
The Landscape of Secondary Genetic Rearrangements in Pediatric Patients with B-Cell Acute Lymphoblastic Leukemia with t(12;21)

 ,
,
Abstract
:1. Introduction
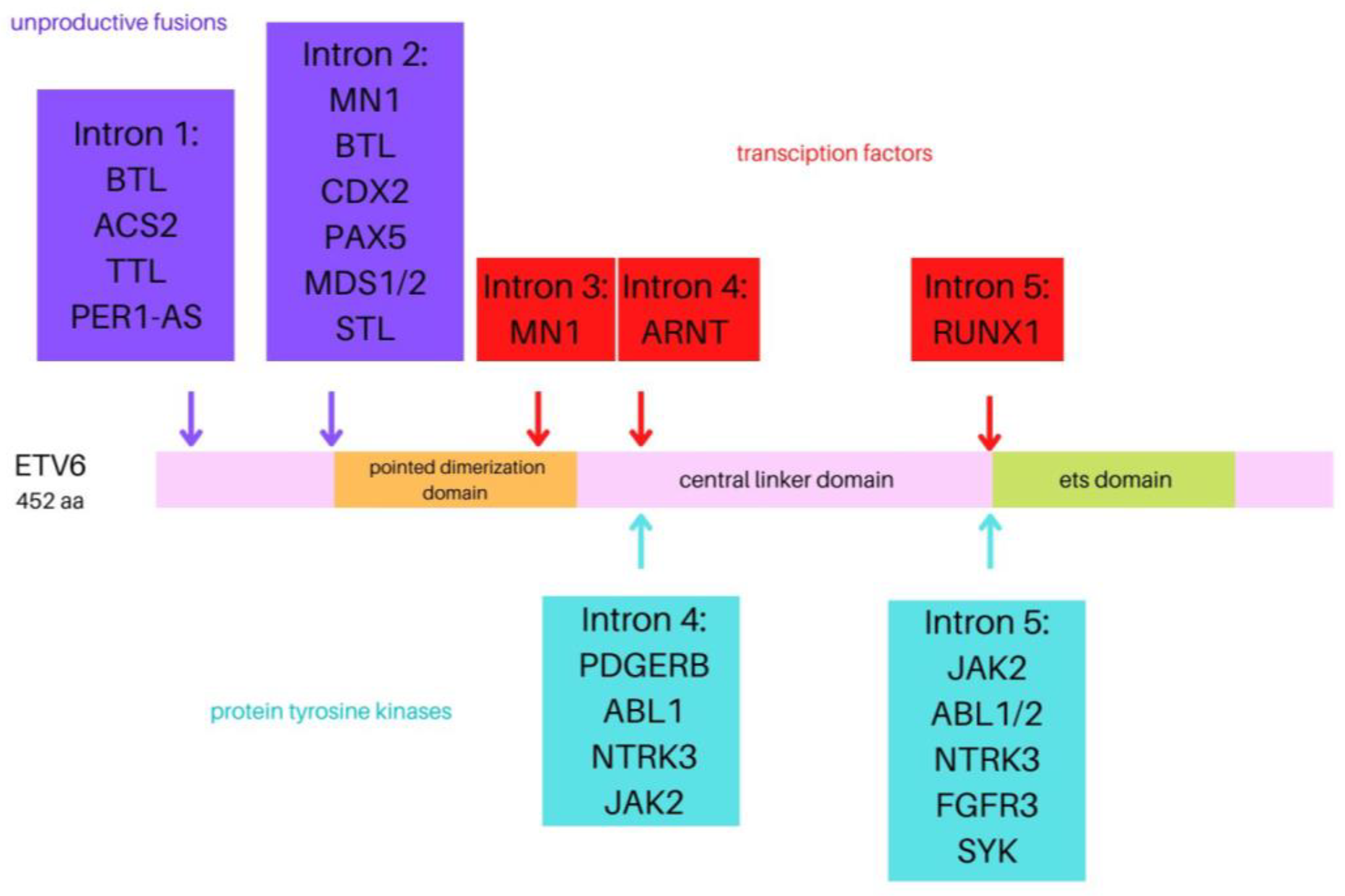
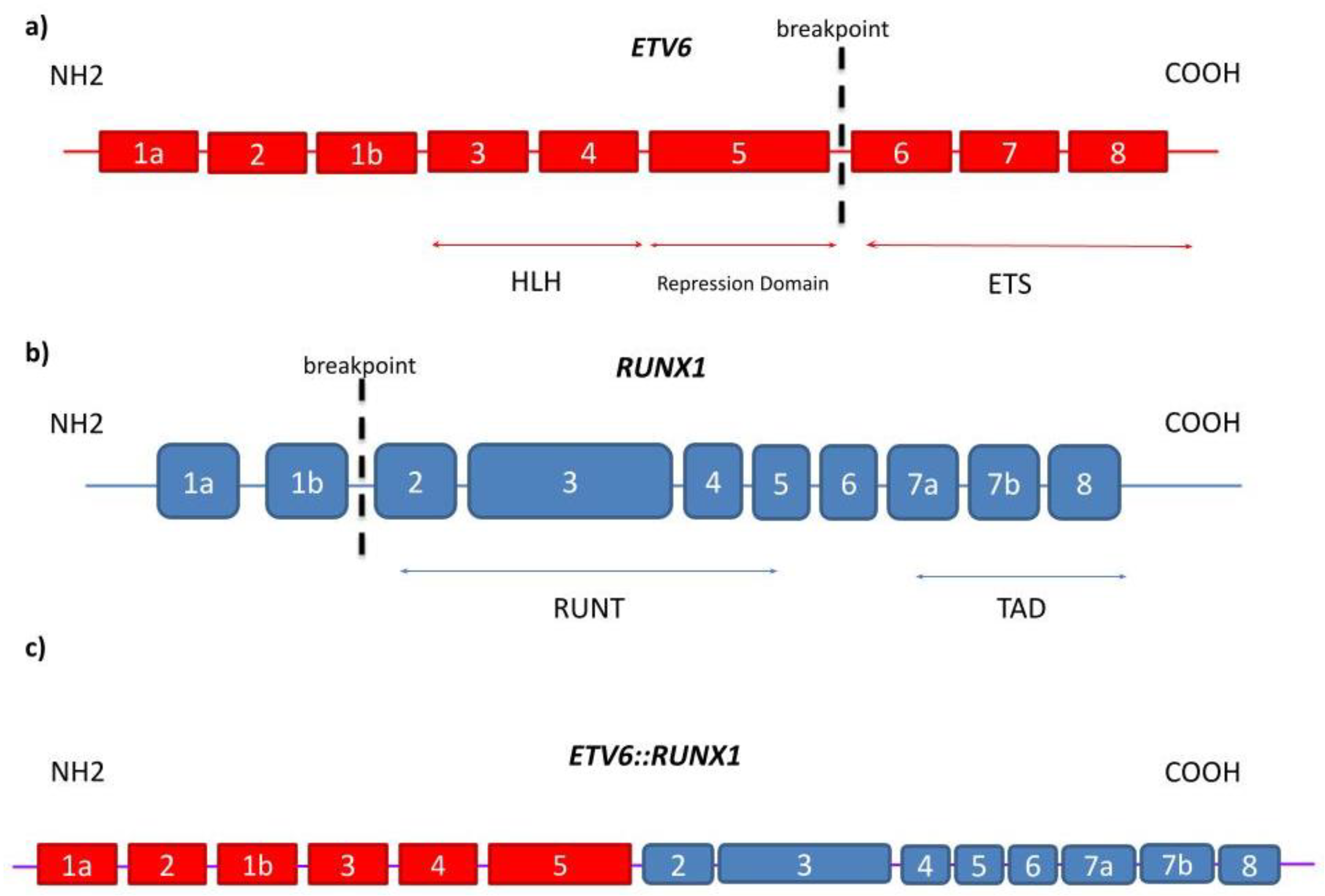
1.1. ETV6
1.2. RUNX1
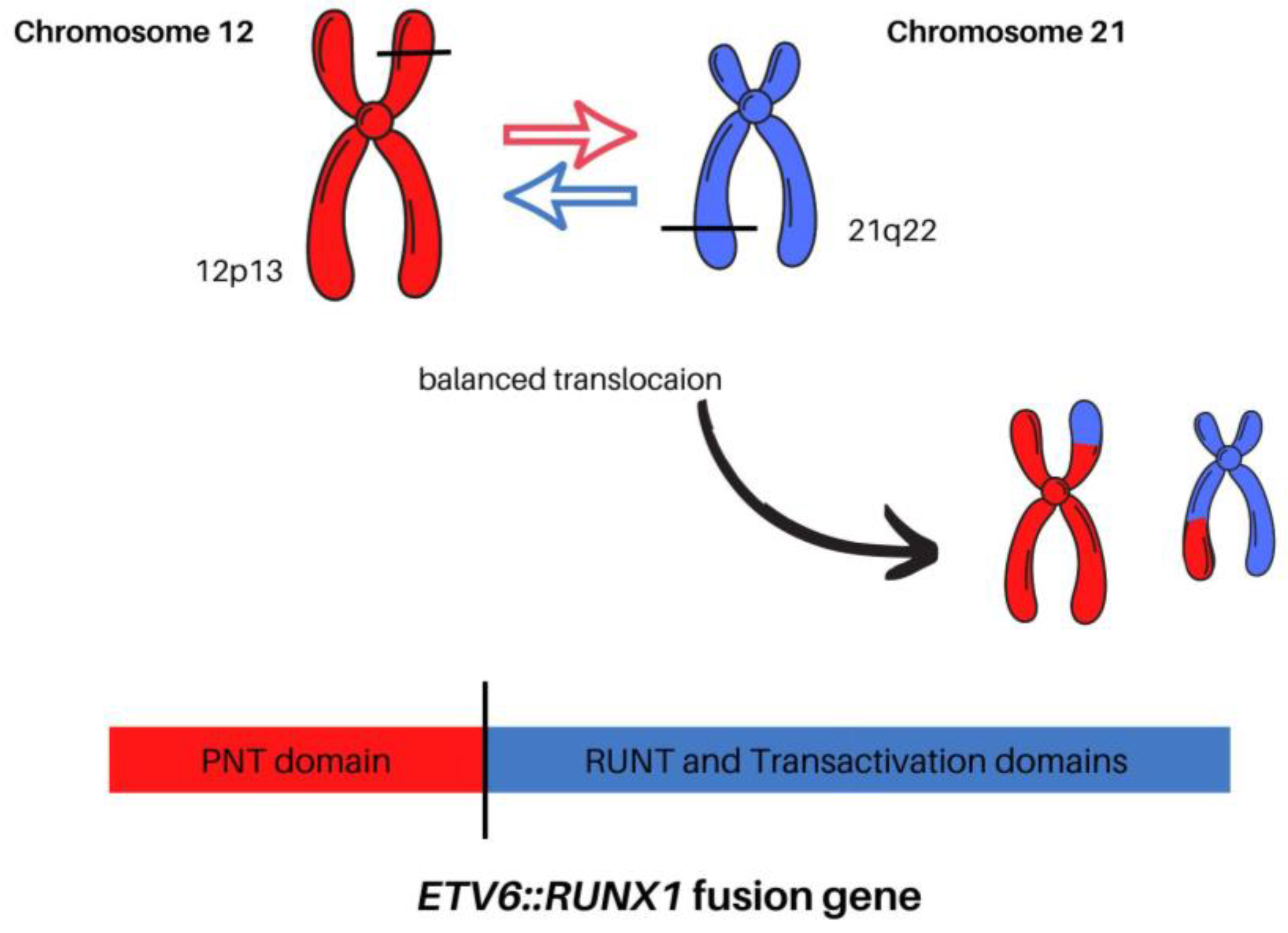
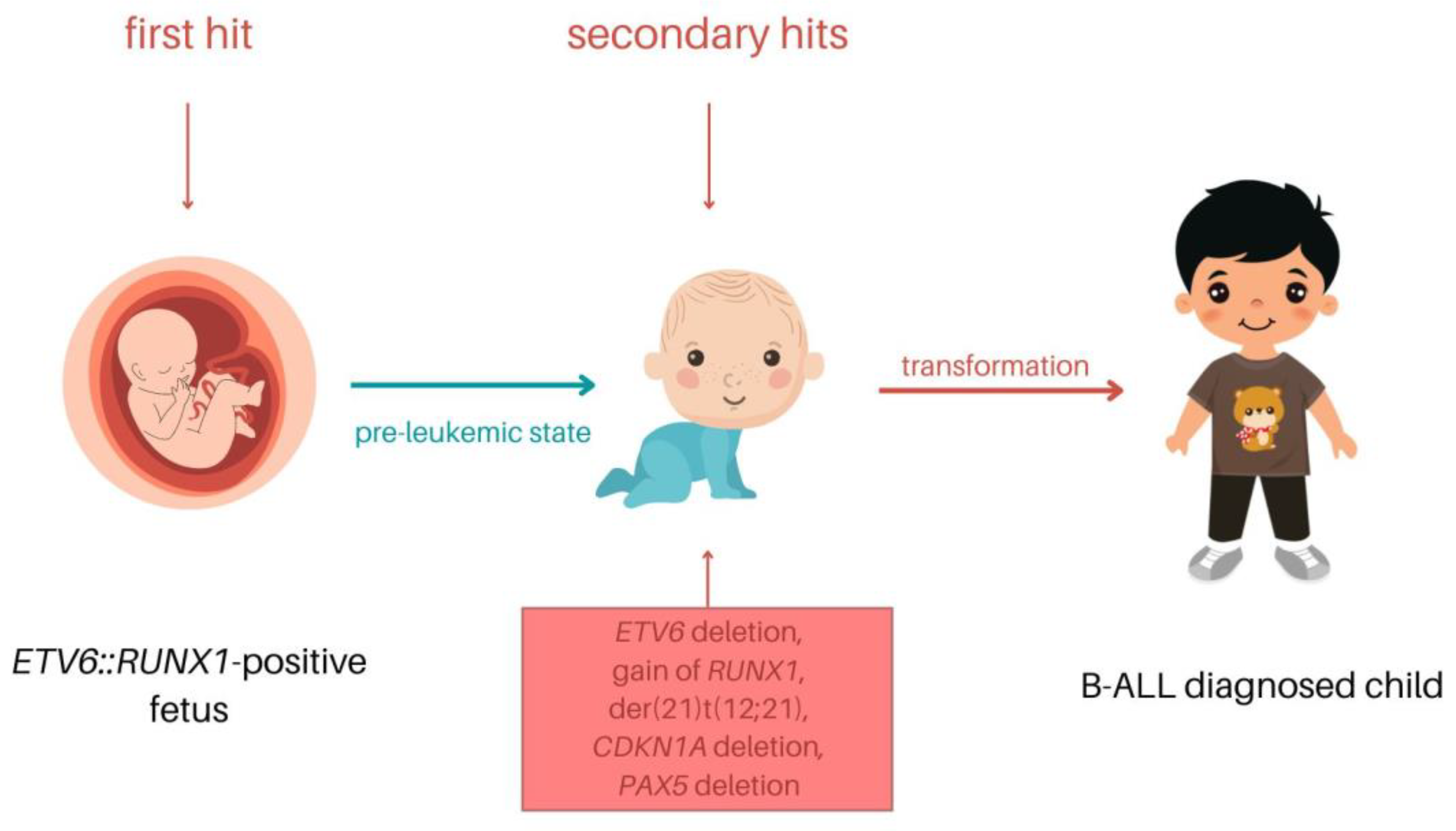
1.3. Detection, Mechanism, and Clinical Significance of t(12;21)
2. Secondary Genetic Rearrangements
2.1. Chromosome 12
2.2. Chromosome 6
2.3. Chromosome 21
2.4. Chromosome 9
2.5. Chromosome 5
2.6. Chromosome 7
2.7. Chromosome 11
3. ETV6::RUNX1+ Prognosis and Treatment
4. ETV6::RUNX1+ Future Treatment Perspective
5. ETV6::RUNX1-like
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AIEOP | AssociazioneItaliana di Ematologia e OncologiaPediatrica |
| AIM1 | Absent in melanoma 1 protein |
| ALL | Acute lymphoblastic leukemia |
| AML | Acute myeloid leukemia |
| ANGPTL2 | Angiopoietin-like 2 |
| ARPP21 | CAMP-regulated phosphoprotein 21 |
| B-ALL | B-cell acute lymphoblastic leukemia |
| BCR::ABL1 | BCR and ABL fusion gene |
| BCR::ABL1-like ALL | BCR and ABL fusion gene-like acute lymphoblastic leukemia |
| BFM | Berlin–Frankfurt–Münster |
| BIRC7 | Baculoviral IAP repeat containing 7 |
| BM | Bone marrow |
| BMF | Bcl2 modifying factor |
| BTG1 | BTG-anti-proliferation factor 1 |
| CD95 | Cluster of differentiation 95 |
| CDKN1A | Cyclin-dependent kinase inhibitor 1A |
| CDKN1B | Cyclin-dependent kinase inhibitor 1B |
| CLIC5 | Chloride intracellular channel 5 |
| CNAs | Copy number alteration |
| CNS1 | CNS1 = patients with white blood cell count in cerebrospinal fluid < 5 and having no blasts in the cerebrospinal fluid |
| DLBCL | Diffuse large B-cell lymphoma |
| EBF1 | EBF transcription factor 1 |
| EFS | Event-free survival |
| ETS | Erythroblast transformation specific |
| ETV6 | ETS variant transcription factor 6 |
| ETV6::RUNX1 | ETS variant transcription factor 6 and RUNX family transcription factor 1 fusion gene |
| ETV6::RUNX1-like ALL | ETS variant transcription factor 6 and RUNX family transcription factor 1 fusion gene-like acute lymphoblastic leukemia |
| ETV6::RUNX1+ | ETV6::RUNX1-positive |
| ETV6::RUNX1− | ETV6::RUNX1-negative |
| FISH | Fluorescence in situ hybridization (FISH) |
| FLT3 | Fms-related receptor tyrosine kinase 3 |
| FOXO3A | Forkheadbox O3 |
| FPD/AML | familial platelet disorder with predisposition to acute myeloid leukemia (FPD/AML) |
| FRALLE 93 | French group for childhood ALL 93 trial (FRALLE 93 |
| GD-ALL-2008 | Guangdong Children’s Leukemia Group-ALL-2008 |
| GDP | Guanosine diphosphate |
| GIPFEL | Genomic inverse PCR for exploration of ligated breakpoints |
| GTP | Bound and inactive guanosine diphosphate |
| HLH | Helix loop helix |
| HRAS | HRasproto-oncogene, GTPase |
| HSCs | Hematopoietic stem cells |
| iAMP21 | Intrachromosomal amplification of chromosome 21 |
| IGF2BP | Insulin-like growth factor 2 mRNA-binding protein |
| IGF2BP1 | Insulin-like growth factor 2 MRNA-Binding Protein 1 |
| IGF2BP2 | Insulin-like growth factor 2 MRNA-Binding Protein 2 |
| IGF2BP3 | Insulin-like growth factor 2 MRNA-Binding Protein 3 |
| IGH::IL3 | Immunoglobulin heavy locus and interleukin 3 fusion gene |
| IL7R | Interleukin 7 receptor |
| KMT2A | lysine methyltransferase 2A |
| KRAS | KRAS proto-oncogene, GTPase |
| L-ASP | L-asparaginase |
| MDS | Myelodysplastic syndrome |
| MLL-r | mixed-lineage leukemia gene rearrangement |
| MLPA | Multiplex ligation probe-dependent amplification |
| MRD | Minimal residual disease |
| NF1 | Neurofibromin 1 |
| NGS | Next-generation sequencing |
| NOPHO | Nordic Society of Pediatric Hematology and Oncology |
| NR3C1 | Nuclear receptor subfamily 3 group C member 1 |
| NRAS | NRAS proto-oncogene, GTPase |
| OS | Overall survival |
| PAX5 | Paired box 5 |
| PTPN11 | Protein tyrosine phosphatase non-receptor type 11 |
| PTPN11 | Protein tyrosine phosphatase non-receptor type 11 |
| RAGs | Recombination-activating genes |
| RAS | Protein family include |
| RHD | DNA-binding Runt-domain |
| RT-PCR | Reverse transcriptase-polymerase chain reaction |
| RUNX1 | RUNX family transcription factor 1 |
| SPI-B | Spi-B transcription factor |
| SYK | Spleen tyrosine kinase |
| T-ALL | T-cell acute lymphoblastic leukemia |
| TCF3::HLF | Transcription factor 3 and HLF transcription factor fusion gene |
| TCF3::PBX1 | Transcription factor 3 and PBX homeobox 1 |
| WBC | White blood cell |
| WBP1L | WW domain-binding protein 1-like |
| WGS/WES | Whole genome/whole exome sequencing |
| WHO-HAEM5 | 5th edition of the World Health Organization Classification of HaematolymphoidTumours |
| ZFP423 | Zinc finger protein 423 |
References
- Ward, E.; DeSantis, C.; Robbins, A.; Kohler, B.; Jemal, A. Childhood and adolescent cancer statistics, 2014. CA: A Cancer J. Clin. 2014, 64, 83–103. [Google Scholar] [CrossRef]
- Lejman, M.; Chałupnik, A.; Chilimoniuk, Z.; Dobosz, M. Genetic Biomarkers and Their Clinical Implications in B-Cell Acute Lymphoblastic Leukemia in Children. Int. J. Mol. Sci. 2022, 23, 2755. [Google Scholar] [CrossRef]
- Inaba, H.; Pui, C.-H. Advances in the Diagnosis and Treatment of Pediatric Acute Lymphoblastic Leukemia. J. Clin. Med. 2021, 10, 1926. [Google Scholar] [CrossRef]
- Jeha, S.; Pei, D.; Choi, J.; Cheng, C.; Sandlund, J.T.; Coustan-Smith, E.; Campana, D.; Inaba, H.; Rubnitz, J.E.; Ribeiro, R.C.; et al. Improved CNS Control of Childhood Acute Lymphoblastic Leukemia Without Cranial Irradiation: St Jude Total Therapy Study 16. J. Clin. Oncol. 2019, 37, 3377–3391. [Google Scholar] [CrossRef]
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.D.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th edition of the World Health Organization Classification of HaematolymphoidTumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [CrossRef]
- Duffield, A.S.; Mullighan, C.G.; Borowitz, M.J. International Consensus Classification of acute lymphoblastic leukemia/lymphoma. Virchows Arch. 2022, 1. Available online: https://link.springer.com/article/10.1007/s00428-022-03448-8#citeas (accessed on 24 November 2022). [CrossRef]
- Szczepański, T.; Harrison, C.J.; van Dongen, J.J. Genetic aberrations in paediatric acute leukaemias and implications for management of patients. Lancet Oncol. 2010, 11, 880–889. [Google Scholar] [CrossRef]
- Tran, T.H.; Tasian, S.K. Has Ph-like ALL Superseded Ph+ ALL as the Least Favorable Subtype? BestPract. Res. Clin. Haematol. 2021, 34, 101331. [Google Scholar] [CrossRef]
- Forestier, E.; Heyman, M.; Andersen, M.K.; Autio, K.; Blennow, E.; Borgström, G.; Golovleva, I.; Heim, S.; Heinonen, K.; Hovland, R.; et al. Outcome of ETV6/RUNX1-positive childhood acute lymphoblastic leukaemia in the NOPHO-ALL-1992 protocol: Frequent late relapses but good overall survival. Br. J. Haematol. 2008, 140, 665–672. [Google Scholar] [CrossRef]
- ETV6 ETS Variant Transcription Factor 6 [Homo Sapiens (Human)]—Gene—NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene?Db=gene&Cmd=DetailsSearch&Term=2120 (accessed on 14 January 2023).
- Bohlander, S.K. ETV6: A versatile player in leukemogenesis. Semin. Cancer Biol. 2005, 15, 162–174. [Google Scholar] [CrossRef]
- Fisher, M.H.; Kirkpatrick, G.D.; Stevens, B.; Jones, C.; Callaghan, M.; Rajpurkar, M.; Fulbright, J.; Cooper, M.A.; Rowley, J.; Porter, C.C.; et al. ETV6 Germline Mutations Cause HDAC3/NCOR2 Mislocalization and Upregulation of Interferon Response Genes. JCI Insight 2020, 5, e140332. [Google Scholar] [CrossRef] [PubMed]
- Neveu, B.; Richer, C.; Cassart, P.; Caron, M.; Jimenez-Cortes, C.; St-Onge, P.; Fuchs, C.; Garnier, N.; Gobeil, S.; Sinnett, D. Identification of new ETV6 modulators through a high-throughput functional screening. Iscience 2022, 25, 103858. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Rispoli, R.; Patient, R.; Ciau-Uitz, A.; Porcher, C. Etv6 activates vegfa expression through positive and negative transcriptional regulatory networks in Xenopus embryos. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Wely, K.H.M.; Meester-Smoor, M.A.; Janssen, M.J.F.W.; Aarnoudse, A.-J.; Grosveld, G.C.; Zwarthoff, E.C. The MN1-TEL myeloid leukemia-associated fusion protein has a dominant-negative effect on RAR-RXR-mediated transcription. Oncogene 2007, 26, 5733–5740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cools, J.; Bilhou-Nabera, C.; Wlodarska, I.; Cabrol, C.; Talmant, P.; Bernard, P.; Hagemeijer, A.; Marynen, P. Fusion of a Novel Gene, BTL, to ETV6 in Acute Myeloid Leukemias with a t(4;12)(Q11-Q12;P13). Blood 1999, 94, 1820–1824. [Google Scholar] [CrossRef]
- Fazio, G.; Palmi, C.; Rolink, A.; Biondi, A.; Cazzaniga, G. PAX5/TEL Acts as a Transcriptional Repressor Causing down-Modulation of CD19, Enhances Migration to CXCL12, and Confers Survival Advantage in Pre-BI Cells. Cancer Res. 2008, 68, 181–189. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; E Churpek, J.; Keel, S.B.; Walsh, T.; Lee, M.K.; Loeb, K.R.; Gulsuner, S.; Pritchard, C.C.; Sanchez-Bonilla, M.; Delrow, J.J.; et al. Germline ETV6 mutations in familial thrombocytopenia and hematologic malignancy. Nat. Genet. 2015, 47, 180–185. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Gu, D.; Du, M.; Xu, Z.; Zhang, S.; Zhu, L.; Lu, J.; Zhang, R.; Xing, J.; Miao, X.; et al. Common genetic variation in ETV6 is associated with colorectal cancer susceptibility. Nat. Commun. 2016, 7, 11478. [Google Scholar] [CrossRef] [Green Version]
- Noetzli, L.; Lo, R.W.; Lee-Sherick, A.B.; Callaghan, M.; Noris, P.; Savoia, A.; Rajpurkar, M.; Jones, K.; Gowan, K.; Balduini, C.; et al. Germline Mutations in ETV6 Are Associated with Thrombocytopenia, Red Cell Macrocytosis and Predisposition to Lymphoblastic Leukemia. Nat. Genet. 2015, 47, 535–538. [Google Scholar] [CrossRef] [Green Version]
- Di Paola, J.; Porter, C.C. ETV6-related thrombocytopenia and leukemia predisposition. Blood 2019, 134, 663–667. [Google Scholar] [CrossRef]
- Melazzini, F.; Palombo, F.; Balduini, A.; De Rocco, D.; Marconi, C.; Noris, P.; Gnan, C.; Pippucci, T.; Bozzi, V.; Faleschini, M.; et al. Clinical and pathogenic features of ETV6 -related thrombocytopenia with predisposition to acute lymphoblastic leukemia. Haematologica 2016, 101, 1333–1342. [Google Scholar] [CrossRef] [Green Version]
- RUNX1 RUNX Family Transcription Factor 1 [Homo Sapiens (Human)]—Gene—NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene?Db=gene&Cmd=DetailsSearch&Term=861 (accessed on 14 January 2023).
- Lie-A-Ling, M.; Mevel, R.; Patel, R.; Blyth, K.; Baena, E.; Kouskoff, V.; Lacaud, G. RUNX1 Dosage in Development and Cancer. Mol. Cells 2020, 43, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Hass, M.R.; Brissette, D.; Parameswaran, S.; Pujato, M.; Donmez, O.; Kottyan, L.C.; Weirauch, M.T.; Kopan, R. Runx1 shapes the chromatin landscape via a cascade of direct and indirect targets. PLoS Genet. 2021, 17, e1009574. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, V.; Kern, W.; Harbich, S.; Alpermann, T.; Jeromin, S.; Schnittger, S.; Haferlach, C.; Haferlach, T.; Kohlmann, A. Prognostic relevance of RUNX1 mutations in T-cell acute lymphoblastic leukemia. Haematologica 2011, 96, 1874–1877. [Google Scholar] [CrossRef] [Green Version]
- Silva, M.C.D.A.; Krepischi, A.C.V.; Kulikowski, L.D.; Zanardo, É.A.; Nardinelli, L.; Leal, A.M.; Costa, S.S.; Muto, N.H.; Rocha, V.; Velloso, E.D.R.P. Deletion of RUNX1 exons 1 and 2 associated with familial platelet disorder with propensity to acute myeloid leukemia. Cancer Genet. 2018, 222–223, 32–37. [Google Scholar] [CrossRef]
- Latger-Cannard, V.; Philippe, C.; Bouquet, A.; Baccini, V.; Alessi, M.-C.; Ankri, A.; Bauters, A.; Bayart, S.; Cornillet-Lefebvre, P.; Daliphard, S.; et al. Haematological spectrum and genotype-phenotype correlations in nine unrelated families with RUNX1 mutations from the French network on inherited platelet disorders. Orphanet J. Rare Dis. 2016, 11, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seitz, V.; Kleo, K.; Dröge, A.; Schaper, S.; Elezkurtaj, S.; Bedjaoui, N.; Dimitrova, L.; Sommerfeld, A.; Berg, E.; von der Wall, E.; et al. Evidence for a role of RUNX1 as recombinase cofactor for TCRβ rearrangements and pathological deletions in ETV6-RUNX1 ALL. Sci. Rep. 2020, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Qiu, K.Y.; Xu, H.G.; Luo, X.Q.; Mai, H.R.; Liao, N.; Yang, L.H.; Zheng, M.C.; Wan, W.Q.; Wu, X.D.; Liu, R.Y.; et al. Prognostic Value and Outcome for ETV6/RUNX1-Positive Pediatric Acute Lymphoblastic Leukemia: A Report From the South China Children’s Leukemia Group. Front. Oncol. 2021, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Kim, S.; Jang, P.-S.; Chung, N.-G.; Cho, B. Differing Outcomes of Patients with High Hyperdiploidy and ETV6-RUNX1 Rearrangement in Korean Pediatric Precursor B Cell Acute Lymphoblastic Leukemia. Cancer Res. Treat. 2021, 53, 567–575. [Google Scholar] [CrossRef]
- Jarosova, M.; Volejnikova, J.; Porizkova, I.; Holzerova, M.; Pospisilova, D.; Novak, Z.; Vrbkova, J.; Mihal, V. Chromosomal aberrations in childhood acute lymphoblastic leukemia: 15-year single center experience. Cancer Genet. 2016, 209, 340–347. [Google Scholar] [CrossRef]
- Ampatzidou, Μ.; Florentin, L.; Papadakis, V.; Paterakis, G.; Tzanoudaki, M.; Bouzarelou, D.; Papadhimitriou, S.I.; Polychronopoulou, S. Copy Number Alteration Profile Provides Additional Prognostic Value for Acute Lymphoblastic Leukemia Patients Treated on BFM Protocols. Cancers 2021, 13, 3289. [Google Scholar] [CrossRef] [PubMed]
- Aydin, C.; Cetin, Z.; Manguoglu, A.E.; Tayfun, F.; Clark, O.A.; Kupesiz, A.; Akkaya, B.; Karauzum, S.B. Evaluation of ETV6/RUNX1 Fusion and Additional Abnormalities Involving ETV6 and/or RUNX1 Genes Using FISH Technique in Patients with Childhood Acute Lymphoblastic Leukemia. Indian J. Hematol. Blood Transfus 2016, 32, 154–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.-J.; Zhu, X.-H.; Yang, Y.; Wu, Y.; Lu, F.-J.; Zhai, X.-W.; Wang, H.-S. Prevalence of ETV6–RUNX1 fusion gene in children with acute lymphoblastic leukemia in China. Cancer Genet. Cytogenet. 2007, 178, 57–60. [Google Scholar] [CrossRef]
- Inamdar, N.; A Kumar, S.; Banavali, S.D.; Advani, S.; Magrath, I.; Bhatia, K. Comparative incidence of the rearrangements of TEL/AML1 and ALL1 genes in pediatric precursor B acute lymphoblastic leukemias in India. Int. J. Oncol. 1998, 13, 1319–1341. [Google Scholar] [CrossRef]
- Piette, C.; Suciu, S.; Clappier, E.; Bertrand, Y.; Drunat, S.; Girard, S.; Yakouben, K.; Plat, G.; Dastugue, N.; Mazingue, F.; et al. Differential impact of drugs on the outcome of ETV6-RUNX1 positive childhood B-cell precursor acute lymphoblastic leukaemia: Results of the EORTC CLG 58881 and 58951 trials. Leukemia 2017, 32, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Gandemer, V.; Chevret, S.; Petit, A.; Vermylen, C.; Leblanc, T.; Michel, G.; Schmitt, C.; Lejars, O.; Schneider, P.; Demeocq, F.; et al. Excellent Prognosis of Late Relapses of ETV6/RUNX1-Positive Childhood Acute Lymphoblastic Leukemia: Lessons from the FRALLE 93 Protocol. Haematologica 2012, 97, 1743–1750. [Google Scholar] [CrossRef] [Green Version]
- Conter, V.; Bartram, C.R.; Valsecchi, M.G.; Schrauder, A.; Panzer-Grümayer, R.; Möricke, A.; Aricò, M.; Zimmermann, M.; Mann, G.; De Rossi, G.; et al. Molecular response to treatment redefines all prognostic factors in children and adolescents with B-cell precursor acute lymphoblastic leukemia: Results in 3184 patients of the AIEOP-BFM ALL 2000 study. Blood 2010, 115, 3206–3214. [Google Scholar] [CrossRef] [Green Version]
- Mata-Rocha, M.; Rangel-López, A.; Jimenez-Hernandez, E.; Nuñez-Enríquez, J.C.; Morales-Castillo, B.A.; Sánchez-Escobar, N.; Sepúlveda-Robles, O.A.; Bravata-Alcántara, J.C.; Nájera-Cortés, A.S.; Pérez-Saldivar, M.L.; et al. Low Prevalence of ETV6::RUNX1 Fusion Gene in a Hispanic Population. Front. Pediatr. 2022, 10, 837656. [Google Scholar] [CrossRef]
- Romana, S.P.; Mauchauffé, M.; Le Coniat, M.; Chumakov, I.; Le Paslier, D.; Berger, R.; Bernard, A.O. The t(12;21) of Acute Lymphoblastic Leukemia Results in a Tel-AML1 Gene Fusion. Blood 1995, 85, 3662–3670. [Google Scholar]
- Vaskova, M.; Fronkova, E.; Starkova, J.; Kalina, T.; Mejstrikova, E.; Hrusak, O. CD44 and CD27 delineate B-precursor stages with different recombination status and with an uneven distribution in nonmalignant and malignant hematopoiesis. Tissue Antigens 2007, 71, 57–66. [Google Scholar] [CrossRef]
- de Zen, L.; Orfao, A.; Cazzaniga, G.; Masiero, L.; Cocito, M.G.; Spinelli, M.; Rivolta, A.; Biondi, A.; Zanesco, L.; Basso, G. Quantitative Multiparametric Immunophenotyping in Acute Lymphoblastic Leukemia: Correlation with Specific Genotype. I. ETV6/AML1 ALLs Identification. Leukemia 2000, 14, 1225–1231. [Google Scholar] [CrossRef] [PubMed]
- Blunck, C.B.; Terra-Granado, E.; Noronha, E.P.; Wajnberg, G.; Passetti, F.; Pombo-De-Oliveira, M.S.; Emerenciano, M. CD9 predicts ETV6-RUNX1 in childhood B-cell precursor acute lymphoblastic leukemia. Hematol. Transfus. Cell Ther. 2019, 41, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, H.; Ohira, M.; Shimizu, K.; Mitani, K.; Hirai, H.; Imai, T.; Yokoyama, K.; Soceda, E.; Ohkl, M. Alternative splicing and genomic structure of the AML1 gene involved in acute myeloid leukemia. Nucleic Acids Res. 1995, 23, 2762–2769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiemels, J.L.; Cazzaniga, G.; Daniotti, M.; Eden, O.B.; Addison, G.M.; Masera, G.; Saha, V.; Biondi, A.; Greaves, M.F. Prenatal origin of acute lymphoblastic leukaemia in children. Lancet 1999, 354, 1499–1503. [Google Scholar] [CrossRef]
- Golub, T.R.; Barker, G.F.; Bohlander, S.K.; Hiebert, S.W.; Ward, D.C.; Bray-Ward, P.; Morgan, E.; Raimondi, S.C.; Rowley, J.D.; Gilliland, D.G. Fusion of the TEL gene on 12p13 to the AML1 gene on 21q22 in acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. USA 1995, 92, 4917–4921. [Google Scholar] [CrossRef] [Green Version]
- Ford, A.M.; Bennett, C.A.; Price, C.M.; Bruin, M.C.A.; Van Wering, E.R.; Greaves, M. Fetal origins of the TEL-AML1 fusion gene in identical twins with leukemia. Proc. Natl. Acad. Sci.USA 1998, 95, 4584–4588. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zeng, H.M.; Zhang, L.P. ETV6/RUNX1-Positive Childhood Acute Lymphoblastic Leukemia in China: Excellent Prognosis with Improved BFM Protocol. Ital. J. Pediatr. 2018, 44, 94. [Google Scholar] [CrossRef]
- Bhojwani, D.; Pei, D.; Sandlund, J.T.; Jeha, S.; Ribeiro, R.C.; Rubnitz, J.E.; Raimondi, S.C.; A Shurtleff, S.; Onciu, M.; Cheng, C.; et al. ETV6-RUNX1-positive childhood acute lymphoblastic leukemia: Improved outcome with contemporary therapy. Leukemia 2011, 26, 265–270. [Google Scholar] [CrossRef]
- Loh, M.L.; Goldwasser, M.A.; Silverman, L.B.; Poon, W.-M.; Vattikuti, S.; Cardoso, A.; Neuberg, D.S.; Shannon, K.M.; Sallan, S.E.; Gilliland, D.G. Prospective analysis of TEL/AML1-positive patients treated on Dana-Farber Cancer Institute Consortium Protocol 95-01. Blood 2006, 107, 4508–4513. [Google Scholar] [CrossRef] [Green Version]
- Li, X.-Y.; Li, J.-Q.; Luo, X.-Q.; Wu, X.-D.; Sun, X.; Xu, H.-G.; Li, C.-G.; Liu, R.-Y.; Sun, X.-F.; Chen, H.-Q.; et al. Reduced intensity of early intensification does not increase the risk of relapse in children with standard risk acute lymphoblastic leukemia—A multi-centric clinical study of GD-2008-ALL protocol. BMC Cancer 2021, 21, 1–11. [Google Scholar] [CrossRef]
- Polak, R.; Bierings, M.B.; Van Der Leije, C.S.; Sanders, M.A.; Roovers, O.; Marchante, J.R.M.; Boer, J.M.; Cornelissen, J.J.; Pieters, R.; den Boer, M.L.; et al. Autophagy inhibition as a potential future targeted therapy for ETV6-RUNX1-driven B-cell precursor acute lymphoblastic leukemia. Haematologica 2018, 104, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Burjanivova, T.; Madzo, J.; Muzikova, K.; Meyer, C.; Schneider, B.; Votava, F.; Marschalek, R.; Stary, J.; Trka, J.; Zuna, J. Prenatal origin of childhood AML occurs less frequently than in childhood ALL. BMC Cancer 2006, 6, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuna, J.; Madzo, J.; Krejci, O.; Zemanova, Z.; Kalinova, M.; Muzikova, K.; Zapotocky, M.; Starkova, J.; Hrusak, O.; Horak, J.; et al. ETV6/RUNX1 (TEL/AML1) Is a Frequent Prenatal First Hit in Childhood Leukemia. Blood 2011, 117, 368–369. [Google Scholar] [CrossRef] [PubMed]
- Fueller, E.; Schaefer, D.; Fischer, U.; Krell, P.F.I.; Stanulla, M.; Borkhardt, A.; Slany, R.K. Genomic Inverse PCR for Exploration of Ligated Breakpoints (GIPFEL), a New Method to Detect Translocations in Leukemia. PLoS ONE 2014, 9, e104419. [Google Scholar] [CrossRef] [Green Version]
- Mori, H.; Colman, S.M.; Xiao, Z.; Ford, A.M.; Healy, L.E.; Donaldson, C.; Hows, J.M.; Navarrete, C.; Greaves, M. Chromosome translocations and covert leukemic clones are generated during normal fetal development. Proc. Natl. Acad. Sci. USA 2002, 99, 8242–8247. [Google Scholar] [CrossRef] [Green Version]
- Zakaria, Z.; Ahid, M.F.M.D.; Ismail, A.; Keoh, T.S.; Nor, N.M.; Kamaluddin, N.R.; Esa, E.; Yuen, L.K.; Rahman, E.J.A.; Osman, R. Chromosomal Aberrations in ETV6/RUNX1-Positive Childhood Acute Lymphoblastic Leukemia Using 244K Oligonucleotide Array Comparative Genomic Hybridization. Mol. Cytogenet. 2012, 5, 41. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Mullighan, C.; Harvey, R.; Wu, G.; Chen, X.; Edmonson, M.; Buetow, K.H.; Carroll, W.L.; Chen, I.-M.; Devidas, M.; et al. Key pathways are frequently mutated in high-risk childhood acute lymphoblastic leukemia: A report from the Children’s Oncology Group. Blood 2011, 118, 3080–3087. [Google Scholar] [CrossRef]
- Attarbaschi, A.; Mann, G.; Strehl, S.; König, M.; Steiner, M.; Jeitler, V.; Lion, T.; Dworzak, M.N.; Gadner, H.; Haas, O.A. Deletion of 11q23 Is a Highly Specific Nonrandom Secondary Genetic Abnormality of ETV6/RUNX1-Rearranged Childhood Acute Lymphoblastic Leukemia. Leukemia 2007, 21, 584–586. [Google Scholar] [CrossRef] [Green Version]
- Borst, L.; Wesolowska, A.; Joshi, T.; Borup, R.; Nielsen, F.C.; Andersen, M.K.; Jonsson, O.G.; Wehner, P.S.; Wesenberg, F.; Frost, B.M.; et al. Genome-Wide Analysis of Cytogenetic Aberrations in ETV6/RUNX1-Positive Childhood Acute Lymphoblastic Leukaemia. Br. J. Haematol. 2012, 157, 476–482. [Google Scholar] [CrossRef] [Green Version]
- Ampatzidou, M.; Papadhimitriou, S.I.; Paterakis, G.; Pavlidis, D.; Tsitsikas; Kostopoulos, I.V.; Papadakis, V.; Vassilopoulos, G.; Polychronopoulou, S. ETV6/RUNX1-Positive Childhood Acute Lymphoblastic Leukemia (ALL): The Spectrum of Clonal Heterogeneity and Its Impact on Prognosis. Cancer Genet. 2018, 224–225, 1–11. [Google Scholar] [CrossRef]
- Rodríguez-Hernández, G.; Casado-García, A.; Isidro-Hernández, M.; Picard, D.; Raboso-Gallego, J.; Alemán-Arteaga, S.; Orfao, A.; Blanco, O.; Riesco, S.; Prieto-Matos, P.; et al. The Second Oncogenic Hit Determines the Cell Fate of ETV6-RUNX1 Positive Leukemia. Front. Cell Dev. Biol. 2021, 9, 1834. [Google Scholar] [CrossRef] [PubMed]
- Ueno, H.; Yoshida, K.; Shiozawa, Y.; Nannya, Y.; Iijima-Yamashita, Y.; Kiyokawa, N.; Shiraishi, Y.; Chiba, K.; Tanaka, H.; Isobe, T.; et al. Landscape of driver mutations and their clinical impacts in pediatric B-cell precursor acute lymphoblastic leukemia. Blood Adv. 2020, 4, 5165–5173. [Google Scholar] [CrossRef]
- Pais, A.P.; Amare Kadam, P.S.; Raje, G.C.; Banavali, S.; Parikh, P.; Kurkure, P.; Arora, B.; Gujral, S.; Kumar, S.A.; Badrinath, Y. RUNX1 Aberrations in ETV6/RUNX1-Positive and ETV6/RUNX1-Negative Patients: Its Hemato-Pathological and Prognostic Significance in a Large Cohort (619 Cases) of ALL. Pediatr. Hematol. Oncol. 2008, 25, 582–597. [Google Scholar] [CrossRef]
- Nordlund, J.; Marincevic-Zuniga, Y.; Cavelier, L.; Raine, A.; Martin, T.; Lundmark, A.; Abrahamsson, J.; Norén-Nyström, U.; Lönnerholm, G.; Syvänen, A.-C. Refined detection and phasing of structural aberrations in pediatric acute lymphoblastic leukemia by linked-read whole-genome sequencing. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Chang, L.; Zhu, X. Pathogenesis of ETV6/RUNX1 -Positive Childhood Acute Lymphoblastic Leukemia and Mechanisms Underlying Its Relapse. Oncotarget 2017, 8, 35445–35459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bokemeyer, A.; Eckert, C.; Meyr, F.; Koerner, G.; von Stackelberg, A.; Ullmann, R.; Türkmen, S.; Henze, G.; Seeger, K. Copy Number Genome Alterations Are Associated with Treatment Response and Outcome in Relapsed Childhood ETV6/RUNX1-Positive Acute Lymphoblastic Leukemia. Haematologica 2014, 99, 706–714. [Google Scholar] [CrossRef] [Green Version]
- Forero, R.M.; Hernández, M.; Hernández- Rivas, J.M. Genetics of Acute Lymphoblastic Leukemia. In Leukemia; Guenova, M., Balatzenko, G., Eds.; IntechOpen: London, UK, 2013. [Google Scholar]
- Dun, K.A.; Vanhaeften, R.; Batt, T.J.; Riley, L.A.; Diano, G.; Williamson, J. BCR-ABL1 gene rearrangement as a subclonal change in ETV6-RUNX1–positive B-cell acute lymphoblastic leukemia. Blood Adv. 2016, 1, 132–138. [Google Scholar] [CrossRef] [Green Version]
- Raynaud, S.; Cavé, H.; Baens, M.; Bastard, C.; Cacheux, V.; Grosgeorge, J.; Guidal-Giroux, C.; Guo, C.; Vilmer, E.; Marynen, P.; et al. The 12;21 translocation involving TEL and deletion of the other TEL allele: Two frequently associated alterations found in childhood acute lymphoblastic leukemia. Blood 1996, 87, 2891–2899. [Google Scholar] [CrossRef]
- Komuro, H.; Valentine, M.B.; Rubnitz, J.E.; Saito, M.; Raimondi, S.C.; Carroll, A.J.; Yi, T.; Sherr, C.J.; Look, A.T. p27KIP1 Deletions in Childhood Acute Lymphoblastic Leukemia. Neoplasia 1999, 1, 253–261. [Google Scholar] [CrossRef] [Green Version]
- Enshaei, A.; Schwab, C.J.; Konn, Z.J.; Mitchell, C.D.; Kinsey, S.E.; Wade, R.; Vora, A.; Harrison, C.J.; Moorman, A.V. Long-term follow-up of ETV6-RUNX1 ALL reveals that NCI risk, rather than secondary genetic abnormalities, is the key risk factor. Leukemia 2013, 27, 2256–2259. [Google Scholar] [CrossRef]
- Guo, B.; Godzik, A.; Reed, J.C. Bcl-G, a Novel Pro-apoptotic Member of the Bcl-2 Family. J. Biol. Chem. 2001, 276, 2780–2785. [Google Scholar] [CrossRef] [PubMed]
- Yuniati, L.; Scheijen, B.; van der Meer, L.T.; van Leeuwen, F.N. Tumor suppressors BTG1 and BTG2: Beyond growth control. J Cell Physiol. 2019, 234, 5379–5389. [Google Scholar] [CrossRef] [Green Version]
- Waanders, E.; Scheijen, B.; van der Meer, L.T.; van Reijmersdal, S.V.; van Emst, L.; Kroeze, Y.; Sonneveld, E.; Hoogerbrugge, P.M.; van Kessel, A.G.; van Leeuwen, F.N.; et al. The origin and nature of tightly clustered BTG1 deletions in precursor B-cell acute lymphoblastic leukemia support a model of multiclonal evolution. PLoS Genet. 2012, 8, e1002533. [Google Scholar] [CrossRef]
- Jerchel, I.S.; Hoogkamer, A.Q.; Ariës, I.M.; Steeghs, E.M.P.; Boer, J.M.; Besselink, N.J.M.; Boeree, A.; Van De Ven, C.; De Groot-Kruseman, H.A.; De Haas, V.; et al. RAS pathway mutations as a predictive biomarker for treatment adaptation in pediatric B-cell precursor acute lymphoblastic leukemia. Leukemia 2018, 32, 931–940. [Google Scholar] [CrossRef]
- Nishii, R.; Baskin-Doerfler, R.; Yang, W.; Oak, N.; Zhao, X.; Yang, W.; Hoshitsuki, K.; Bloom, M.; Verbist, K.C.; Burns, M.A.; et al. Molecular basis of ETV6-mediated predisposition to childhood acute lymphoblastic leukemia. Blood 2021, 137, 364–373. [Google Scholar] [CrossRef] [PubMed]
- van Delft, F.W.; Horsley, S.; Colman, S.; Anderson, K.; Bateman, C.; Kempski, H.; Zuna, J.; Eckert, C.; Saha, V.; Kearney, L.; et al. Clonal origins of relapse in ETV6-RUNX1 acute lymphoblastic leukemia. Blood 2011, 117, 6247–6254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, N.K.; Foster, S.D.; Wang, X.; Knezevic, K.; Schütte, J.; Kaimakis, P.; Chilarska, P.M.; Kinston, S.; Ouwehand, W.H.; Dzierzak, E.; et al. Combinatorial Transcriptional Control In Blood Stem/Progenitor Cells: Genome-wide Analysis of Ten Major Transcriptional Regulators. Cell Stem Cell 2010, 7, 532–544. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.S.; Francis, A.; Turkistany, S.; Shukla, D.; Wong, A.; Batista, C.R.; DeKoter, R.P. ETV6-RUNX1 interacts with a region in SPIB intron 1 to regulate gene expression in pre-B-cell acute lymphoblastic leukemia. Exp. Hematol. 2019, 73, 50–63.e2. [Google Scholar] [CrossRef] [PubMed]
- Pang, M.; Minnich, M.; Gangatirkar, P.; Zheng, Z.; Ebert, A.; Song, G.; Dickins, R.; Corcoran, L.M.; Mullighan, C.G.; Busslinger, M.; et al. PU.1 cooperates with IRF4 and IRF8 to suppress pre-B-cell leukemia. Leukemia 2016, 30, 1375–1387. [Google Scholar] [CrossRef] [Green Version]
- Jakobczyk, H.; Jiang, Y.; Debaize, L.; Soubise, B.; Avner, S.; Sérandour, A.A.; Rouger-Gaudichon, J.; Rio, A.-G.; Carroll, J.S.; Raslova, H.; et al. ETV6-RUNX1 and RUNX1 directly regulate RAG1 expression: One more step in the understanding of childhood B-cell acute lymphoblastic leukemia leukemogenesis. Leukemia 2021, 36, 549–554. [Google Scholar] [CrossRef]
- Lilljebjörn, H.; Soneson, C.; Andersson, A.; Heldrup, J.; Behrendtz, M.; Kawamata, N.; Ogawa, S.; Koeffler, H.P.; Mitelman, F.; Johansson, B.; et al. The Correlation Pattern of Acquired Copy Number Changes in 164 ETV6/RUNX1-Positive Childhood Acute Lymphoblastic Leukemias. Hum. Mol. Genet. 2010, 19, 3150. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Gu, Z. PAX5 alterations in B-cell acute lymphoblastic leukemia. Front. Oncol. 2022, 12. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, M.; Shukla, R.; Dwivedi, S.; Saxena, R.; Luthra, K.; Kabra, M.; Seth, R. Gene copy number alterations in Indian children with B-acute Lymphoblastic Leukemia: Correlation with survival outcome. Pediatr. Hematol. Oncol. J. 2021, 6, 151–157. [Google Scholar] [CrossRef]
- Kuster, L.; Grausenburger, R.; Fuka, G.; Kaindl, U.; Krapf, G.; Inthal, A.; Mann, G.; Kauer, M.; Rainer, J.; Kofler, R.; et al. ETV6/RUNX1-Positive Relapses Evolve from an Ancestral Clone and Frequently Acquire Deletions of Genes Implicated in Glucocorticoid Signaling. Blood 2011, 117, 2658–2667. [Google Scholar] [CrossRef]
- Grausenburger, R.; Bastelberger, S.; Eckert, C.; Kauer, M.; Stanulla, M.; Frech, C.; Bauer, E.; Stoiber, D.; von Stackelberg, A.; Attarbaschi, A.; et al. Genetic Alterations in Glucocorticoid Signaling Pathway Components Are Associated with Adverse Prognosis in Children with Relapsed ETV6/RUNX1-Positive Acute Lymphoblastic Leukemia. Leuk Lymphoma 2016, 57, 1163–1173. [Google Scholar] [CrossRef] [PubMed]
- Kelley, C.M.; Ikeda, T.; Koipally, J.; Avitahl, N.; Wu, L.; Georgopoulos, K.; Morgan, B.A. Helios, a novel dimerization partner of Ikaros expressed in the earliest hematopoietic progenitors. Curr. Biol. 1998, 8, 508-S1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullighan, C.G.; Su, X.; Zhang, J.; Zhang, J.; Radtke, I.; Phillips, L.A.A.; Miller, C.B.; Ma, J.; Liu, W.; Cheng, C. Children’s Oncology Group: Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N. Engl. J. Med. 2009, 360, 470–480. [Google Scholar] [CrossRef]
- Ramesh-Kumar, D.; Guil, S. The IGF2BP family of RNA binding proteins links epitranscriptomics to cancer. Semin. Cancer Biol. 2022, 86, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Boby, E.; Nidhi, T.; Jain, A.; Singh, J.; Singh, A.; Chattopadhyay, P.; Bakhshi, S.; Chopra, A.; Palanichamy, J.K. Diagnostic Utility of IGF2BP1 and Its Targets as Potential Biomarkers in ETV6-RUNX1 Positive B-Cell Acute Lymphoblastic Leukemia. Front. Oncol. 2021, 11, 588101. [Google Scholar] [CrossRef]
- Stoskus, M.; Gineikiene, E.; Valceckiene, V.; Valatkaite, B.; Pileckyte, R.; Griskevicius, L. Identification of characteristic IGF2BP expression patterns in distinct B-ALL entities. Blood Cells Mol. Dis. 2011, 46, 321–326. [Google Scholar] [CrossRef]
- Mäkinen, A.; Nikkilä, A.; Haapaniemi, T.; Oksa, L.; Mehtonen, J.; Vänskä, M.; Heinäniemi, M.; Paavonen, T.; Lohi, O. IGF2BP3 Associates with Proliferative Phenotype and Prognostic Features in B-Cell Acute Lymphoblastic Leukemia. Cancers 2021, 13, 1505. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, M.-F.; Smadbeck, J.B.; Sharma, N.; Blackburn, P.R.; Benevides, J.D.; Akkari, Y.M.N.; Jaroscak, J.J.; Znoyko, I.; Wolff, D.J.; Schandl, C.A.; et al. Apparent coexistence of ETV6::RUNX1 and KMT2A::MLLT3 fusions due to a nonproductive KMT2A rearrangement in B-ALL. Leuk. Lymphoma 2022, 63, 2243–2246. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Rapado, I.; Li, Y.; E Potter, N.; Wedge, D.; Tubio, J.; Alexandrov, L.B.; Van Loo, P.; Cooke, S.L.; Marshall, J.; et al. RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nat. Genet. 2014, 46, 116–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeger, K.; Adams, H.P.; Buchwald, D.; Beyermann, B.; Kremens, B.; Niemeyer, C.; Ritter, J.; Schwabe, D.; Harms, D.; Schrappe, M.; et al. TEL-AML1 Fusion Transcript in Relapsed Childhood Acute Lymphoblastic Leukemia The Berlin-Frankfurt-Münster Study 1035 Group. Blood 1998, 91, 1716–1722. [Google Scholar] [CrossRef] [Green Version]
- Pui, C.H.; Sandlund, J.T.; Pei, D.; Campana, D.; Rivera, G.K.; Ribeiro, R.C.; Rubnitz, J.E.; Razzouk, B.I.; Howard, S.C.; Hudson, M.M.; et al. Improved outcome for children with acute lymphoblastic leukemia: Results of Total Therapy Study XIIIB at St Jude Children’s Research Hospital. Blood 2004, 104, 2690–2696. [Google Scholar] [CrossRef]
- Narla, R.K.; Navara, C.; Sarquis, M.; Uckun, F.M. Chemosensitivity of TEL-AML1 Fusion Transcript Positive Acute Lymphoblastic Leukemia Cells. Leuk. Lymphoma 2001, 41, 615–623. [Google Scholar] [CrossRef]
- Ramakers-van Woerden, N.L.; Pieters, R.; Loonen, A.H.; Hubeek, I.; van Drunen, E.; Beverloo, H.B.; Slater, R.M.; Harbott, J.; Seyfarth, J.; van Wering, E.R.; et al. TEL/AML1 gene fusion is related to in vitro drug sensitivity for L-asparaginase in childhood acute lymphoblastic leukemia. Blood 2000, 96, 1094–1099. [Google Scholar]
- Teachey, D.T.; Hunger, S.P.; Loh, M.M.L. Optimizing therapy in the modern age: Differences in length of maintenance therapy in acute lymphoblastic leukemia. Blood 2021, 137, 168–177. [Google Scholar] [CrossRef]
- Inaba, H.; Mullighan, C.G. Pediatric Acute Lymphoblastic Leukemia. Haematologica 2020, 105, 2524. [Google Scholar] [CrossRef]
- Kato, M.; Ishimaru, S.; Seki, M.; Yoshida, K.; Shiraishi, Y.; Chiba, K.; Kakiuchi, N.; Sato, Y.; Ueno, H.; Tanaka, H.; et al. Long-term outcome of 6-month maintenance chemotherapy for acute lymphoblastic leukemia in children. Leukemia 2017, 31, 580–584. [Google Scholar] [CrossRef]
- Schrappe, M.; Bleckmann, K.; Zimmermann, M.; Biondi, A.; Möricke, A.; Locatelli, F.; Cario, G.; Rizzari, C.; Attarbaschi, A.; Valsecchi, M.G.; et al. Reduced-Intensity Delayed Intensification in Standard-Risk Pediatric Acute Lymphoblastic Leukemia Defined by Undetectable Minimal Residual Disease: Results of an International Randomized Trial (AIEOP-BFM ALL 2000). J. Clin. Oncol. 2018, 36, 244–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serafin, V.; Porcù, E.; Cortese, G.; Mariotto, E.; Veltri, G.; Bresolin, S.; Basso, G.; Accordi, B. SYK Targeting Represents a Potential Therapeutic Option for Relapsed Resistant Pediatric ETV6-RUNX1 B-Acute Lymphoblastic Leukemia Patients. Int. J. Mol. Sci. 2019, 20, 6175. [Google Scholar] [CrossRef] [PubMed]
- Akbari, M.; Ebrahimabadi, S.; Golalipour, M.; Shahbazi, M.; Farazmandfar, T. The correction of ETV6/RUNX1 translocation in acute lymphocytic leukemia cells: A new gene targeting system by homologous recombination mechanism. J. Appl. Genet. 2020, 61, 67–73. [Google Scholar] [CrossRef]
- Lilljebjörn, H.; Fioretos, T. New oncogenic subtypes in pediatric B-cell precursor acute lymphoblastic leukemia. Blood 2017, 130, 1395–1401. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Dai, Y.; Wu, L.; Zhang, M.; Ouyang, W.; Huang, J.; Chen, S. Emerging molecular subtypes and therapeutic targets in B-cell precursor acute lymphoblastic leukemia. Front. Med. 2021, 15, 347–371. [Google Scholar] [CrossRef]
- Chen, D.; Camponeschi, A.; Nordlund, J.; Marincevic-Zuniga, Y.; Abrahamsson, J.; Lönnerholm, G.; Fogelstrand, L.; Mårtensson, I.L. RAG1 Co-Expression Signature Identifies ETV6-RUNX1-like B-Cell Precursor Acute Lymphoblastic Leukemia in Children. Cancer Med. 2021, 10, 3997–4003. [Google Scholar] [CrossRef]
- Zaliova, M.; Kotrova, M.; Bresolin, S.; Stuchly, J.; Stary, J.; Hrusak, O.; teKronnie, G.; Trka, J.; Zuna, J.; Vaskova, M. ETV6/RUNX1-like Acute Lymphoblastic Leukemia: A Novel B-Cell Precursor Leukemia Subtype Associated with the CD27/CD44 Immunophenotype. Genes Chromosomes Cancer 2017, 56, 608–616. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chromosome Number | Observed Abnormality | Possibly Covered Genes | Frequency of Occurrence | Reference |
|---|---|---|---|---|
| 12 | deletion of 12p | ETV6, CDKN1B, BCL2L14, BTG1, KRAS | 12–39% | [58,66,67] |
| 6 | deletion of 6q | AIM1, PRDM1, FOXO3, CCNC3, FYN, CDKN1A | 13–33% | [66,68] |
| 21 | gain of normal chromosome 21 gain of the der(21)t(12;21)(p13;q22) | RUNX1 | 25% | [61,67] |
| 9 | deletion of 9p | CDKN2A/B, PAX5, MTAP, JAK2, P14ARF, P16IKN4a/ARF | 10–25% | [58,66] |
| 5 | deletion of 5q | NR3C1, EBF1 | 23% | [69] |
| 3 | deletion of 3p deletion of 3q | LIMDI, ARPP-21, ULK4, FHIT, CD200, BTLA, TBL1XR1 | 3–21% | [66,67] |
| 14 | deletion of 14q | DPF3 | 3–21% | [58] |
| 7 | deletion of 7q deletion of 7p | IKZF1, IGF2BP | 3–18% | [2,58] |
| 4 | duplication of 4q deletion of 4q | NR3C2, YIPF7, ARHGAP10 | 6–17% | [66] |
| 19 | deletion of 19q | CEBPA, UHRF1, GRLF1, NPAS1, TMEM160 | 6–13% | [67] |
| X | monosomy X in females/ gain of X in males gain of Xp duplication of Xq | SPANXB, HMGB3, FAM50A, HTATSF1 | 4–11% | [66] |
| 11 | deletion of 11q deletion of 11p | CD44, RAG1/2, BACL2, GNG3, HNRPUL2, TTC9C, ATM, KMT2A, HRAS | 10% | [67,68] |
| 1 | deletion of 1q | TROVE2, GLRX2, CDC73, B3GALT2, PDE4B, NRAS, | 10% | [69] |
| 15 | deletion of 15q | LTK, MIRN626 | 10% | [70] |
| 13 | deletion of 13q/monosomy | BTG1, RB1, SERP2, DLEU1/2/7, STBP4, TRIM3, KCNRG, MIRN16-1, MIRN15A | 5–10% | [58] |
| 8 | deletion of 8p | CTSB, LOXL2, NKX3-1, WHSC1L1,FGFR1, IDO1, IDO2, KAT6A | 6–8% | [58,68] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaczmarska, A.; Derebas, J.; Pinkosz, M.; Niedźwiecki, M.; Lejman, M. The Landscape of Secondary Genetic Rearrangements in Pediatric Patients with B-Cell Acute Lymphoblastic Leukemia with t(12;21). Cells 2023, 12, 357. https://doi.org/10.3390/cells12030357
Kaczmarska A, Derebas J, Pinkosz M, Niedźwiecki M, Lejman M. The Landscape of Secondary Genetic Rearrangements in Pediatric Patients with B-Cell Acute Lymphoblastic Leukemia with t(12;21). Cells. 2023; 12(3):357. https://doi.org/10.3390/cells12030357
Chicago/Turabian StyleKaczmarska, Agnieszka, Justyna Derebas, Michalina Pinkosz, Maciej Niedźwiecki, and Monika Lejman. 2023. "The Landscape of Secondary Genetic Rearrangements in Pediatric Patients with B-Cell Acute Lymphoblastic Leukemia with t(12;21)" Cells 12, no. 3: 357. https://doi.org/10.3390/cells12030357