krauss, R.M.; Packard, C.J.; Bentzon, J.F.; Binder, C.J.; Daemen, M.J.; Demer, L.L.; Hegele, R.A.; Nicholls, S.J.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: Pathophysiological, genetic, and therapeutic insights: A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2020, 41, 2313–2330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, X.; Chen, Y.; Liu, Q.; Mei, X.; Liu, J.; Tang, Y.; Luo, R.; Sun, D.; Ma, Y.; Wu, W.; et al. LDLR dysfunction induces LDL accumulation and promotes pulmonary fibrosis. Clin. Transl. Med. 2022, 12, e711. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, J.; Engel, D.F.; de Paula, G.C.; Melo, H.M.; Lopes, S.C.; Ribeiro, C.T.; Delanogare, E.; Moreira, J.C.F.; Gelain, D.P.; Prediger, R.D.; et al. LDL Receptor Deficiency Does not Alter Brain Amyloid-beta Levels but Causes an Exacerbation of Apoptosis. J. Alzheimers Dis. 2020, 73, 585–596. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sreekumar, P.G.; Su, F.; Spee, C.; Araujo, E.; Nusinowitz, S.; Reddy, S.T.; Kannan, R. Oxidative Stress and Lipid Accumulation Augments Cell Death in LDLR-Deficient RPE Cells and Ldlr−/− Mice. Cells 2023, 12, 43. https://doi.org/10.3390/cells12010043
Sreekumar PG, Su F, Spee C, Araujo E, Nusinowitz S, Reddy ST, Kannan R. Oxidative Stress and Lipid Accumulation Augments Cell Death in LDLR-Deficient RPE Cells and Ldlr−/− Mice. Cells. 2023; 12(1):43. https://doi.org/10.3390/cells12010043
Chicago/Turabian StyleSreekumar, Parameswaran Gangadharan, Feng Su, Christine Spee, Eduardo Araujo, Steven Nusinowitz, Srinivasa T Reddy, and Ram Kannan. 2023. "Oxidative Stress and Lipid Accumulation Augments Cell Death in LDLR-Deficient RPE Cells and Ldlr−/− Mice" Cells 12, no. 1: 43. https://doi.org/10.3390/cells12010043