
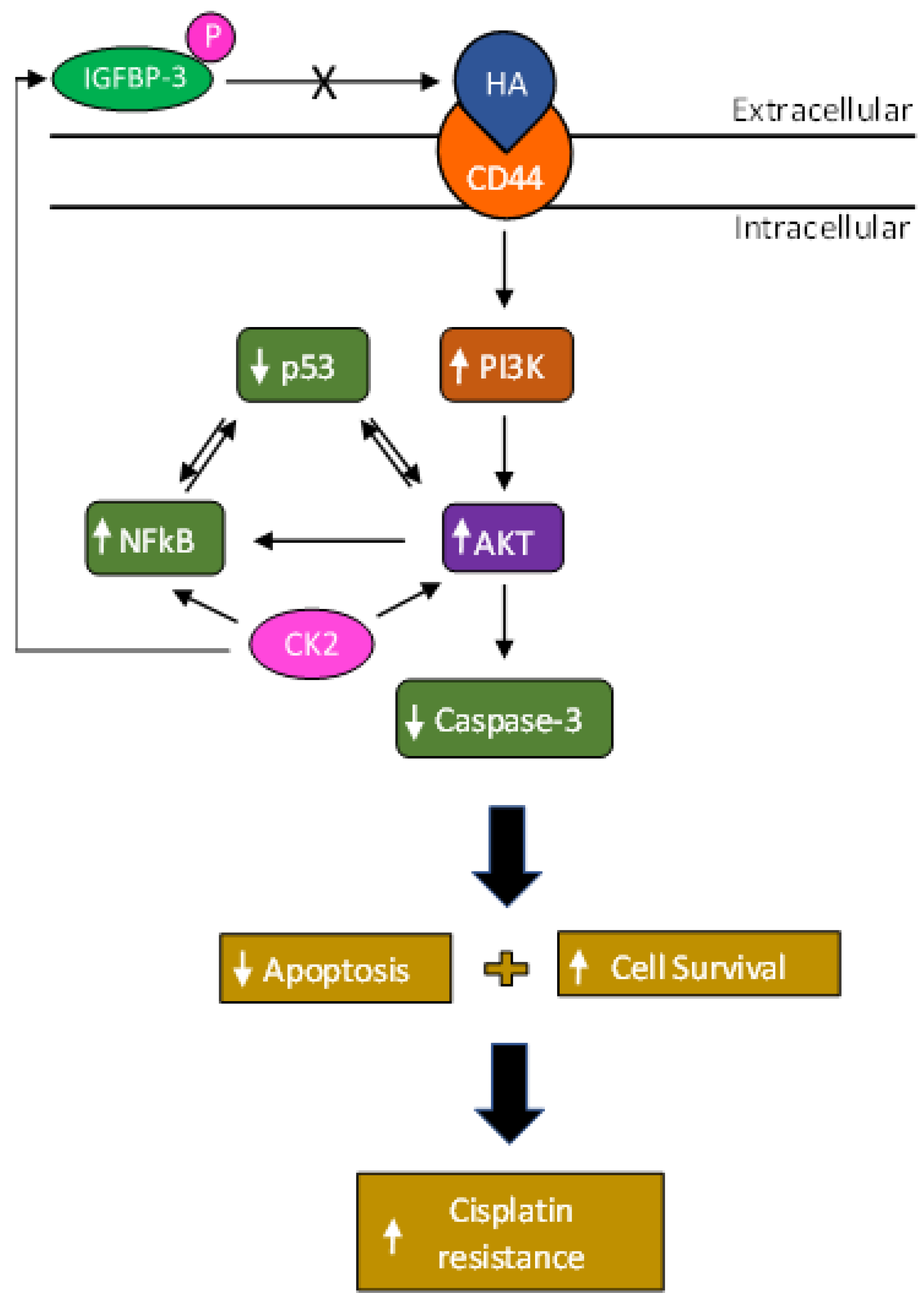
Phosphorylation of IGFBP-3 by Casein Kinase 2 Blocks Its Interaction with Hyaluronan, Enabling HA-CD44 Signaling Leading to Increased NSCLC Cell Survival and Cisplatin Resistance


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. MTT Assay
2.4. Apoptosis Assay
2.5. ELISA
2.6. Western Blotting
2.7. PI3K Assay
2.8. AKT assay
2.9. NFκB Assay
2.10. p53 Transcription Factor Activity Assay
2.11. Statistical Analysis
3. Results
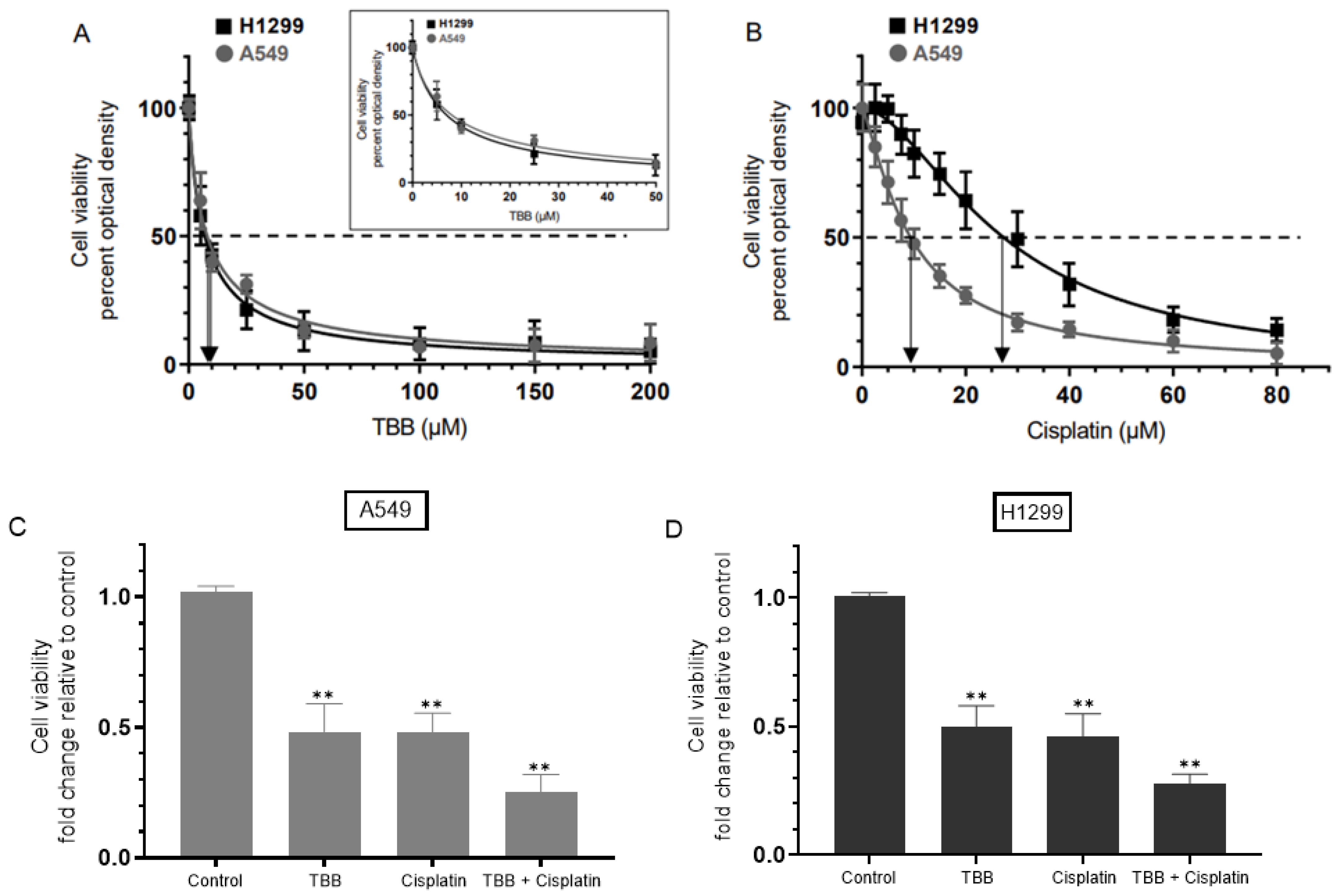
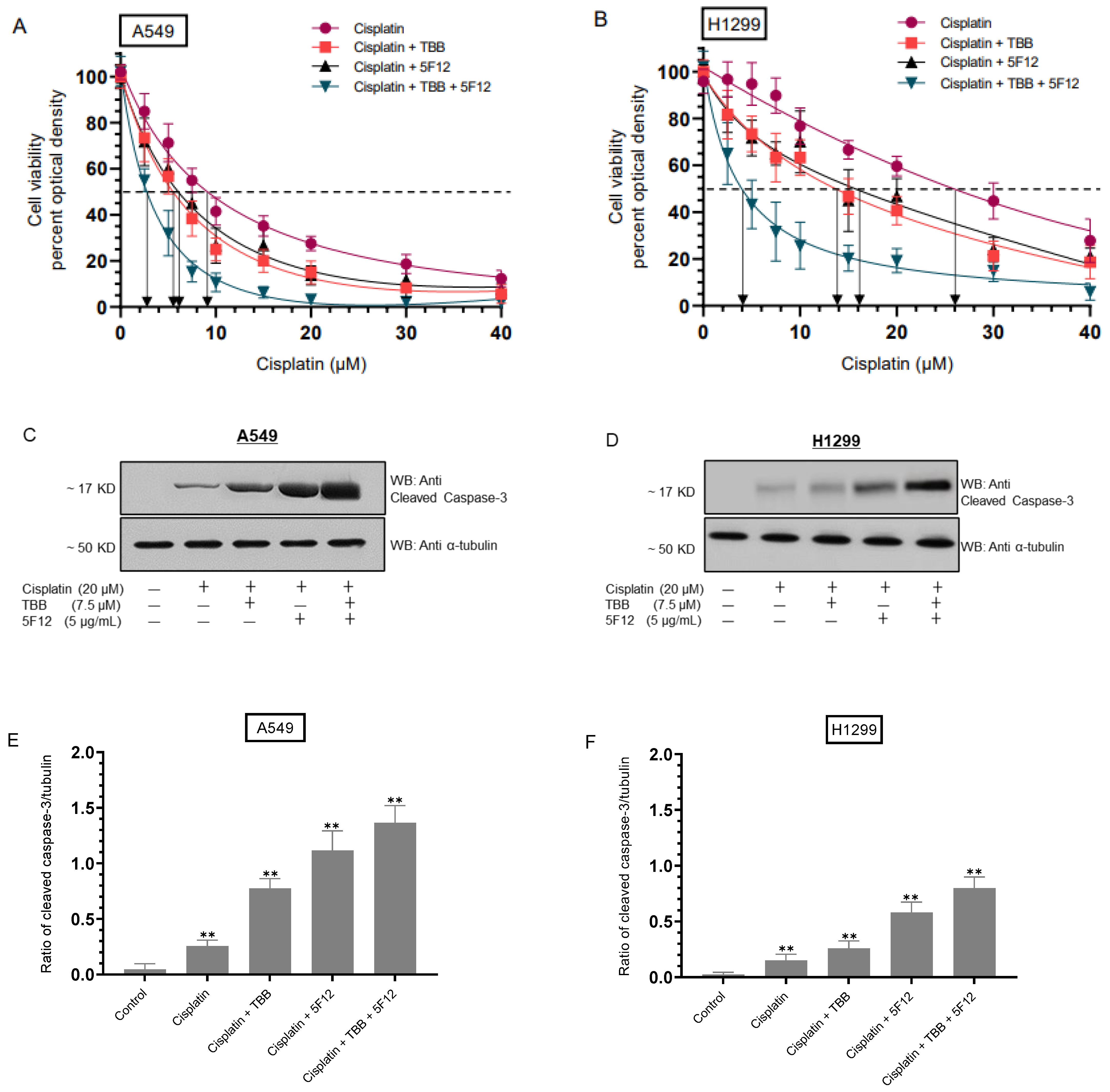
3.1. Treatment with the CK2 Inhibitor (TBB) and Cisplatin Diminished Cell Viability More Effectively than Treatment with Either TBB or Cisplatin Alone
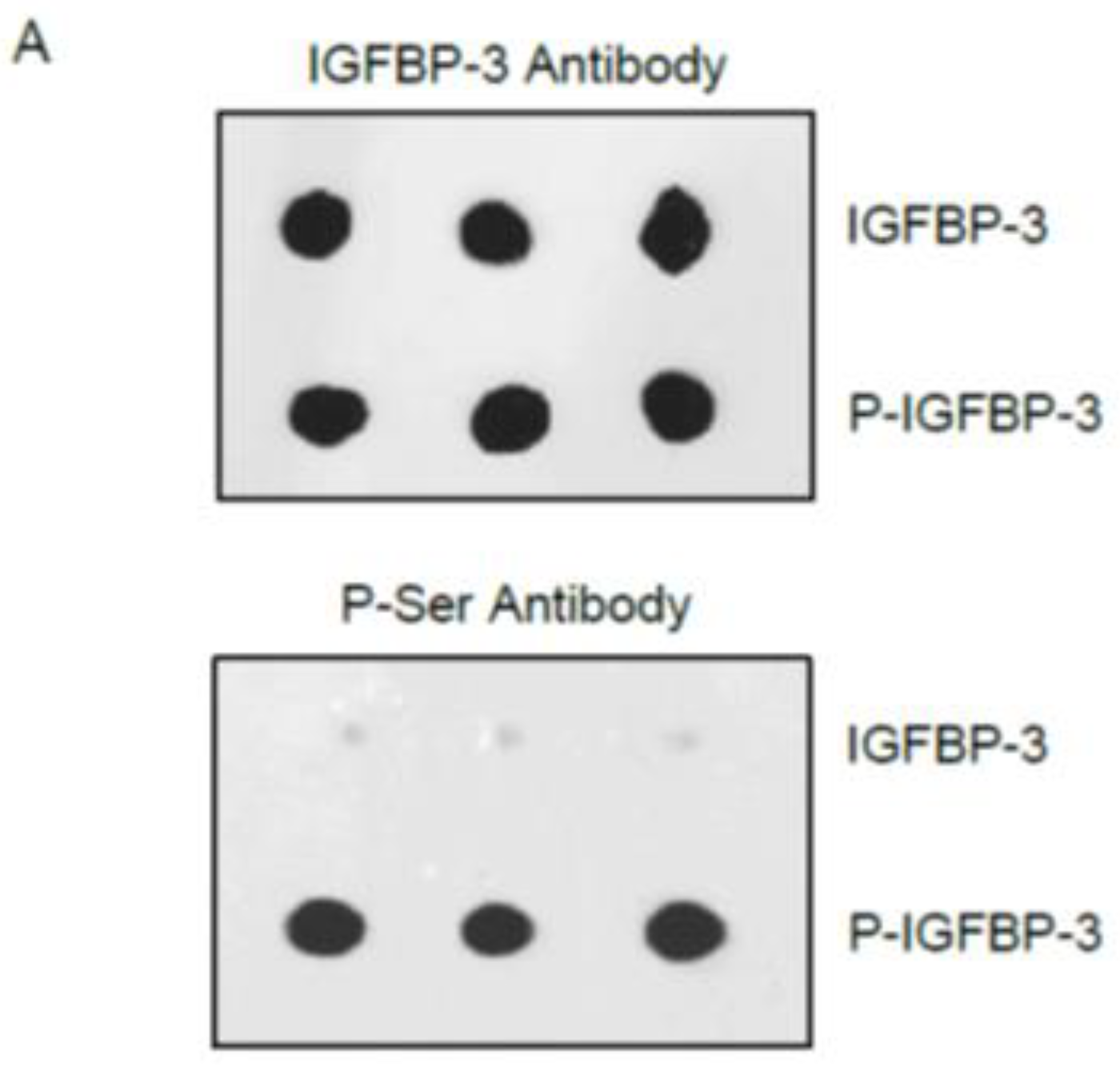
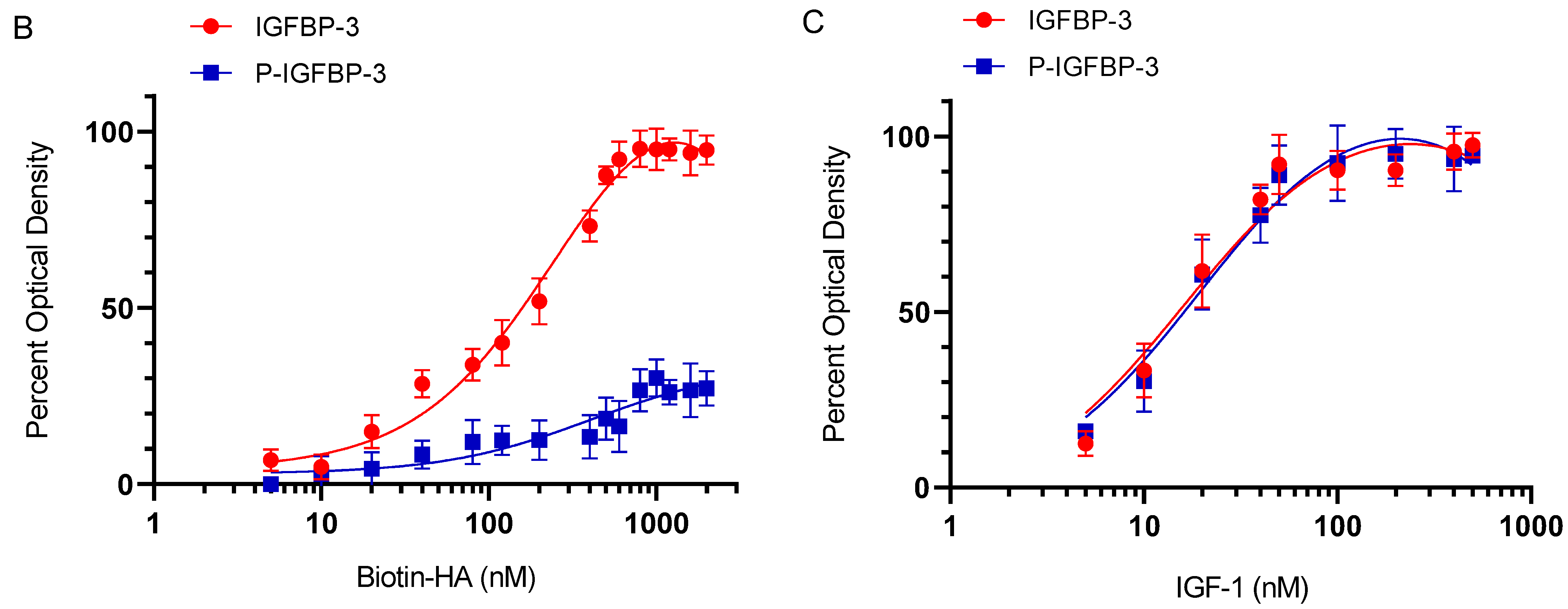
3.2. Phosphorylation of IGFBP-3 by CK2 Decreases Its Binding to HA but Not to IGF-1
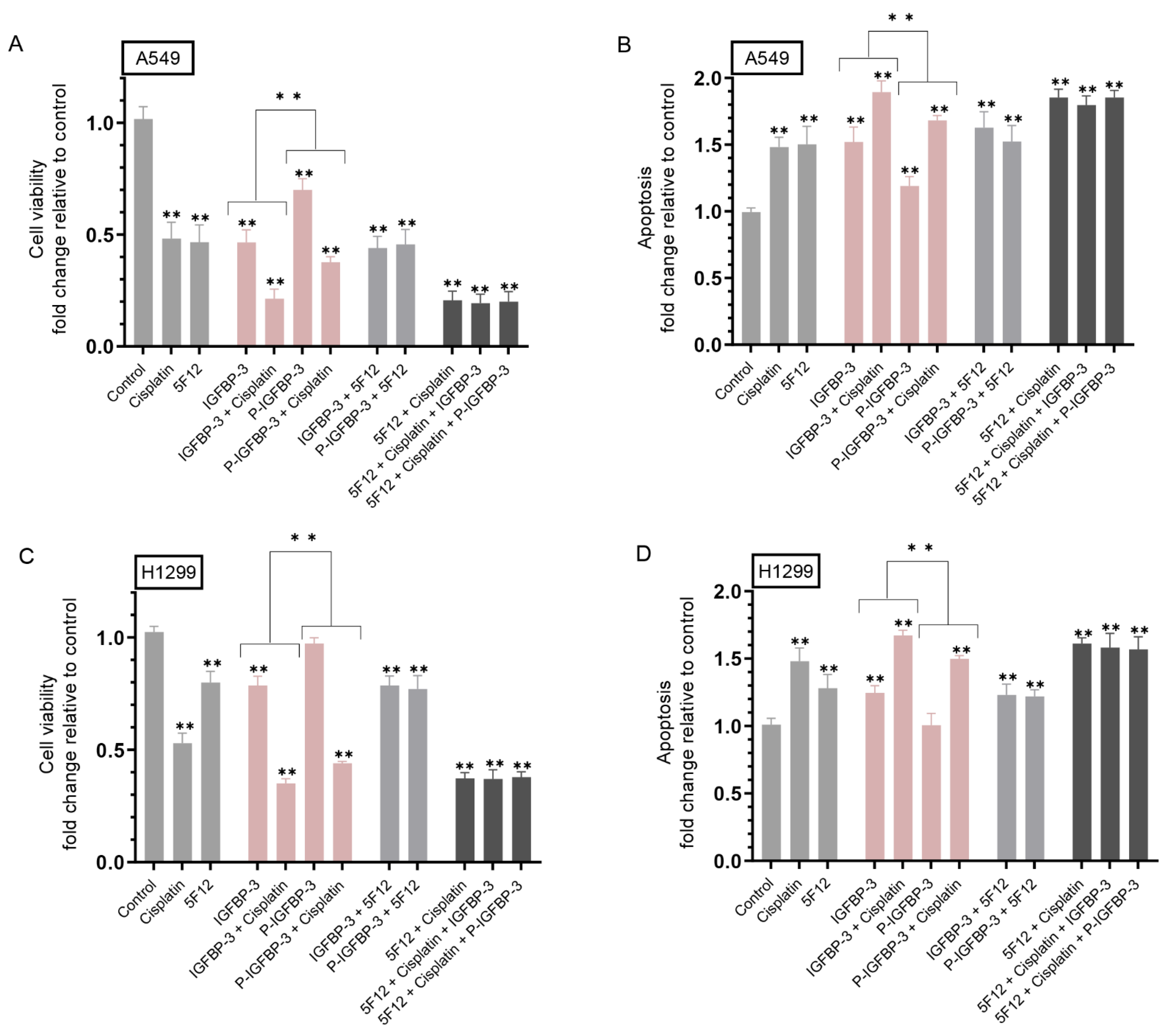
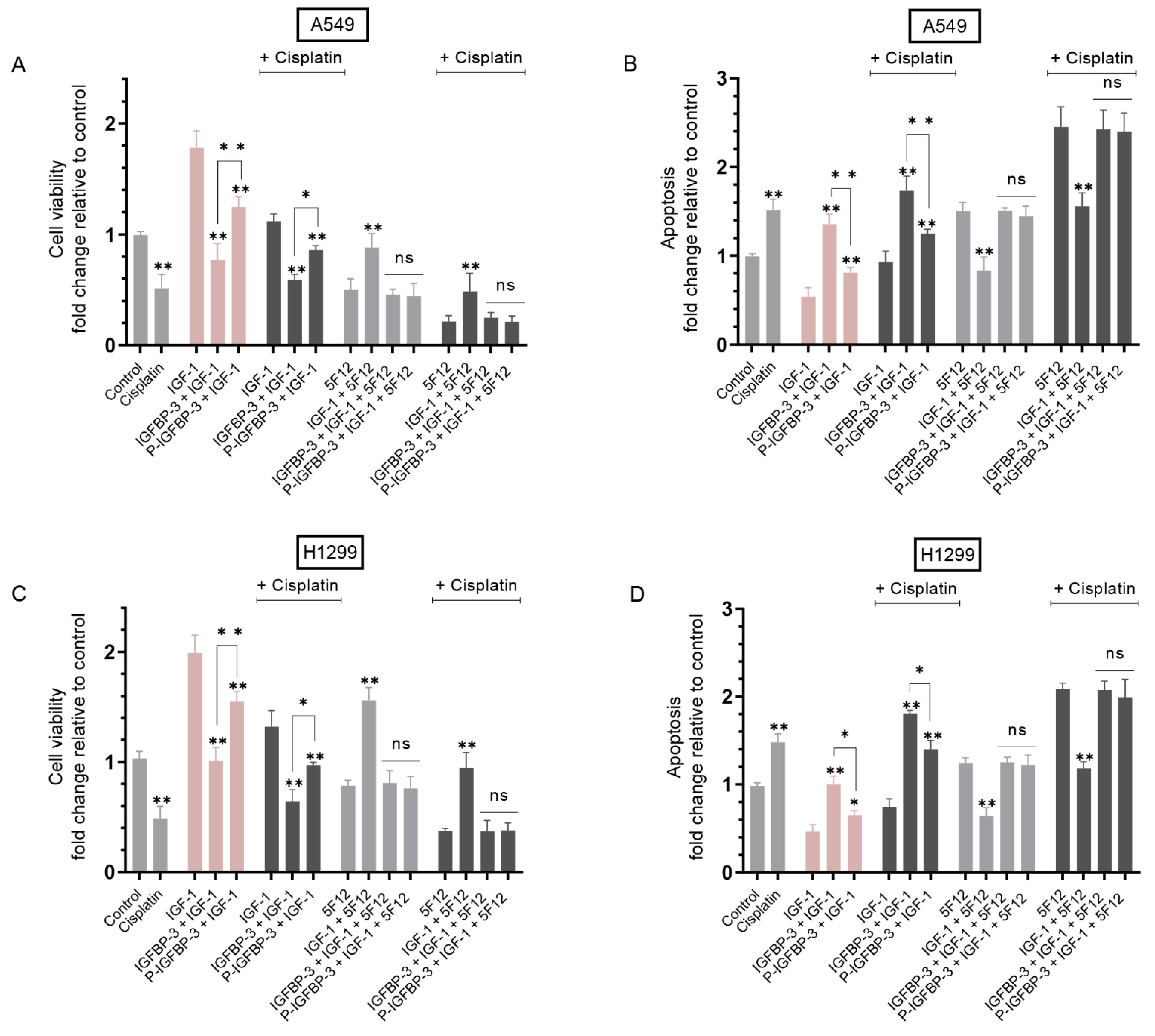
3.3. IGFBP-3 Phosphorylated by CK2 Was Less Effective than IGFBP-3 at Decreasing Cell Viability and Increasing Apoptosis in the Absence or Presence of Cisplatin
3.4. Treatment of Cells with IGF-1 and Either IGFBP-3 or P-IGFBP-3 Had the Same Effect on Cell Viability and Apoptosis in the Presence of the CD44 Antibody, 5F12, with or without Cisplatin
3.5. Sensitivity of A549 and H1299 Cells to Cisplatin Increased upon Co-Treatment with TBB and 5F12
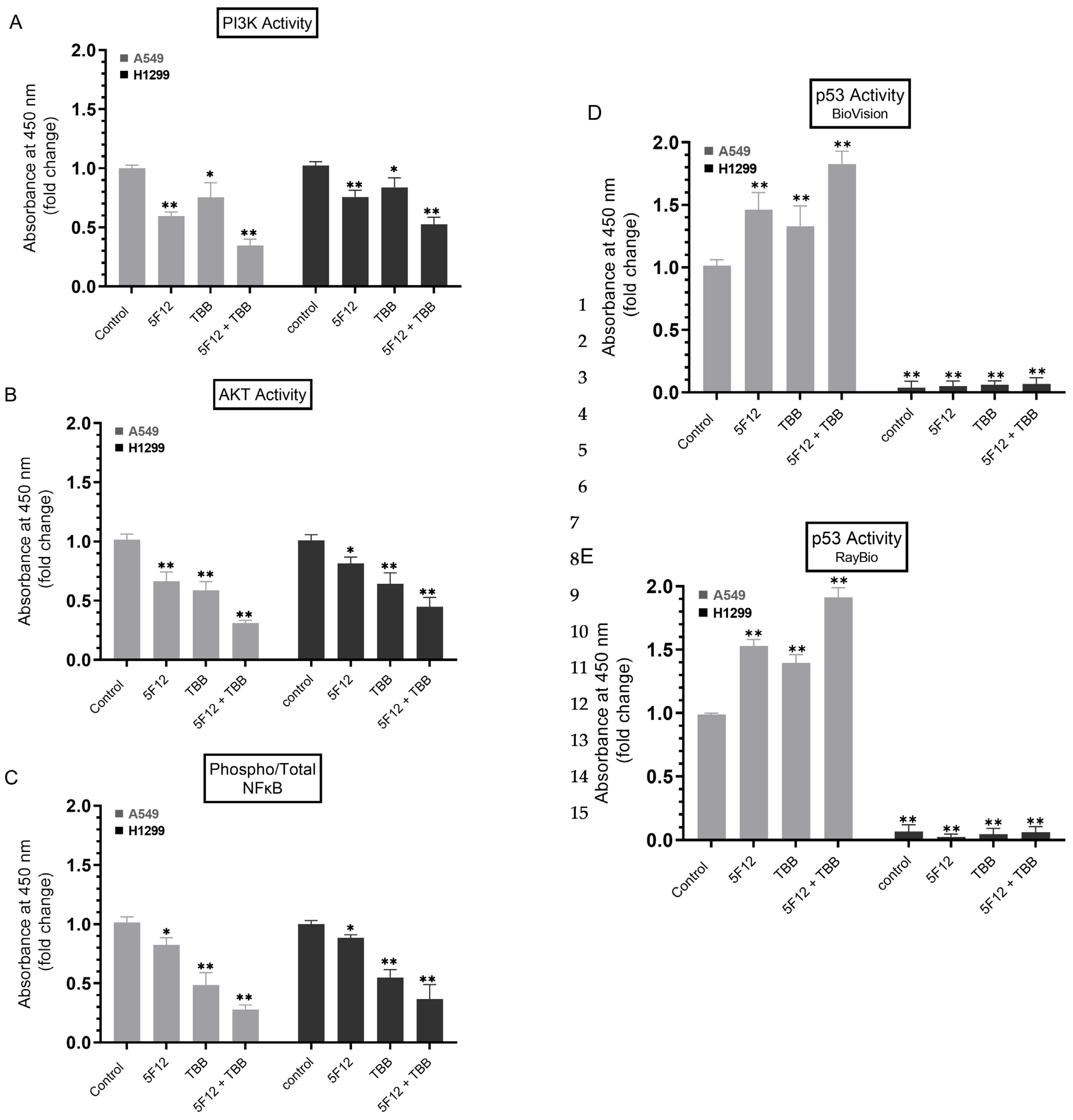
3.6. A549 and H1299 Cell Co-Treatment with 5F12 and TBB Was Most Effective at Abolishing the PI3K and AKT Activities and the Phospho/Total NFκB Ratio and at Increasing the p53 Activity in A549 Cells Only
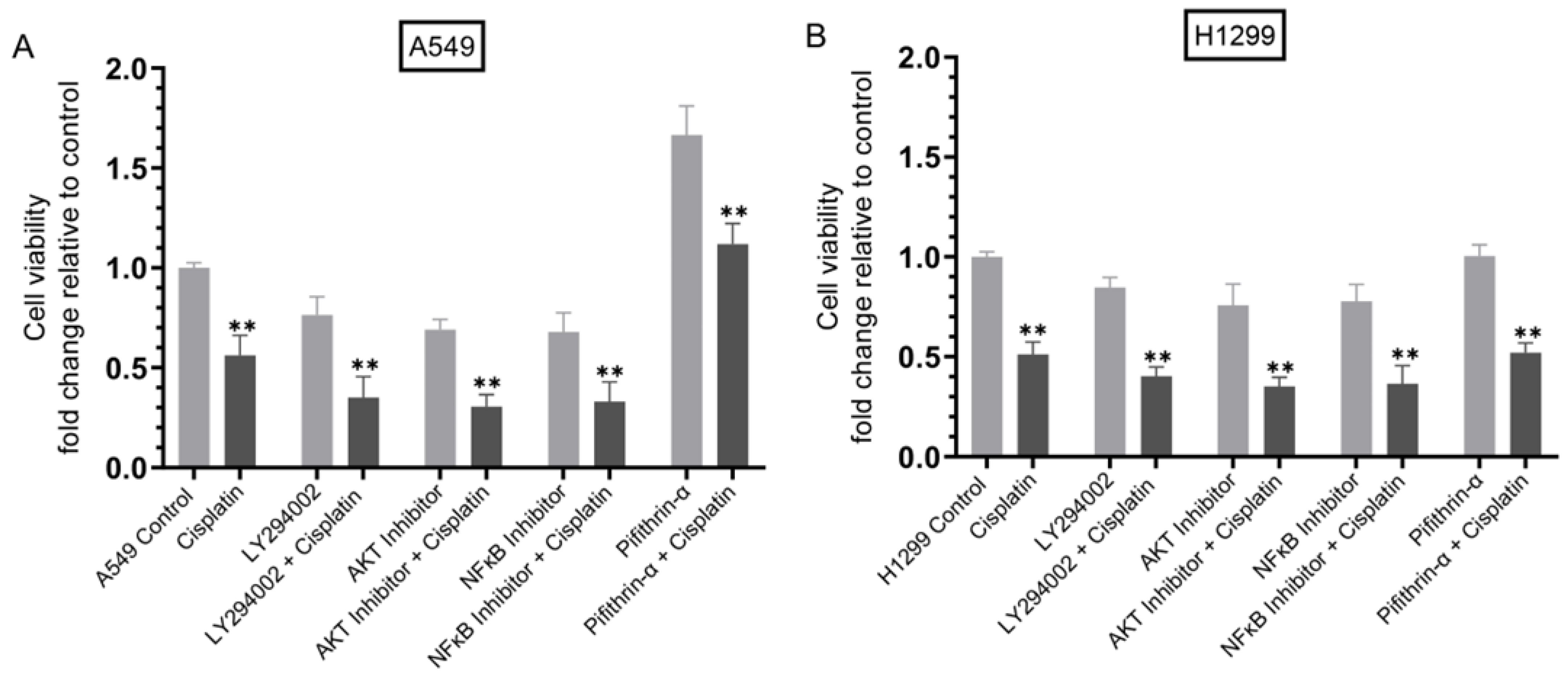
3.7. Cisplatin Sensitivity Increased upon Co-Treatment of Cells with Inhibitors Targeted against PI3K, AKT, or NFκB but Decreased in A549 Cells Treated with the p53 Inhibitor
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siddiqui, F.; Siddiqui, A.H. Lung Cancer. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Tchounwou, P.B.; Dasari, S.; Noubissi, F.K.; Ray, P.; Kumar, S. Advances in Our Understanding of the Molecular Mechanisms of Action of Cisplatin in Cancer Therapy. J. Exp. Pharmacol. 2021, 13, 303–328. [Google Scholar] [CrossRef]
- Chen, S.-H.; Chang, J.-Y. New Insights into Mechanisms of Cisplatin Resistance: From Tumor Cell to Microenvironment. Int. J. Mol. Sci. 2019, 20, 4136. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; Yu, H.; Shi, X.; Sun, L.; Zhou, Q.; Zheng, D.; Shi, H.; Li, N.; Zhang, X.; Shao, G. Cisplatin Sensitivity Is Enhanced in Non-Small Cell Lung Cancer Cells by Regulating Epithelial-Mesenchymal Transition through Inhibition of Eukaryotic Translation Initiation Factor 5A2. BMC Pulm. Med. 2014, 14, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.Y.; Chung, Y.J.; Park, S.W.; Kim, J.S.; Rhyu, M.G.; Kim, H.K.; Lee, K.S. The Relationship between Cisplatin-Induced Apoptosis and P53, Bcl-2 and Bax Expression in Human Lung Cancer Cells. Korean J. Intern. Med. 1999, 14, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Ray, R.; Al Khashali, H.; Haddad, B.; Wareham, J.; Coleman, K.-L.; Alomari, D.; Ranzenberger, R.; Guthrie, J.; Heyl, D.; Evans, H.G. Regulation of Cisplatin Resistance in Lung Cancer Cells by Nicotine, BDNF, and a β-Adrenergic Receptor Blocker. Int. J. Mol. Sci. 2022, 23, 12829. [Google Scholar] [CrossRef]
- Borgo, C.; D’Amore, C.; Sarno, S.; Salvi, M.; Ruzzene, M. Protein Kinase CK2: A Potential Therapeutic Target for Diverse Human Diseases. Sig. Transduct. Target Ther. 2021, 6, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Yalak, G.; Olsen, B.R. Proteomic Database Mining Opens up Avenues Utilizing Extracellular Protein Phosphorylation for Novel Therapeutic Applications. J. Transl. Med. 2015, 13, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yalak, G.; Vogel, V. Extracellular Phosphorylation and Phosphorylated Proteins: Not Just Curiosities but Physiologically Important. Sci. Signal. 2012, 5, re7. [Google Scholar] [CrossRef]
- Ruzzene, M.; Penzo, D.; Pinna, L.A. Protein Kinase CK2 Inhibitor 4,5,6,7-Tetrabromobenzotriazole (TBB) Induces Apoptosis and Caspase-Dependent Degradation of Haematopoietic Lineage Cell-Specific Protein 1 (HS1) in Jurkat Cells. Biochem. J. 2002, 364, 41–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez, F.; Allende, C.C.; Allende, J.E. Protein Kinase Casein Kinase 2 Holoenzyme Produced Ectopically in Human Cells Can Be Exported to the External Side of the Cellular Membrane. Proc. Natl. Acad. Sci. USA 2005, 102, 4718–4723. [Google Scholar] [CrossRef] [PubMed]
- Cobb, L.J.; Mehta, H.; Cohen, P. Enhancing the Apoptotic Potential of Insulin-Like Growth Factor-Binding Protein-3 in Prostate Cancer by Modulation of CK2 Phosphorylation. Mol. Endocrinol. 2009, 23, 1624–1633. [Google Scholar] [CrossRef] [Green Version]
- Coverley, J.A.; Martin, J.L.; Baxter, R.C. The Effect of Phosphorylation by Casein Kinase 2 on the Activity of Insulin-like Growth Factor-Binding Protein-3. Endocrinology 2000, 141, 564–570. [Google Scholar] [CrossRef]
- Montenarh, M.; Götz, C. Ecto-Protein Kinase CK2, the Neglected Form of CK2. Biomed. Rep. 2018, 8, 307–313. [Google Scholar] [CrossRef] [Green Version]
- Scaglioni, P.P.; Yung, T.M.; Cai, L.F.; Erdjument-Bromage, H.; Kaufman, A.J.; Singh, B.; Teruya-Feldstein, J.; Tempst, P.; Pandolfi, P.P. A CK2-Dependent Mechanism for Degradation of the PML Tumor Suppressor. Cell 2006, 126, 269–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva-Pavez, E.; Tapia, J.C. Protein Kinase CK2 in Cancer Energetics. Front. Oncol. 2020, 10, 893. [Google Scholar] [CrossRef] [PubMed]
- Raman, P.S.; Alves, C.S.; Wirtz, D.; Konstantopoulos, K. Distinct Kinetic and Molecular Requirements Govern CD44 Binding to Hyaluronan versus Fibrin(Ogen). Biophys. J. 2012, 103, 415–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misra, S.; Heldin, P.; Hascall, V.C.; Karamanos, N.K.; Skandalis, S.S.; Markwald, R.R.; Ghatak, S. HA/CD44 Interactions as Potential Targets for Cancer Therapy. FEBS J. 2011, 278, 1429–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misra, S.; Hascall, V.C.; Markwald, R.R.; Ghatak, S. Interactions between Hyaluronan and Its Receptors (CD44, RHAMM) Regulate the Activities of Inflammation and Cancer. Front. Immunol. 2015, 6, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherman, L.S.; Matsumoto, S.; Su, W.; Srivastava, T.; Back, S.A. Hyaluronan Synthesis, Catabolism, and Signaling in Neurodegenerative Diseases. Int. J. Cell Biol. 2015, 2015, e368584. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Tolg, C.; Turley, E. Dissecting the Dual Nature of Hyaluronan in the Tumor Microenvironment. Front. Immunol. 2019, 10, 947. [Google Scholar] [CrossRef]
- Toole, B.P. Hyaluronan: From Extracellular Glue to Pericellular Cue. Nat. Rev. Cancer 2004, 4, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Toole, B.P.; Slomiany, M.G. Hyaluronan, CD44 and Emmprin: Partners in Cancer Cell Chemoresistance. Drug Resist. Updat. 2008, 11, 110–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chanmee, T.; Ontong, P.; Kimata, K.; Itano, N. Key Roles of Hyaluronan and Its CD44 Receptor in the Stemness and Survival of Cancer Stem Cells. Front. Oncol. 2015, 5, 180. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Zhao, S.; Karnad, A.; Freeman, J.W. The Biology and Role of CD44 in Cancer Progression: Therapeutic Implications. J. Hematol. Oncol. 2018, 11, 64. [Google Scholar] [CrossRef] [Green Version]
- Kultti, A.; Li, X.; Jiang, P.; Thompson, C.B.; Frost, G.I.; Shepard, H.M. Therapeutic Targeting of Hyaluronan in the Tumor Stroma. Cancers 2012, 4, 873–903. [Google Scholar] [CrossRef] [Green Version]
- Torre, C.; Wang, S.J.; Xia, W.; Bourguignon, L.Y.W. Reduction of Hyaluronan-CD44-Mediated Growth, Migration, and Cisplatin Resistance in Head and Neck Cancer Due to Inhibition of Rho Kinase and PI-3 Kinase Signaling. Arch. Otolaryngol. Head Neck Surg. 2010, 136, 493–501. [Google Scholar] [CrossRef] [Green Version]
- Nagy, N.; Kuipers, H.F.; Frymoyer, A.R.; Ishak, H.D.; Bollyky, J.B.; Wight, T.N.; Bollyky, P.L. 4-Methylumbelliferone Treatment and Hyaluronan Inhibition as a Therapeutic Strategy in Inflammation, Autoimmunity, and Cancer. Front. Immunol. 2015, 6, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lokeshwar, V.B.; Lopez, L.E.; Munoz, D.; Chi, A.; Shirodkar, S.P.; Lokeshwar, S.D.; Escudero, D.O.; Dhir, N.; Altman, N. ANTITUMOR ACTIVITY OF HYALURONIC ACID SYNTHESIS INHIBITOR 4-METHYLUMBELLIFERONE IN PROSTATE CANCER CELLS. Cancer Res. 2010, 70, 2613–2623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yates, T.J.; Lopez, L.E.; Lokeshwar, S.D.; Ortiz, N.; Kallifatidis, G.; Jordan, A.; Hoye, K.; Altman, N.; Lokeshwar, V.B. Dietary Supplement 4-Methylumbelliferone: An Effective Chemopreventive and Therapeutic Agent for Prostate Cancer. J. Natl. Cancer Inst. 2015, 107, djv085. [Google Scholar] [CrossRef] [Green Version]
- Ban, H.; Uchakina, O.; McKallip, R.J. Hyaluronic Acid Inhibitor 4-Methylumbelliferone Activates the Intrinsic Apoptosis Pathway in K562 Chronic Myelogenous Leukemia Cells. Anticancer Res. 2015, 35, 5231–5240. [Google Scholar]
- Yoshida, E.; Kudo, D.; Nagase, H.; Suto, A.; Shimoda, H.; Suto, S.; Kakizaki, I.; Endo, M.; Hakamada, K. 4-Methylumbelliferone Decreases the Hyaluronan-Rich Extracellular Matrix and Increases the Effectiveness of 5-Fluorouracil. Anticancer Res. 2018, 38, 5799–5804. [Google Scholar] [CrossRef] [PubMed]
- Firth, S.M.; Baxter, R.C. Cellular Actions of the Insulin-like Growth Factor Binding Proteins. Endocr. Rev. 2002, 23, 824–854. [Google Scholar] [CrossRef] [PubMed]
- Baxter, R.C. IGF Binding Proteins in Cancer: Mechanistic and Clinical Insights. Nat. Rev. Cancer 2014, 14, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Allard, J.B.; Duan, C. IGF-Binding Proteins: Why Do They Exist and Why Are There So Many? Front Endocrinol (Lausanne) 2018, 9, 117–128. [Google Scholar] [CrossRef] [Green Version]
- Butt, A.J.; Williams, A.C. IGFBP-3 and Apoptosis—A Licence to Kill? Apoptosis 2001, 6, 199–205. [Google Scholar] [CrossRef]
- Forbes, B.E.; McCarthy, P.; Norton, R.S. Insulin-Like Growth Factor Binding Proteins: A Structural Perspective. Front. Endocrinol. 2012, 3, 38. [Google Scholar] [CrossRef] [Green Version]
- Jogie-Brahim, S.; Feldman, D.; Oh, Y. Unraveling Insulin-like Growth Factor Binding Protein-3 Actions in Human Disease. Endocr. Rev. 2009, 30, 417–437. [Google Scholar] [CrossRef] [Green Version]
- Martin, J.L.; Coverley, J.A.; Pattison, S.T.; Baxter, R.C. Insulin-like Growth Factor-Binding Protein-3 Production by MCF-7 Breast Cancer Cells: Stimulation by Retinoic Acid and Cyclic Adenosine Monophosphate and Differential Effects of Estradiol. Endocrinology 1995, 136, 1219–1226. [Google Scholar] [CrossRef]
- Rajah, R.; Valentinis, B.; Cohen, P. Insulin-like Growth Factor (IGF)-Binding Protein-3 Induces Apoptosis and Mediates the Effects of Transforming Growth Factor-Beta1 on Programmed Cell Death through a P53- and IGF-Independent Mechanism. J. Biol. Chem. 1997, 272, 12181–12188. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Lee, H.Y.; Weinzimer, S.A.; Powell, D.R.; Clifford, J.L.; Kurie, J.M.; Cohen, P. Direct Functional Interactions between Insulin-like Growth Factor-Binding Protein-3 and Retinoid X Receptor-Alpha Regulate Transcriptional Signaling and Apoptosis. J. Biol. Chem. 2000, 275, 33607–33613. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-Y.; Moon, H.; Chun, K.-H.; Chang, Y.-S.; Hassan, K.; Ji, L.; Lotan, R.; Khuri, F.R.; Hong, W.K. Effects of Insulin-like Growth Factor Binding Protein-3 and Farnesyltransferase Inhibitor SCH66336 on Akt Expression and Apoptosis in Non–Small-Cell Lung Cancer Cells. J. Natl. Cancer Inst. 2004, 96, 1536–1548. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.S.; Wang, L.; Liu, D.; Mao, L.; Hong, W.K.; Khuri, F.R.; Lee, H.-Y. Correlation between Insulin-like Growth Factor-Binding Protein-3 Promoter Methylation and Prognosis of Patients with Stage I Non-Small Cell Lung Cancer. Clin. Cancer Res. 2002, 8, 3669–3675. [Google Scholar] [PubMed]
- Põld, M.; Krysan, K.; Põld, A.; Dohadwala, M.; Heuze-Vourc’h, N.; Mao, J.T.; Riedl, K.L.; Sharma, S.; Dubinett, S.M. Cyclooxygenase-2 Modulates the Insulin-Like Growth Factor Axis in Non–Small-Cell Lung Cancer. Cancer Res. 2004, 64, 6549–6555. [Google Scholar] [CrossRef] [Green Version]
- Ho, G.Y.F.; Zheng, S.L.; Cushman, M.; Perez-Soler, R.; Kim, M.; Xue, X.; Wang, T.; Schlecht, N.F.; Tinker, L.; Rohan, T.E.; et al. Associations of Insulin and IGFBP-3 with Lung Cancer Susceptibility in Current Smokers. J. Natl. Cancer Inst. 2016, 108, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarthy, K.; Laban, C.; McVittie, C.J.; Ogunkolade, W.; Khalaf, S.; Bustin, S.; Carpenter, R.; Jenkins, P.J. The Expression and Function of IGFBP-3 in Normal and Malignant Breast Tissue. Anticancer Res. 2009, 29, 3785–3790. [Google Scholar]
- Marzec, K.A.; Baxter, R.C.; Martin, J.L. Targeting Insulin-Like Growth Factor Binding Protein-3 Signaling in Triple-Negative Breast Cancer. Biomed. Res. Int. 2015, 2015, 638526. [Google Scholar] [CrossRef]
- Lee, H.-Y.; Chun, K.-H.; Liu, B.; Wiehle, S.A.; Cristiano, R.J.; Hong, W.K.; Cohen, P.; Kurie, J.M. Insulin-like Growth Factor Binding Protein-3 Inhibits the Growth of Non-Small Cell Lung Cancer. Cancer Res. 2002, 62, 3530–3537. [Google Scholar]
- Wang, Y.A.; Sun, Y.; Palmer, J.; Solomides, C.; Huang, L.-C.; Shyr, Y.; Dicker, A.P.; Lu, B. IGFBP3 Modulates Lung Tumorigenesis and Cell Growth through IGF1 Signaling. Mol. Cancer Res. 2017, 15, 896–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muterspaugh, R.; Price, D.; Esckilsen, D.; McEachern, S.; Guthrie, J.; Heyl, D.; Evans, H.G. Interaction of Insulin-Like Growth Factor-Binding Protein 3 With Hyaluronan and Its Regulation by Humanin and CD44. Biochemistry 2018, 57, 5726–5737. [Google Scholar] [CrossRef]
- Ibanez de Caceres, I.; Cortes-Sempere, M.; Moratilla, C.; Machado-Pinilla, R.; Rodriguez-Fanjul, V.; Manguán-García, C.; Cejas, P.; López-Ríos, F.; Paz-Ares, L.; de CastroCarpeño, J.; et al. IGFBP-3 Hypermethylation-Derived Deficiency Mediates Cisplatin Resistance in Non-Small-Cell Lung Cancer. Oncogene 2010, 29, 1681–1690. [Google Scholar] [CrossRef] [Green Version]
- Borgo, C.; Ruzzene, M. Role of Protein Kinase CK2 in Antitumor Drug Resistance. J. Exp. Clin. Cancer Res. 2019, 38, 287. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Song, P.; Pang, J. The CK2 Inhibitor CX4945 Reverses Cisplatin Resistance in the A549/DDP Human Lung Adenocarcinoma Cell Line. Oncol. Lett. 2019, 18, 3845–3856. [Google Scholar] [CrossRef] [Green Version]
- Castello, J.; Ragnauth, A.; Friedman, E.; Rebholz, H. CK2—An Emerging Target for Neurological and Psychiatric Disorders. Pharmaceuticals (Basel) 2017, 10, 7. [Google Scholar] [CrossRef]
- Dorandish, S.; Devos, J.; Clegg, B.; Price, D.; Muterspaugh, R.; Guthrie, J.; Heyl, D.L.; Evans, H.G. Biochemical Determinants of the IGFBP-3-Hyaluronan Interaction. FEBS Open Bio. 2020, 10, 1668–1684. [Google Scholar] [CrossRef] [PubMed]
- Price, D.; Dorandish, S.; Williams, A.; Iwaniec, B.; Stephens, A.; Marshall, K.; Guthrie, J.; Heyl, D.; Evans, H.G. Humanin Blocks the Aggregation of Amyloid-β Induced by Acetylcholinesterase, an Effect Abolished in the Presence of IGFBP-3. Biochemistry 2020, 59, 1981–2002. [Google Scholar] [CrossRef]
- Price, D.; Muterspaugh, R.; Clegg, B.; Williams, A.; Stephens, A.; Guthrie, J.; Heyl, D.; Guy Evans, H. IGFBP-3 Blocks Hyaluronan-CD44 Signaling, Leading to Increased Acetylcholinesterase Levels in A549 Cell Media and Apoptosis in a P53-Dependent Manner. Sci. Rep. 2020, 10, 5083–5099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorandish, S.; Atali, S.; Ray, R.; Al Khashali, H.; Coleman, K.-L.; Guthrie, J.; Heyl, D.; Evans, H.G. Differences in the Relative Abundance of ProBDNF and Mature BDNF in A549 and H1299 Human Lung Cancer Cell Media. Int. J. Mol. Sci. 2021, 22, 7059. [Google Scholar] [CrossRef] [PubMed]
- Al Khashali, H.; Ray, R.; Coleman, K.-L.; Atali, S.; Haddad, B.; Wareham, J.; Guthrie, J.; Heyl, D.; Evans, H.G. Regulation of the Soluble Amyloid Precursor Protein α (SAPPα) Levels by Acetylcholinesterase and Brain-Derived Neurotrophic Factor in Lung Cancer Cell Media. Int. J. Mol. Sci. 2022, 23, 10746. [Google Scholar] [CrossRef]
- Atali, S.; Dorandish, S.; Devos, J.; Williams, A.; Price, D.; Taylor, J.; Guthrie, J.; Heyl, D.; Evans, H.G. Interaction of Amyloid Beta with Humanin and Acetylcholinesterase Is Modulated by ATP. FEBS Open Bio. 2020, 10, 2805–2823. [Google Scholar] [CrossRef]
- Dorandish, S.; Williams, A.; Atali, S.; Sendo, S.; Price, D.; Thompson, C.; Guthrie, J.; Heyl, D.; Evans, H.G. Regulation of Amyloid-β Levels by Matrix Metalloproteinase-2/9 (MMP2/9) in the Media of Lung Cancer Cells. Sci. Rep. 2021, 11, 9708. [Google Scholar] [CrossRef]
- Evans, H.G.; Guthrie, J.W.; Jujjavarapu, M.; Hendrickson, N.; Eitel, A.; Park, Y.; Garvey, J.; Newman, R.; Esckilsen, D.; Heyl, D.L. D-Amino Acid Analogues of the Antimicrobial Peptide CDT Exhibit Anti- Cancer Properties in A549, a Human Lung Adenocarcinoma Cell Line. Protein Pept. Lett. 2017, 24, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Morohaku, K.; Hoshino, Y.; Sasada, H.; Sato, E. Incorporation of Phosphatase Inhibitor in Culture Prompts Growth Initiation of Isolated Non-Growing Oocytes. PLoS ONE 2013, 8, e77533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zawadzka, A.M.; Schilling, B.; Cusack, M.P.; Sahu, A.K.; Drake, P.; Fisher, S.J.; Benz, C.C.; Gibson, B.W. Phosphoprotein Secretome of Tumor Cells as a Source of Candidates for Breast Cancer Biomarkers in Plasma. Mol. Cell Proteomics 2014, 13, 1034–1049. [Google Scholar] [CrossRef] [PubMed]
- Njomen, E.; Evans, H.G.; Gedara, S.H.; Heyl, D.L. Humanin Peptide Binds to Insulin-Like Growth Factor-Binding Protein 3 (IGFBP3) and Regulates Its Interaction with Importin-β. Protein Pept. Lett. 2015, 22, 869–876. [Google Scholar] [CrossRef]
- Heyl, D.L.; Iwaniec, B.; Esckilsen, D.; Price, D.; Guttikonda, P.; Cooper, J.; Lombardi, J.; Milletti, M.; Evans, H.G. Using Small Peptide Segments of Amyloid-β and Humanin to Examine Their Physical Interactions. Protein Pept. Lett. 2019, 26, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Al Khashali, H.; Wareham, J.; Ray, R.; Haddad, B.; Coleman, K.-L.; Ranzenberger, R.; McCombs, P.; Guthrie, J.; Heyl, D.; Evans, H.G. Opposing Roles of IGFBP-3 and Heparanase in Regulating A549 Lung Cancer Cell Survival. Cells 2022, 11, 3533. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Long, H.; Yang, Y.-L.; Wang, Y.; Hsieh, D.; Li, W.; Au, A.; Stoppler, H.J.; Xu, Z.; Jablons, D.M.; et al. Inhibition of CK2α Down-Regulates Notch1 Signalling in Lung Cancer Cells. J. Cell Mol. Med. 2013, 17, 854–862. [Google Scholar] [CrossRef] [PubMed]
- Hwa, V.; Oh, Y.; Rosenfeld, R.G. The Insulin-like Growth Factor-Binding Protein (IGFBP) Superfamily. Endocr. Rev. 1999, 20, 761–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikonen, M.; Liu, B.; Hashimoto, Y.; Ma, L.; Lee, K.-W.; Niikura, T.; Nishimoto, I.; Cohen, P. Interaction between the Alzheimer’s Survival Peptide Humanin and Insulin-like Growth Factor-Binding Protein 3 Regulates Cell Survival and Apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 13042–13047. [Google Scholar] [CrossRef] [Green Version]
- Fowlkes, J.L.; Serra, D.M. Characterization of Glycosaminoglycan-Binding Domains Present in Insulin-like Growth Factor-Binding Protein-3. J. Biol. Chem. 1996, 271, 14676–14679. [Google Scholar] [CrossRef] [Green Version]
- Bach, L.A.; Headey, S.J.; Norton, R.S. IGF-Binding Proteins--the Pieces Are Falling into Place. Trends Endocrinol. Metab. 2005, 16, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Wheatcroft, S.B.; Kearney, M.T. IGF-Dependent and IGF-Independent Actions of IGF-Binding Protein-1 and -2: Implications for Metabolic Homeostasis. Trends Endocrinol. Metab. 2009, 20, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Mattheolabakis, G.; Milane, L.; Singh, A.; Amiji, M.M. Hyaluronic Acid Targeting of CD44 for Cancer Therapy: From Receptor Biology to Nanomedicine. J. Drug Target. 2015, 23, 605–618. [Google Scholar] [CrossRef]
- Nikitovic, D.; Kouvidi, K.; Kavasi, R.-M.; Berdiaki, A.; Tzanakakis, G.N. Hyaluronan/Hyaladherins—A Promising Axis for Targeted Drug Delivery in Cancer. Curr. Drug Deliv. 2016, 13, 500–511. [Google Scholar] [CrossRef] [PubMed]
- Sarris, E.G.; Saif, M.W.; Syrigos, K.N. The Biological Role of PI3K Pathway in Lung Cancer. Pharmaceuticals 2012, 5, 1236–1264. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, A.; Das, K.; Lerner, N.; Sathe, S.; Cicek, M.; Casey, G.; Sizemore, N. The AKT/I Kappa B Kinase Pathway Promotes Angiogenic/Metastatic Gene Expression in Colorectal Cancer by Activating Nuclear Factor-Kappa B and Beta-Catenin. Oncogene 2005, 24, 1021–1031. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Zhang, Q.; Peng, X.; Zhou, C.; Zhong, Y.; Chen, X.; Qiu, Y.; Jin, M.; Gong, M.; Kong, D. Stellettin B Induces G1 Arrest, Apoptosis and Autophagy in Human Non-Small Cell Lung Cancer A549 Cells via Blocking PI3K/Akt/MTOR Pathway. Sci. Rep. 2016, 6, 27071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, D.; Ueno, L.; Vogt, P.K. Akt-Mediated Regulation of NFκB and the Essentialness of NFκB for the Oncogenicity of PI3K and Akt. Int. J. Cancer 2009, 125, 2863–2870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozes, O.N.; Mayo, L.D.; Gustin, J.A.; Pfeffer, S.R.; Pfeffer, L.M.; Donner, D.B. NF-KappaB Activation by Tumour Necrosis Factor Requires the Akt Serine-Threonine Kinase. Nature 1999, 401, 82–85. [Google Scholar] [CrossRef]
- Chou, Y.-E.; Hsieh, M.-J.; Chiou, H.-L.; Lee, H.-L.; Yang, S.-F.; Chen, T.-Y. CD44 Gene Polymorphisms on Hepatocellular Carcinoma Susceptibility and Clinicopathologic Features. Biomed. Res. Int. 2014, 2014, 231474. [Google Scholar] [CrossRef] [Green Version]
- Kundu, B.; Saha, P.; Datta, K.; Kundu, S.C. A Silk Fibroin Based Hepatocarcinoma Model and the Assessment of the Drug Response in Hyaluronan-Binding Protein 1 Overexpressed HepG2 Cells. Biomaterials 2013, 34, 9462–9474. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Getz, G.; Wheeler, D.A.; Mardis, E.R.; McLellan, M.D.; Cibulskis, K.; Sougnez, C.; Greulich, H.; Muzny, D.M.; Morgan, M.B.; et al. Somatic Mutations Affect Key Pathways in Lung Adenocarcinoma. Nature 2008, 455, 1069–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Han, C.Y.; Duan, F.G.; Fan, X.-X.; Yao, X.-J.; Parks, R.J.; Tang, Y.-J.; Wang, M.-F.; Liu, L.; Tsang, B.K.; et al. P53 Sensitizes Chemoresistant Non-Small Cell Lung Cancer via Elevation of Reactive Oxygen Species and Suppression of EGFR/PI3K/AKT Signaling. Cancer Cell Int. 2019, 19, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Leroy, B.; Girard, L.; Hollestelle, A.; Minna, J.D.; Gazdar, A.F.; Soussi, T. Analysis of TP53 Mutation Status in Human Cancer Cell Lines: A Reassessment. Hum. Mutat. 2014, 35, 756–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How Does P53 Induce Apoptosis and How Does This Relate to P53-Mediated Tumour Suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [Green Version]
- Levine, A.J.; Oren, M. The First 30 Years of P53: Growing Ever More Complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef] [Green Version]
- Webster, G.A.; Perkins, N.D. Transcriptional Cross Talk between NF-ΚB and P53. Mol. Cell Biol. 1999, 19, 3485–3495. [Google Scholar] [CrossRef] [Green Version]
- Meylan, E.; Dooley, A.L.; Feldser, D.M.; Shen, L.; Turk, E.; Ouyang, C.; Jacks, T. Requirement for NF-KappaB Signalling in a Mouse Model of Lung Adenocarcinoma. Nature 2009, 462, 104–107. [Google Scholar] [CrossRef] [Green Version]
- Pavlakis, E.; Stiewe, T. P53′s Extended Reach: The Mutant P53 Secretome. Biomolecules 2020, 10, 307. [Google Scholar] [CrossRef] [Green Version]
- Cortés-Sempere, M.; de Miguel, M.P.; Pernía, O.; Rodriguez, C.; de Castro Carpeño, J.; Nistal, M.; Conde, E.; López-Ríos, F.; Belda-Iniesta, C.; Perona, R.; et al. IGFBP-3 Methylation-Derived Deficiency Mediates the Resistance to Cisplatin through the Activation of the IGFIR/Akt Pathway in Non-Small Cell Lung Cancer. Oncogene 2013, 32, 1274–1283. [Google Scholar] [CrossRef] [Green Version]
- Lesley, J.; English, N.M.; Gál, I.; Mikecz, K.; Day, A.J.; Hyman, R. Hyaluronan Binding Properties of a CD44 Chimera Containing the Link Module of TSG-6. J. Biol. Chem. 2002, 277, 26600–26608. [Google Scholar] [CrossRef] [Green Version]
- Bajorath, J.; Greenfield, B.; Munro, S.B.; Day, A.J.; Aruffo, A. Identification of CD44 Residues Important for Hyaluronan Binding and Delineation of the Binding Site. J. Biol. Chem. 1998, 273, 338–343. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Alonso, M. del C.; Díaz, D.; Berbis, M.Á.; Marcelo, F.; Cañada, J.; Jiménez-Barbero, J. Protein-Carbohydrate Interactions Studied by NMR: From Molecular Recognition to Drug Design. Curr. Protein Pept. Sci. 2012, 13, 816–830. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Narula, N.; Azzopardi, S.; Smith, R.S.; Nasar, A.; Altorki, N.K.; Mittal, V.; Somwar, R.; Stiles, B.M.; Du, Y.-C.N. Expression of the Receptor for Hyaluronic Acid Mediated Motility (RHAMM) Is Associated with Poor Prognosis and Metastasis in Non-Small Cell Lung Carcinoma. Oncotarget 2016, 7, 39957–39969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madsen, K.L.; Gerke, O.; Høilund-Carlsen, P.F.; Olsen, B.B. Cisplatin-Resistant CD44+ Lung Cancer Cells Are Sensitive to Auger Electrons. Int. J. Mol. Sci. 2022, 23, 7131. [Google Scholar] [CrossRef]
- Leung, E.L.-H.; Fiscus, R.R.; Tung, J.W.; Tin, V.P.-C.; Cheng, L.C.; Sihoe, A.D.-L.; Fink, L.M.; Ma, Y.; Wong, M.P. Non-Small Cell Lung Cancer Cells Expressing CD44 Are Enriched for Stem Cell-like Properties. PLoS ONE 2010, 5, e14062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.-Y.; Vadhan, A.; Chen, P.-H.; Lee, Y.-L.; Chao, C.-Y.; Cheng, K.-H.; Chang, Y.-C.; Hu, S.C.-S.; Yuan, S.-S.F. CD44 Promotes Lung Cancer Cell Metastasis through ERK-ZEB1 Signaling. Cancers 2021, 13, 4057. [Google Scholar] [CrossRef]
- Yu, H.-G.; Ai, Y.-W.; Yu, L.-L.; Zhou, X.-D.; Liu, J.; Li, J.-H.; Xu, X.-M.; Liu, S.; Chen, J.; Liu, F.; et al. Phosphoinositide 3-Kinase/Akt Pathway Plays an Important Role in Chemoresistance of Gastric Cancer Cells against Etoposide and Doxorubicin Induced Cell Death. Int. J. Cancer 2008, 122, 433–443. [Google Scholar] [CrossRef]
- Briassouli, P.; Chan, F.; Savage, K.; Reis-Filho, J.S.; Linardopoulos, S. Aurora-A Regulation of Nuclear Factor-KappaB Signaling by Phosphorylation of IkappaBalpha. Cancer Res. 2007, 67, 1689–1695. [Google Scholar] [CrossRef] [Green Version]
- Heavey, S.; Godwin, P.; Baird, A.-M.; Barr, M.P.; Umezawa, K.; Cuffe, S.; Finn, S.P.; O’Byrne, K.J.; Gately, K. Strategic Targeting of the PI3K-NFκB Axis in Cisplatin-Resistant NSCLC. Cancer Biol. Ther. 2014, 15, 1367–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sancho-Martínez, S.M.; Piedrafita, F.J.; Cannata-Andía, J.B.; López-Novoa, J.M.; López-Hernández, F.J. Necrotic Concentrations of Cisplatin Activate the Apoptotic Machinery but Inhibit Effector Caspases and Interfere with the Execution of Apoptosis. Toxicol. Sci. 2011, 122, 73–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murnyák, B.; Hortobágyi, T. Immunohistochemical Correlates of TP53 Somatic Mutations in Cancer. Oncotarget 2016, 7, 64910–64920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant P53 as a Guardian of the Cancer Cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef]
- Freed-Pastor, W.A.; Prives, C. Mutant P53: One Name, Many Proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.; Zhou, Y.; Huang, L.; Ou, W.; Wu, J.; Li, S.; Xu, J.; Feng, J.; Liu, B. TP53 Mutation Is Associated with a Poor Clinical Outcome for Non-Small Cell Lung Cancer: Evidence from a Meta-Analysis. Mol. Clin. Oncol. 2016, 5, 705–713. [Google Scholar] [CrossRef] [Green Version]
- Saleh, M.M.; Scheffler, M.; Merkelbach-Bruse, S.; Scheel, A.H.; Ulmer, B.; Wolf, J.; Buettner, R. Comprehensive Analysis of TP53 and KEAP1 Mutations and Their Impact on Survival in Localized- and Advanced-Stage NSCLC. J. Thorac. Oncol. 2022, 17, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Guntur, V.P.; Waldrep, J.C.; Guo, J.J.; Selting, K.; Dhand, R. Increasing P53 Protein Sensitizes Non-Small Cell Lung Cancer to Paclitaxel and Cisplatin in Vitro. Anticancer Res. 2010, 30, 3557–3564. [Google Scholar]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coleman, K.-l.; Chiaramonti, M.; Haddad, B.; Ranzenberger, R.; Henning, H.; Al Khashali, H.; Ray, R.; Darweesh, B.; Guthrie, J.; Heyl, D.; et al. Phosphorylation of IGFBP-3 by Casein Kinase 2 Blocks Its Interaction with Hyaluronan, Enabling HA-CD44 Signaling Leading to Increased NSCLC Cell Survival and Cisplatin Resistance. Cells 2023, 12, 405. https://doi.org/10.3390/cells12030405
Coleman K-l, Chiaramonti M, Haddad B, Ranzenberger R, Henning H, Al Khashali H, Ray R, Darweesh B, Guthrie J, Heyl D, et al. Phosphorylation of IGFBP-3 by Casein Kinase 2 Blocks Its Interaction with Hyaluronan, Enabling HA-CD44 Signaling Leading to Increased NSCLC Cell Survival and Cisplatin Resistance. Cells. 2023; 12(3):405. https://doi.org/10.3390/cells12030405
Chicago/Turabian StyleColeman, Kai-ling, Michael Chiaramonti, Ben Haddad, Robert Ranzenberger, Heather Henning, Hind Al Khashali, Ravel Ray, Ban Darweesh, Jeffrey Guthrie, Deborah Heyl, and et al. 2023. "Phosphorylation of IGFBP-3 by Casein Kinase 2 Blocks Its Interaction with Hyaluronan, Enabling HA-CD44 Signaling Leading to Increased NSCLC Cell Survival and Cisplatin Resistance" Cells 12, no. 3: 405. https://doi.org/10.3390/cells12030405