
Investigation of Rare Non-Coding Variants in Familial Multiple Myeloma

 , , and
, , and
Abstract
:1. Introduction
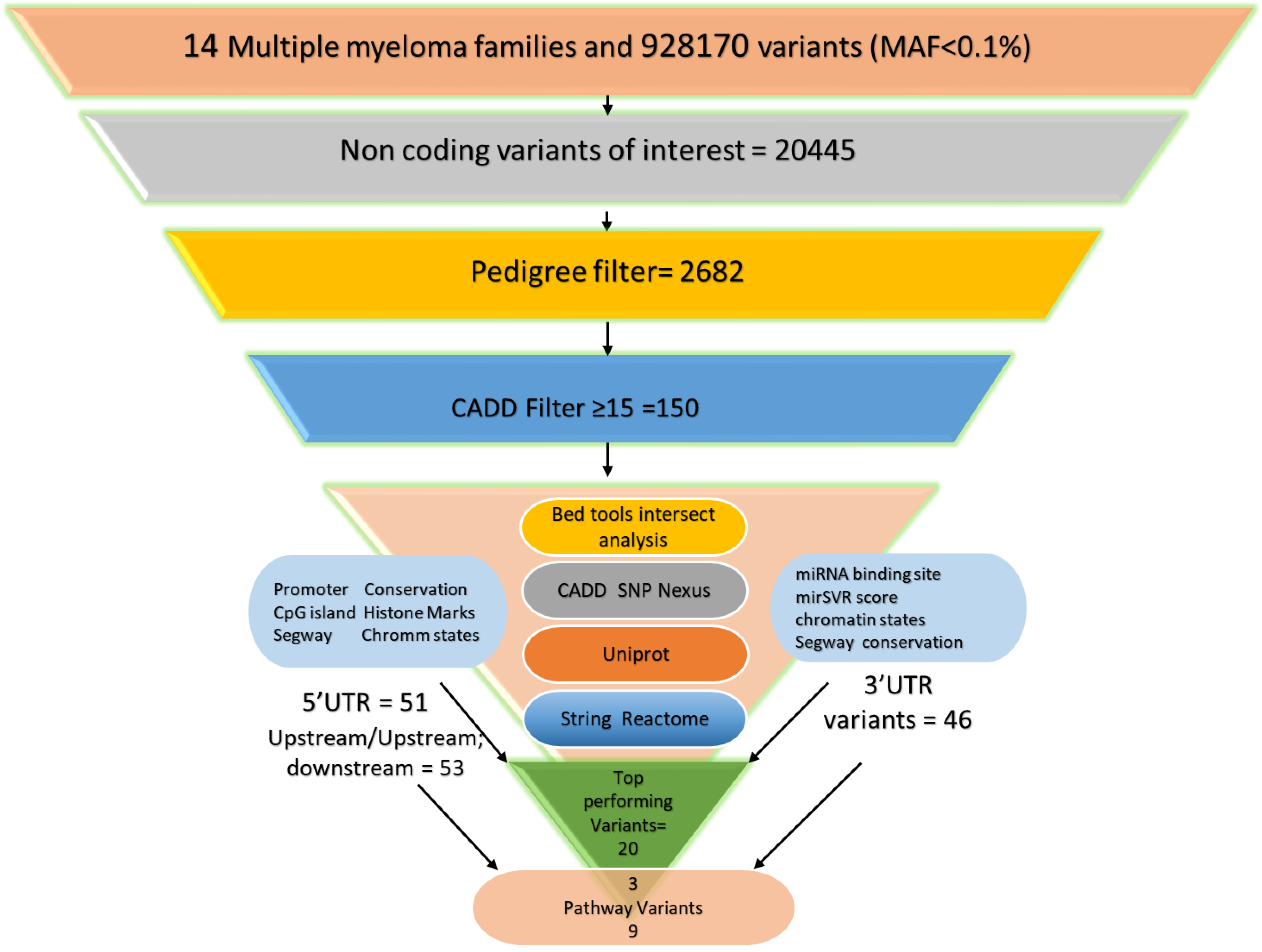
2. Materials and Methods
2.1. Multiple Myeloma Families and Whole-Genome Sequencing
2.2. Prioritization through FCVPPv2
2.3. Conservation
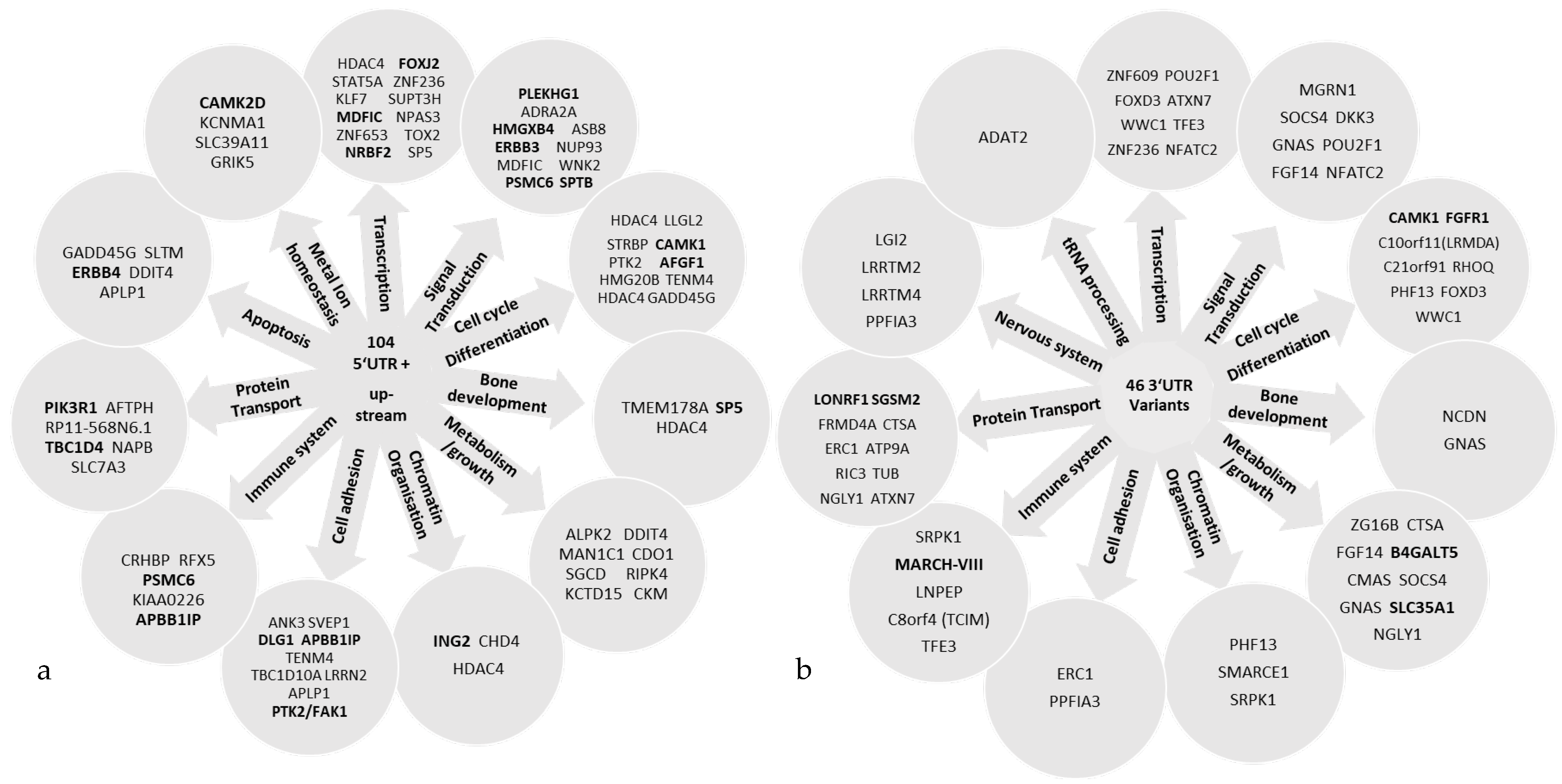
2.4. Analysis of Upstream and 5′ UTR Variants
2.5. TFs/TF Binding Sites
2.6. Graphic Visualization
2.7. Analysis of 3′ UTR Variants
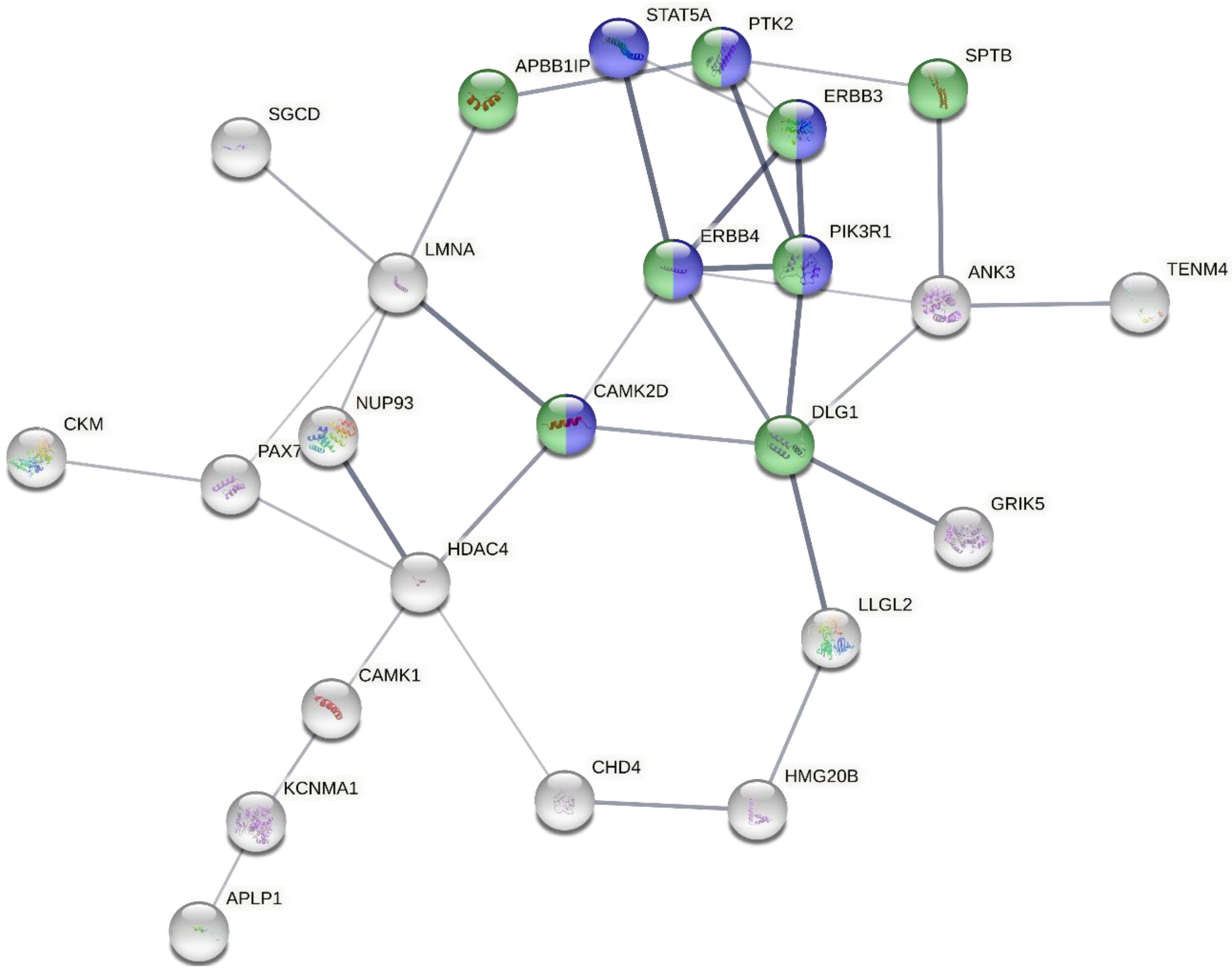
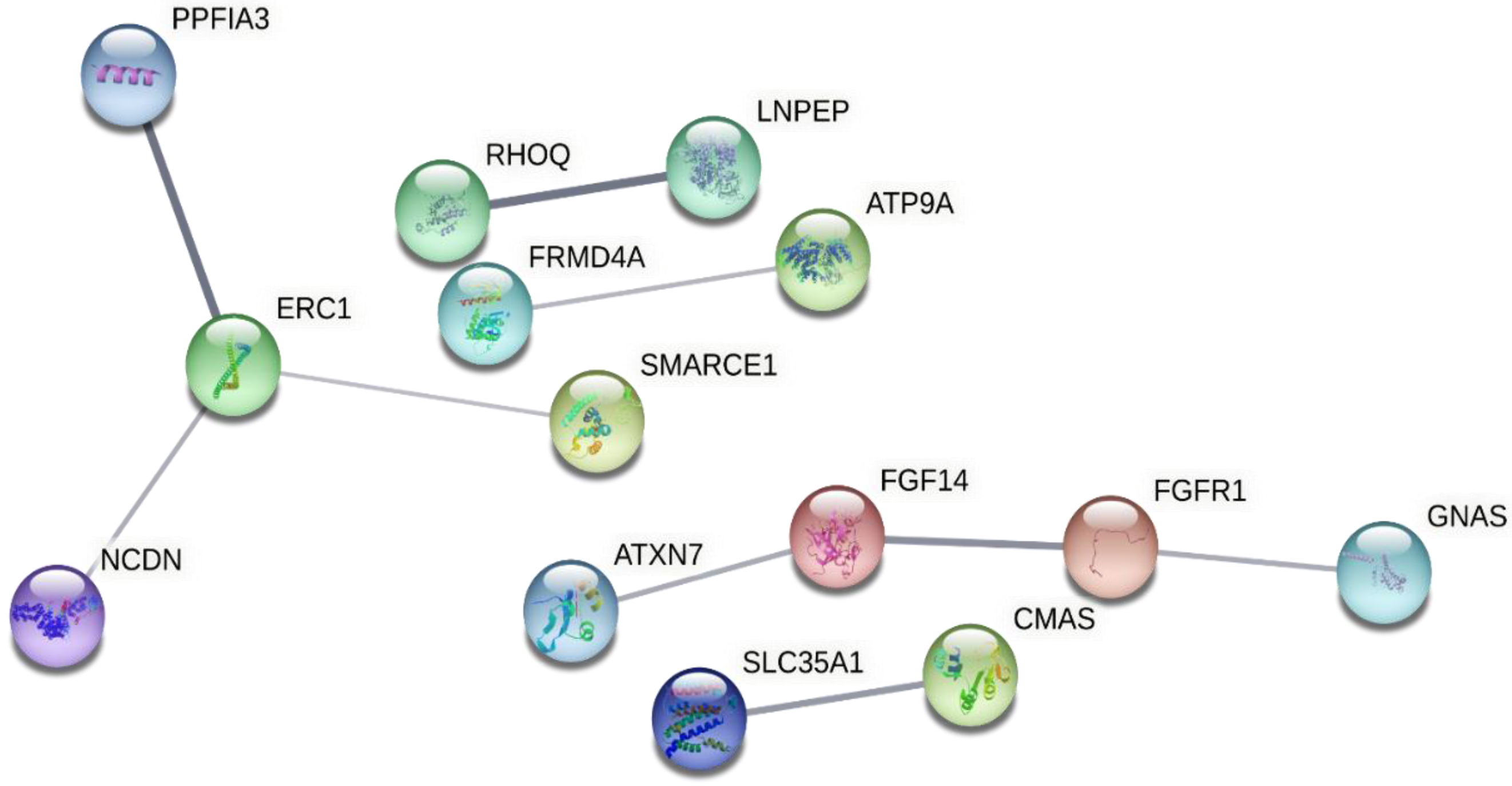
2.8. Biological Function and Pathway Enrichment Analysis
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- van de Donk, N.W.C.J.; Pawlyn, C.; Yong, K.L. Multiple myeloma. Lancet 2021, 397, 410–427. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Becker, N. Epidemiology of Multiple Myeloma. In Multiple Myeloma; Moehler, T., Goldschmidt, H., Eds.; Springer: Berlin, Heidelberg, Germany, 2011; pp. 25–35. [Google Scholar]
- Waller, R.G.; Darlington, T.M.; Wei, X.; Madsen, M.J.; Thomas, A.; Curtin, K.; Coon, H.; Rajamanickam, V.; Musinsky, J.; Jayabalan, D.; et al. Novel pedigree analysis implicates DNA repair and chromatin remodeling in multiple myeloma risk. PLOS Genet. 2018, 14, e1007111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertesi, M.; Vallée, M.; Wei, X.; Revuelta, M.V.; Galia, P.; Demangel, D.; Oliver, J.; Foll, M.; Chen, S.; Perrial, E.; et al. Exome sequencing identifies germline variants in DIS3 in familial multiple myeloma. Leukemia 2019, 33, 2324–2330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catalano, C.; Paramasivam, N.; Blocka, J.; Giangiobbe, S.; Huhn, S.; Schlesner, M.; Weinhold, N.; Sijmons, R.; de Jong, M.; Langer, C.; et al. Characterization of rare germline variants in familial multiple myeloma. Blood Cancer J. 2021, 11, 33. [Google Scholar] [CrossRef]
- Went, M.; Sud, A.; Försti, A.; Halvarsson, B.-M.; Weinhold, N.; Kimber, S.; van Duin, M.; Thorleifsson, G.; Holroyd, A.; Johnson, D.C.; et al. Identification of multiple risk loci and regulatory mechanisms influencing susceptibility to multiple myeloma. Nat. Commun. 2018, 9, 3707. [Google Scholar] [CrossRef]
- Pertesi, M.; Went, M.; Hansson, M.; Hemminki, K.; Houlston, R.S.; Nilsson, B. Genetic predisposition for multiple myeloma. Leukemia 2020, 34, 697–708. [Google Scholar] [CrossRef]
- Frank, C.; Fallah, M.; Chen, T.; Mai, E.K.; Sundquist, J.; Försti, A.; Hemminki, K. Search for familial clustering of multiple myeloma with any cancer. Leukemia 2016, 30, 627–632. [Google Scholar] [CrossRef]
- Ellingford, J.M.; Ahn, J.W.; Bagnall, R.D.; Baralle, D.; Barton, S.; Campbell, C.; Downes, K.; Ellard, S.; Duff-Farrier, C.; FitzPatrick, D.R.; et al. Recommendations for clinical interpretation of variants found in non-coding regions of the genome. Genome Med. 2022, 14, 73. [Google Scholar] [CrossRef]
- Kumar, A.; Bandapalli, O.R.; Paramasivam, N.; Giangiobbe, S.; Diquigiovanni, C.; Bonora, E.; Eils, R.; Schlesner, M.; Hemminki, K.; Försti, A. Familial Cancer Variant Prioritization Pipeline version 2 (FCVPPv2) applied to a papillary thyroid cancer family. Sci. Rep. 2018, 8, 11635. [Google Scholar] [CrossRef]
- Rentzsch, P.; Schubach, M.; Shendure, J.; Kircher, M. CADD-Splice—Improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med. 2021, 13, 31. [Google Scholar] [CrossRef] [PubMed]
- Dayem Ullah, A.Z.; Oscanoa, J.; Wang, J.; Nagano, A.; Lemoine, N.R.; Chelala, C. SNPnexus: Assessing the functional relevance of genetic variation to facilitate the promise of precision medicine. Nucleic Acids Res. 2018, 46, W109–W113. [Google Scholar] [CrossRef] [Green Version]
- Blocka, J.; Durie, B.G.M.; Huhn, S.; Mueller-Tidow, C.; Försti, A.; Hemminki, K.; Goldschmidt, H. Familial Cancer: How to Successfully Recruit Families for Germline Mutations Studies? Multiple Myeloma as an Example. Clin. Lymphoma Myeloma Leuk. 2019, 19, 635–644.e632. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Tarasov, A.; Vilella, A.J.; Cuppen, E.; Nijman, I.J.; Prins, P. Sambamba: Fast processing of NGS alignment formats. Bioinformatics 2015, 31, 2032–2034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rimmer, A.; Phan, H.; Mathieson, I.; Iqbal, Z.; Twigg, S.R.F.; Wilkie, A.O.M.; McVean, G.; Lunter, G.; Consortium, W.G.S. Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat. Genet. 2014, 46, 912–918. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; Abecasis, G.R. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Smigielski, E.M.; Sirotkin, K.; Ward, M.; Sherry, S.T. dbSNP: A database of single nucleotide polymorphisms. Nucleic Acids Res. 2000, 28, 352–355. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wu, C.; Li, C.; Boerwinkle, E. dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum. Mutat. 2016, 37, 235–241. [Google Scholar] [CrossRef] [Green Version]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Ionita-Laza, I.; McCallum, K.; Xu, B.; Buxbaum, J.D. A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nat. Genet. 2016, 48, 214–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Liu, Z.; Lou, S.; Bedford, J.; Mu, X.J.; Yip, K.Y.; Khurana, E.; Gerstein, M. FunSeq2: A framework for prioritizing noncoding regulatory variants in cancer. Genome Biol. 2014, 15, 480. [Google Scholar] [CrossRef]
- Rogers, M.F.; Shihab, H.A.; Mort, M.; Cooper, D.N.; Gaunt, T.R.; Campbell, C. FATHMM-XF: Accurate prediction of pathogenic point mutations via extended features. Bioinformatics 2017, 34, 511–513. [Google Scholar] [CrossRef] [Green Version]
- Smedley, D.; Schubach, M.; Jacobsen, J.O.B.; Köhler, S.; Zemojtel, T.; Spielmann, M.; Jäger, M.; Hochheiser, H.; Washington, N.L.; McMurry, J.A.; et al. A Whole-Genome Analysis Framework for Effective Identification of Pathogenic Regulatory Variants in Mendelian Disease. Am. J. Hum. Genet. 2016, 99, 595–606. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Troyanskaya, O.G. Predicting effects of noncoding variants with deep learning-based sequence model. Nat. Methods 2015, 12, 931–934. [Google Scholar] [CrossRef] [Green Version]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2012, 14, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Cooper, G.M.; Stone, E.A.; Asimenos, G.; Green, E.D.; Batzoglou, S.; Sidow, A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005, 15, 901–913. [Google Scholar] [CrossRef] [Green Version]
- Siepel, A.; Bejerano, G.; Pedersen, J.S.; Hinrichs, A.S.; Hou, M.; Rosenbloom, K.; Clawson, H.; Spieth, J.; Hillier, L.W.; Richards, S.; et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005, 15, 1034–1050. [Google Scholar] [CrossRef] [Green Version]
- Pollard, K.S.; Hubisz, M.J.; Rosenbloom, K.R.; Siepel, A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010, 20, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Lizio, M.; Harshbarger, J.; Shimoji, H.; Severin, J.; Kasukawa, T.; Sahin, S.; Abugessaisa, I.; Fukuda, S.; Hori, F.; Ishikawa-Kato, S.; et al. Gateways to the FANTOM5 promoter level mammalian expression atlas. Genome Biol. 2015, 16, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Encode. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fornes, O.; Castro-Mondragon, J.A.; Khan, A.; van der Lee, R.; Zhang, X.; Richmond, P.A.; Modi, B.P.; Correard, S.; Gheorghe, M.; Baranašić, D.; et al. JASPAR 2020: Update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2020, 48, D87–D92. [Google Scholar] [CrossRef]
- Tyner, C.; Barber, G.P.; Casper, J.; Clawson, H.; Diekhans, M.; Eisenhart, C.; Fischer, C.M.; Gibson, D.; Gonzalez, J.N.; Guruvadoo, L.; et al. The UCSC Genome Browser database: 2017 update. Nucleic Acids Res. 2017, 45, D626–D634. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Ernst, J.; Kellis, M. ChromHMM: Automating chromatin-state discovery and characterization. Nat. Methods 2012, 9, 215–216. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, M.M.; Buske, O.J.; Wang, J.; Weng, Z.; Bilmes, J.A.; Noble, W.S. Unsupervised pattern discovery in human chromatin structure through genomic segmentation. Nat. Methods 2012, 9, 473–476. [Google Scholar] [CrossRef] [Green Version]
- Betel, D.; Koppal, A.; Agius, P.; Sander, C.; Leslie, C. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 2010, 11, R90. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Gillespie, M.; Jassal, B.; Stephan, R.; Milacic, M.; Rothfels, K.; Senff-Ribeiro, A.; Griss, J.; Sevilla, C.; Matthews, L.; Gong, C.; et al. The reactome pathway knowledgebase 2022. Nucleic Acids Res. 2021, 50, D687–D692. [Google Scholar] [CrossRef] [PubMed]
- The UniProt, C. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef] [PubMed]
- Ajore, R.; Niroula, A.; Pertesi, M.; Cafaro, C.; Thodberg, M.; Went, M.; Bao, E.L.; Duran-Lozano, L.; Lopez de Lapuente Portilla, A.; Olafsdottir, T.; et al. Functional dissection of inherited non-coding variation influencing multiple myeloma risk. Nat. Commun. 2022, 13, 151. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Anderson, K.C. Signaling Pathway Mediating Myeloma Cell Growth and Survival. Cancers 2021, 13, 216. [Google Scholar] [CrossRef]
- Hideshima, T.; Mitsiades, C.; Tonon, G.; Richardson, P.G.; Anderson, K.C. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat. Rev. Cancer 2007, 7, 585–598. [Google Scholar] [CrossRef]
- Platanias, L.C. Map kinase signaling pathways and hematologic malignancies. Blood 2003, 101, 4667–4679. [Google Scholar] [CrossRef] [Green Version]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [Green Version]
- Arteaga, C.L.; Engelman, J.A. ERBB receptors: From oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell 2014, 25, 282–303. [Google Scholar] [CrossRef] [Green Version]
- Wandinger, S.K.; Lahortiga, I.; Jacobs, K.; Klammer, M.; Jordan, N.; Elschenbroich, S.; Parade, M.; Jacoby, E.; Linders, J.T.; Brehmer, D.; et al. Quantitative Phosphoproteomics Analysis of ERBB3/ERBB4 Signaling. PLoS ONE 2016, 11, e0146100. [Google Scholar] [CrossRef] [Green Version]
- Sudhesh Dev, S.; Zainal Abidin, S.A.; Farghadani, R.; Othman, I.; Naidu, R. Receptor Tyrosine Kinases and Their Signaling Pathways as Therapeutic Targets of Curcumin in Cancer. Front. Pharmacol. 2021, 12, 772510. [Google Scholar] [CrossRef]
- Krens, S.F.; Spaink, H.P.; Snaar-Jagalska, B.E. Functions of the MAPK family in vertebrate-development. FEBS Lett. 2006, 580, 4984–4990. [Google Scholar] [CrossRef] [PubMed]
- Aghajanian, C.; Soignet, S.; Dizon, D.S.; Pien, C.S.; Adams, J.; Elliott, P.J.; Sabbatini, P.; Miller, V.; Hensley, M.L.; Pezzulli, S.; et al. A Phase I Trial of the Novel Proteasome Inhibitor PS341 in Advanced Solid Tumor Malignancies1. Clin. Cancer Res. 2002, 8, 2505–2511. [Google Scholar] [PubMed]
- Shi, C.X.; Kortüm, K.M.; Zhu, Y.X.; Bruins, L.A.; Jedlowski, P.; Votruba, P.G.; Luo, M.; Stewart, R.A.; Ahmann, J.; Braggio, E.; et al. CRISPR Genome-Wide Screening Identifies Dependence on the Proteasome Subunit PSMC6 for Bortezomib Sensitivity in Multiple Myeloma. Mol. Cancer Ther. 2017, 16, 2862–2870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marziali, F.; Dizanzo, M.P.; Cavatorta, A.L.; Gardiol, D. Differential expression of DLG1 as a common trait in different human diseases: An encouraging issue in molecular pathology. Biol. Chem. 2019, 400, 699–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moser-Katz, T.; Gavile, C.M.; Barwick, B.G.; Lee, K.P.; Boise, L.H. PDZ Proteins SCRIB and DLG1 Regulate Myeloma Cell Surface CD86 Expression, Growth, and Survival. Mol. Cancer Res. 2022, 20, 1122–1136. [Google Scholar] [CrossRef]
- Bhutani, M.; Foureau, D.M.; Atrash, S.; Voorhees, P.M.; Usmani, S.Z. Extramedullary multiple myeloma. Leukemia 2020, 34, 1–20. [Google Scholar] [CrossRef]
- Dai, S.; Zhou, Z.; Chen, Z.; Xu, G.; Chen, Y. Fibroblast Growth Factor Receptors (FGFRs): Structures and Small Molecule Inhibitors. Cells 2019, 8, 614. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Mikse, O.; Liao, R.G.; Li, Y.; Tan, L.; Janne, P.A.; Gray, N.S.; Wong, K.K.; Hammerman, P.S. Ligand-associated ERBB2/3 activation confers acquired resistance to FGFR inhibition in FGFR3-dependent cancer cells. Oncogene 2015, 34, 2167–2177. [Google Scholar] [CrossRef] [Green Version]
- Salazar, L.; Kashiwada, T.; Krejci, P.; Muchowski, P.; Donoghue, D.; Wilcox, W.R.; Thompson, L.M. A novel interaction between fibroblast growth factor receptor 3 and the p85 subunit of phosphoinositide 3-kinase: Activation-dependent regulation of ERK by p85 in multiple myeloma cells. Hum. Mol. Genet. 2009, 18, 1951–1961. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.Y.; Zal, T.; Ch’en, I.L.; Gascoigne, N.R.; Hedrick, S.M. A pivotal role for the multifunctional calcium/calmodulin-dependent protein kinase II in T cells: From activation to unresponsiveness. J. Immunol. 2005, 174, 5583–5592. [Google Scholar] [CrossRef] [Green Version]
- Sui, Y.; Li, X.; Oh, S.; Zhang, B.; Freeman, W.M.; Shin, S.; Janknecht, R. Opposite Roles of the JMJD1A Interaction Partners MDFI and MDFIC in Colorectal Cancer. Sci. Rep. 2020, 10, 8710. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Guo, Y.; Ge, X.; Itoh, H.; Watanabe, A.; Fujiwara, T.; Kodama, T.; Aburatani, H. Elevated expression and potential roles of human Sp5, a member of Sp transcription factor family, in human cancers. Biochem. Biophys. Res. Commun. 2006, 340, 758–766. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Sánchez, C.; Arias-de-la-Fuente, C.; Gómez-Ferrería, M.A.A.; Granadino, B.; Rey-Campos, J. FHX.L and FHX.S, two isoforms of the human fork-head factor FHX (FOXJ2) with differential activity11Edited by M. Yaniv. J. Mol. Biol. 2000, 301, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Wang, Y.; Abi Saab, W.F.; Yang, F.; Pessin, J.E.; Backer, J.M. NRBF2 regulates macroautophagy as a component of Vps34 Complex I. Biochem. J. 2014, 461, 315–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huggins, I.J.; Bos, T.; Gaylord, O.; Jessen, C.; Lonquich, B.; Puranen, A.; Richter, J.; Rossdam, C.; Brafman, D.; Gaasterland, T.; et al. The WNT target SP5 negatively regulates WNT transcriptional programs in human pluripotent stem cells. Nat. Commun. 2017, 8, 1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, S.; Nishikawa, M.; Kobayashi, T.; Harlin, E.W.; Ito, T.; Sato, K.; Sugiyama, T.; Yamakawa, H.; Nagase, T.; Ueda, H. The Rho guanine nucleotide exchange factor PLEKHG1 is activated by interaction with and phosphorylation by Src family kinase member FYN. J. Biol. Chem. 2022, 298, 101579. [Google Scholar] [CrossRef]
- Yamada, M.; Ohkawara, B.; Ichimura, N.; Hyodo-Miura, J.; Urushiyama, S.; Shirakabe, K.; Shibuya, H. Negative regulation of Wnt signalling by HMG2L1, a novel NLK-binding protein. Genes Cells Devoted Mol. Cell. Mech. 2003, 8, 677–684. [Google Scholar] [CrossRef]
- Swaminathan, B.; Thorleifsson, G.; Jöud, M.; Ali, M.; Johnsson, E.; Ajore, R.; Sulem, P.; Halvarsson, B.M.; Eyjolfsson, G.; Haraldsdottir, V.; et al. Variants in ELL2 influencing immunoglobulin levels associate with multiple myeloma. Nat. Commun. 2015, 6, 7213. [Google Scholar] [CrossRef] [Green Version]
- Cai, C.; Tang, Y.-D.; Zhai, J.; Zheng, C. The RING finger protein family in health and disease. Signal. Transduct. Target. Ther. 2022, 7, 300. [Google Scholar] [CrossRef]
- Nottingham, R.M.; Ganley, I.G.; Barr, F.A.; Lambright, D.G.; Pfeffer, S.R. RUTBC1 protein, a Rab9A effector that activates GTP hydrolysis by Rab32 and Rab33B proteins. J. Biol. Chem. 2011, 286, 33213–33222. [Google Scholar] [CrossRef] [Green Version]
- Szulc, B.; Zadorozhna, Y.; Olczak, M.; Wiertelak, W.; Maszczak-Seneczko, D. Novel Insights into Selected Disease-Causing Mutations within the SLC35A1 Gene Encoding the CMP-Sialic Acid Transporter. Int. J. Mol. Sci. 2020, 22, 304. [Google Scholar] [CrossRef] [PubMed]
- Lapaque, N.; Jahnke, M.; Trowsdale, J.; Kelly, A.P. The HLA-DRalpha chain is modified by polyubiquitination. J. Biol. Chem. 2009, 284, 7007–7016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Yang, X.; Liu, M.; Zhang, Z.; Xing, E. Roles of miRNA dysregulation in the pathogenesis of multiple myeloma. Cancer Gene Ther. 2021, 28, 1256–1268. [Google Scholar] [CrossRef] [PubMed]
- Mallampalli, R.K.; Kaercher, L.; Snavely, C.; Pulijala, R.; Chen, B.B.; Coon, T.; Zhao, J.; Agassandian, M. Fbxl12 triggers G1 arrest by mediating degradation of calmodulin kinase I. Cell. Signal. 2013, 25, 2047–2059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sewify, E.M.; Afifi, O.A.; Mosad, E.; Zaki, A.H.; El Gammal, S.A. Cyclin D1 amplification in multiple myeloma is associated with multidrug resistance expression. Clin. Lymphoma Myeloma Leuk. 2014, 14, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Weinhold, N.; Johnson, D.C.; Chubb, D.; Chen, B.; Försti, A.; Hosking, F.J.; Broderick, P.; Ma, Y.P.; Dobbins, S.E.; Hose, D.; et al. The CCND1 c.870G>A polymorphism is a risk factor for t(11;14)(q13;q32) multiple myeloma. Nat. Genet. 2013, 45, 522–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaty, T.H.; Ruczinski, I.; Murray, J.C.; Marazita, M.L.; Munger, R.G.; Hetmanski, J.B.; Murray, T.; Redett, R.J.; Fallin, M.D.; Liang, K.Y.; et al. Evidence for gene-environment interaction in a genome wide study of nonsyndromic cleft palate. Genet. Epidemiol. 2011, 35, 469–478. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
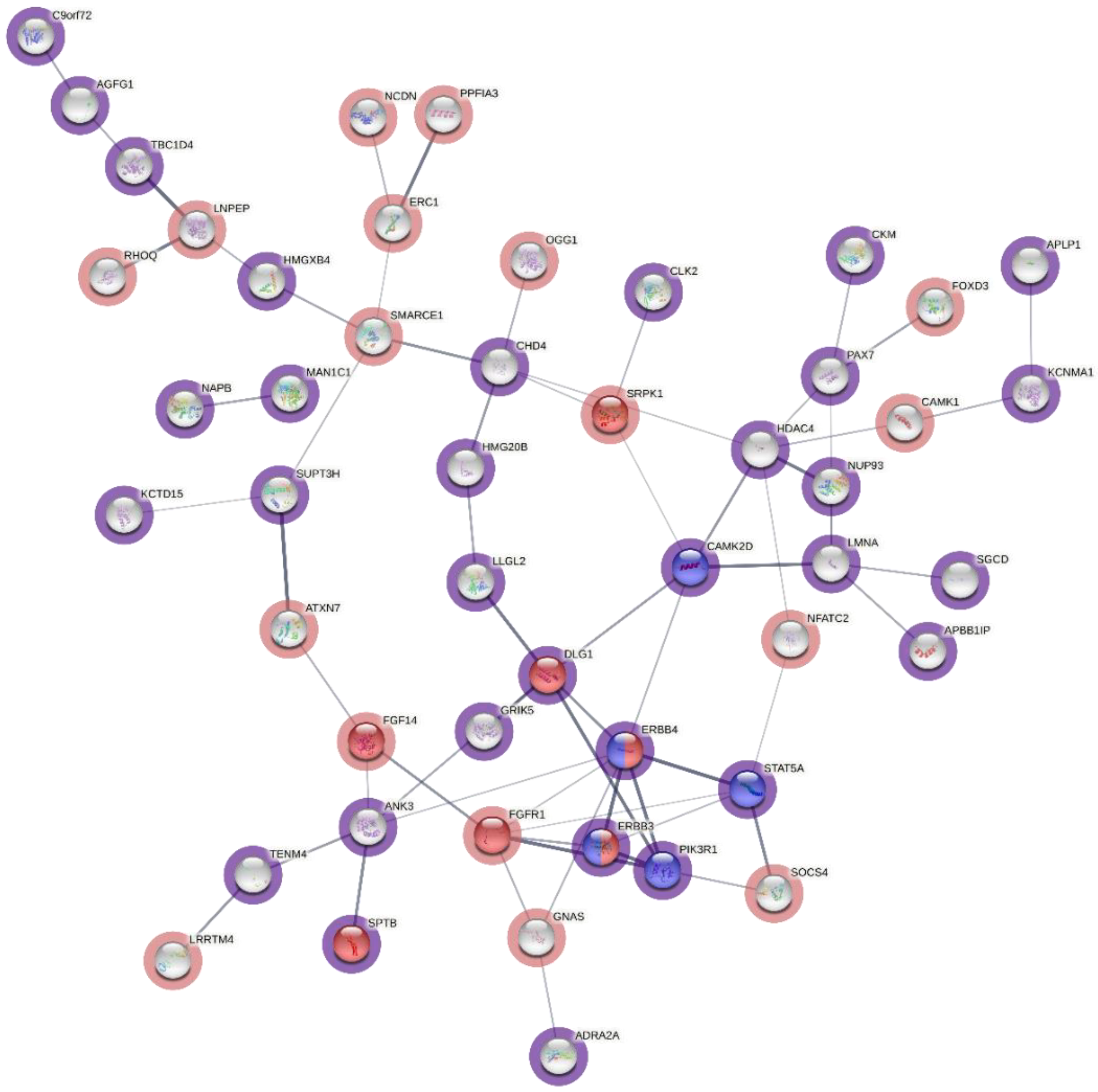{kind=link}
| Family | Gene | Gene Name | Chrom_Pos_Ref_Alt | CADD | Conservation Score/3 | CpG Island (yes/no) | Segway | cHmm | Histone Marks >20 | No. of TFs | Conserved TFBSs | Encode TFs in GM12878/GM12878 ENCSR447YYN | Overall Function |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| >20% | |||||||||||||
| Family_1 | SP5 † | transcription factor Sp5 | 2_171571426_G_A | 16.65 | 2 | Yes | GS | TssA/TssAFlnk: Tx/TxFlnk/TxWk | EncH3K27Ac/K4Me1/K4Me3 | 13 | NR2F1, HDAC6 | DNA-binding transcription factor, bone morphogenesis, metal ion binding | |
| Family_1 | FNDC3B † | fibronectin type III domain containing 3B | 3_171757553_C_A | 15.8 | 2 | Yes | TSS | TssA/TssAFlnk | EncH3K27Ac/K4Me3 | 40 | BCLAF1, Yy1, Pax5, ETS1, TAF1, Tcf12, Egr1, POU2F2, ELF1, RUNX3 | Adipogenesis | |
| Family_1 | CAMK2D * | calcium/calmodulin-dependent protein kinase II delta | 4_114682943_TCCTCCTCCGGCG_T | 19.58 | 3 | No | TF2 | ReprPC/RepPCWk/Quies | EncH3K27Ac/K4Me3 | 2 | CTCF, BCL11A, EBF1, IRF4, BCLAF1, Pax5, Yy1, ELF1, TAF1, Egr1 | Regulation of Ca2+ homeostasis | |
| Family_1 | FOXJ2 † | forkhead box J2 | 12_8185317_GGAGCC_G | 21.9 | 2 | Yes | TSS | TssA/TssAFlnk: TssBiv/EnhBiv | EncH3K27Ac/K4Me3 | 29 | Egr1, SP1 | Transcriptional activator | |
| Family_1 | SPTB * | spectrin, beta, erythrocytic | 14_65346721_C_A | 20.5 | 2 | Yes | TSS | ReprPC/RepPCWk/Quies | NA | E47, Tal-1, ITF-2, Tal-1beta, GATA-1, AP-2alphaA, AP-2gamma | Egr1, HDAC6 | Cytoskeleton network | |
| Family_2 | NRBF2 † | nuclear receptor binding factor 2 | 10_64893005_T_C | 17.12 | 2 | Yes | TSS | ReprPC/RepPCWk/Quies | EncH3K4Me3 | 1 | ATF3, POU2F2, TAF1, ZBTB33, SP1, BCLAF1, Egr1, Tcf12, ELF1, Yy1 | Autophagy, transcription regulation | |
| Family_4 | HMGXB4 † | HMG box domain containing 4 | 22_35653479_C_A | 20.3 | 2 | Yes | TSS | TssA/TssAFlnk | EncH3K27Ac/K4Me3 | 15 | IRF-1 | ELF1, ETS1, SP1, POU2F2, TAF1, BCLAF1, Yy1, Egr1, Tr4, Srf | Wnt signaling |
| Family_6 | ERBB4 * | v-erb-a erythroblastic leukemia viral oncogene homolog 4 (avian) | 2_213404066_C_T | 17.09 | 2 | No | D | TssA/TssAFlnk | 7 | p300 | HDAC6 | Tyrosine kinase, apoptosis, development | |
| Family_6 | AGFG1 † | ArfGAP with FG repeats 1 | 2_228337132_G_A | 16.16 | 2 | Yes | GS | TssA/TssAFlnk | EncH3K27Ac/K4Me3 | 8 | Yy1, ELF1, BCLAF1, Pax5, Egr1, ETS1, BHLHE40, IKZF1, ZNF217, BACH1 | Differentiation, mRNA transport | |
| Family_6 | ING2 † | inhibitor of growth family member 2 | 4_184425877_C_A | 15.71 | 3 | Yes | TSS | TssA/TssAFlnk | EncH3K4Me3 | 12 | Yy1, BCLAF1, ELF1, Egr1, Tcf12, Pax5, SP1, POU2F2, Srf, MEF2A | Chromatin organization, histone deacetylation | |
| Family_6 | PIK3R1 * | phosphoinositide-3-kinase, regulatory subunit 1 (alpha) | 5_67511017_G_C | 16.81 | 1 | Yes | TSS | Enh: ReprPC/RepPCWk/Quies | EncH3K27Ac/K4Me3 | 3 | BCLAF1, ELF1, CTCF, MEF2A, Yy1, TAF1, Egr1, EBF1, Pax5, POU2F2 | Protein transport, stress response | |
| Family_6 | MDFIC † | MyoD family inhibitor domain containing | 7_114562322_C_G | 21.1 | 2 | Yes | TF0 | TssBiv/Biv/EnhBiv:TssA/TssAFlnk | EncH3K27Ac/K4Me3 | 7 | POU2F2, Egr1, BCLAF1, ETS1, Yy1, MEF2A, TAF1, ELF1, RB1, IKZF1 | Transcription regulation, Wnt signaling | |
| Family_6 | TBC1D4 † | TBC1 domain family, member 4 | 13_76056522_G_A | 18.11 | 2 | Yes | GS | TssA/TssAFlnk:ReprPC/RepPCWk/Quies | EncH3K27Ac/K4Me3 | 1 | NF-1 | PU1, ELF1, POU2F2, Egr1, ETS1, Yy1, BCLAF1, CTCF, IRF4, Rad21 | GTPase activator |
| Family_7 | ERBB3 *† | v-erb-b2 erythroblastic leukemia viral oncogene homolog 3 (avian) | 12_56473408_C_T | 18.89 | 3 | Yes | GS | TssA/TssAFlnk | EncH3K27Ac/K4Me1/K4Me3 | 69 | CTCF, IKZF1, TRIM22, RB1, TCF3 | Kinase, signal transduction regulation | |
| Family_7 | PSMC6 *† | proteasome (prosome, macropain) 26S subunit, ATPase, 6 | 14_53173885_C_G | 18.51 | 3 | Yes | TSS | TssA/TssAFlnk | EncH3K27Ac/K4Me3 | 13 | Yy1, TAF1, POU2F2, ELF1, Srf, Gabp, SP1, SIN3A, RB1, PKNOX1, ZNF207, TBP, ELK1 | Ubiquitination, immune system, Wnt signaling | |
| Family_9 | CAMK1 † | calcium/calmodulin-dependent protein kinase I | 3_9811535_G_A | 21.1 | 2 | Yes | TSS | TssA/TssAFlnk:TssBiv/EnhBiv | EncH3K4Me3 | 18 | Pax-5, MIF-1, AP-2gamma, USF1 | RB1 | Cell cycle, differentiation |
| Family_9 | PLEKHG1 † | pleckstrin homology domain containing, family G (with RhoGef domain) member 1 | 6_150921086_G_A | 15.37 | 3 | Yes | TF0 | TssA/TssAFlnk:TssBiv/EnhBiv | EncH3K4Me3 | 11 | IKZF1, NR2F1, ZNF217 ELF1, BACH1, Tcf12 PU1, HDAC6, SP1 | G nucleotide exchange factor | |
| Family_10 | PTK2/FAK1 * | protein tyrosine kinase 2/Focal Adhesion Kinase 1 | 8_142012766_C_T | 15.65 | 1 | No | GS | TssA/TssAFlnk | EncH3K27Ac/K4Me1 | 39 | Cell cycle, migration, adhesion | ||
| Family_11 | DLG1 *† | discs, large homolog 1 (Drosophila) | 3_197024641_C_T | 20.2 | 2 | Yes | TSS | Enh | EncH3K27Ac/K4Me3 | 1 | Yy1, Egr1, ELF1, POU2F2, TAF1, Tcf12, Pax5, HDAC6, ZNF24, BHLHE40 | Host–virus interaction, cadherin binding | |
| Family_11 | APBB1IP * | amyloid beta (A4) precursor protein-binding, family B, member 1 interacting protein | 10_26727608_C_G | 15.3 | 2 | Yes | TF0 | ReprPC/RepPCWk/Quies | EncH3K27Ac/K4Me3 | NA | Yy1, BCLAF1, Pax5, ELF1, PU1, Rad21, RUNX3, IKZF1 MEF2B, BACH1 | Cell adhesion, immune system |
| Family | Gene | Gene Name | Chrom_Pos_Ref_Alt | CADD | Conservation Score | miRNA Binding yes/no | Mir SVR Score | Segway | cHmm > 20 | Overall Function |
|---|---|---|---|---|---|---|---|---|---|---|
| (bold if context++>90) | ||||||||||
| Family_1 | LONRF1 * | LON peptidase N-terminal domain and ring finger 1 | 8_12580093_G_C | 19.75 | 3 | Yes | −1.26 | GE0 | cHmm:Tx/TxWk | Protein polyubiquitination, metal ion binding |
| Family_2 | SLC35A1 * | solute carrier family 35 (CMP-sialic acid transporter), member A1 | 6_88222026_A_G | 16.88 | 2 | Yes | −1.23 | GE0 | cHmm:Tx/TxWk | Transmembrane transport, carbohydrate metabolism |
| Family_6 | MARCHF8 * | membrane-associated ring finger (C3HC4) 8, E3 ubiquitin-protein ligase | 10_45952965_T_C | 16.16 | 3 | Yes | −0.78 | GE0 | cHmm:Tx/TxWk | Immune response, antigen processing MHC class II |
| Family_10 | B4GALT5 * | UDP-Gal:betaGlcNAc beta 1,4- galactosyltransferase, polypeptide 5 | 20_48250790_A_G | 16.44 | 3 | Yes | −0.41 | GE1 | cHmm:Tx/TxWk | Galactosyltransferase, lipid metabolism, regulation of embryonic development |
| Family_12 | FAM76B * | family with sequence similarity 76, member B | 11_95504039_CA_C | 15.82 | 3 | Yes | −0.26 | GE0 | cHmm:Tx/TxWk | Unknown function |
| Family_13 | SGSM2 * | small G protein signaling modulator 2 | 17_2284327_C_T | 21.5 | 2 | Yes | −1.22 | GE1 | cHmm:Tx/TxWk | GTPase activation, intracellular transport |
| Family_2 | FGFR1 † | fibroblast growth factor receptor 1 | 8_38270114_C_T | 17.46 | 1 | Yes | −0.11 | R5 | cHmm:Tx/TxWk/ReprPC/PCWk/Quies | Cell migration, differentiation, proliferation, MAPK pathway |
| Reactome Pathway | Ratio of Proteins in Pathway | Number of Proteins in Pathway | Proteins from Gene Set | p-Value | FDR | Hit Genes |
|---|---|---|---|---|---|---|
| RAF/MAP kinase cascade | 0.0253 | 276 | 10 | 1.19 × 10−5 | 3.88 × 10−3 | PIK3R1,PTK2,DLG1,FGFR1,SPTB,APBB1IP,CAMK2D,ERBB3,ERBB4,PSMC6 |
| MAPK1/MAPK3 signaling | 0.0258 | 282 | 10 | 1.43 × 10−5 | 3.88 × 10−3 | PIK3R1,PTK2,DLG1,FGFR1,SPTB,APBB1IP,CAMK2D,ERBB3,ERBB4,PSMC6 |
| MAPK family signaling cascades | 0.0298 | 326 | 10 | 4.88 × 10−5 | 8.78 × 10−3 | PIK3R1,PTK2,DLG1,FGFR1,SPTB,APBB1IP,CAMK2D,ERBB3,ERBB4,PSMC6 |
| Asparagine N-linked glycosylation | 0.0262 | 286 | 9 | 9.90 × 10−5 | 0.01 | CMAS,ANK3,CTSA,NGLY1,SPTB,B4GALT5,NAPB,MAN1C1,SLC35A1 |
| PI3K events in ERBB2 signaling | 0.0015 | 16 | 3 | 1.66 × 10−4 | 0.02 | PIK3R1,ERBB3,ERBB4 |
| Negative regulation of NMDA receptor-mediated neuronal transmission | 0.0019 | 21 | 3 | 3.68 × 10−4 | 0.03 | DLG1,CAMK2D,CAMK1 |
| Long-term potentiation | 0.0021 | 23 | 3 | 4.79 × 10−4 | 0.04 | DLG1,CAMK2D,ERBB4 |
| Signaling by ERBB4 | 0.0053 | 58 | 4 | 5.81 × 10−4 | 0.04 | PIK3R1,STAT5A,ERBB3,ERBB4 |
| Post NMDA receptor activation events | 0.0057 | 62 | 4 | 7.43 × 10−4 | 0.04 | DLG1,CAMK2D,ERBB4,CAMK1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niazi, Y.; Paramasivam, N.; Blocka, J.; Kumar, A.; Huhn, S.; Schlesner, M.; Weinhold, N.; Sijmons, R.; De Jong, M.; Durie, B.; et al. Investigation of Rare Non-Coding Variants in Familial Multiple Myeloma. Cells 2023, 12, 96. https://doi.org/10.3390/cells12010096
Niazi Y, Paramasivam N, Blocka J, Kumar A, Huhn S, Schlesner M, Weinhold N, Sijmons R, De Jong M, Durie B, et al. Investigation of Rare Non-Coding Variants in Familial Multiple Myeloma. Cells. 2023; 12(1):96. https://doi.org/10.3390/cells12010096
Chicago/Turabian StyleNiazi, Yasmeen, Nagarajan Paramasivam, Joanna Blocka, Abhishek Kumar, Stefanie Huhn, Matthias Schlesner, Niels Weinhold, Rolf Sijmons, Mirjam De Jong, Brian Durie, and et al. 2023. "Investigation of Rare Non-Coding Variants in Familial Multiple Myeloma" Cells 12, no. 1: 96. https://doi.org/10.3390/cells12010096