
The Generation of Human iPSC Lines from Three Individuals with Dravet Syndrome and Characterization of Neural Differentiation Markers in iPSC-Derived Ventral Forebrain Organoid Model

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reprogramming
2.2. Sanger Sequencing
2.3. RNA Isolation, Quantitative Real-Time PCR (RT-PCR)
2.4. Immunofluorescence
2.5. Differentiation into Three Germ Lineages
2.6. Generation of Ventral Forebrain Organoids
2.7. Karyotyping
2.8. Mycoplasma
2.9. Ethical Approval
2.10. Statistical Analysis
3. Results
3.1. Isolation of Human Dermal Fibroblasts and Generation of iPSC Lines
3.2. Classification and Validation of iPSC Lines Generated from DRVT/PS Donors
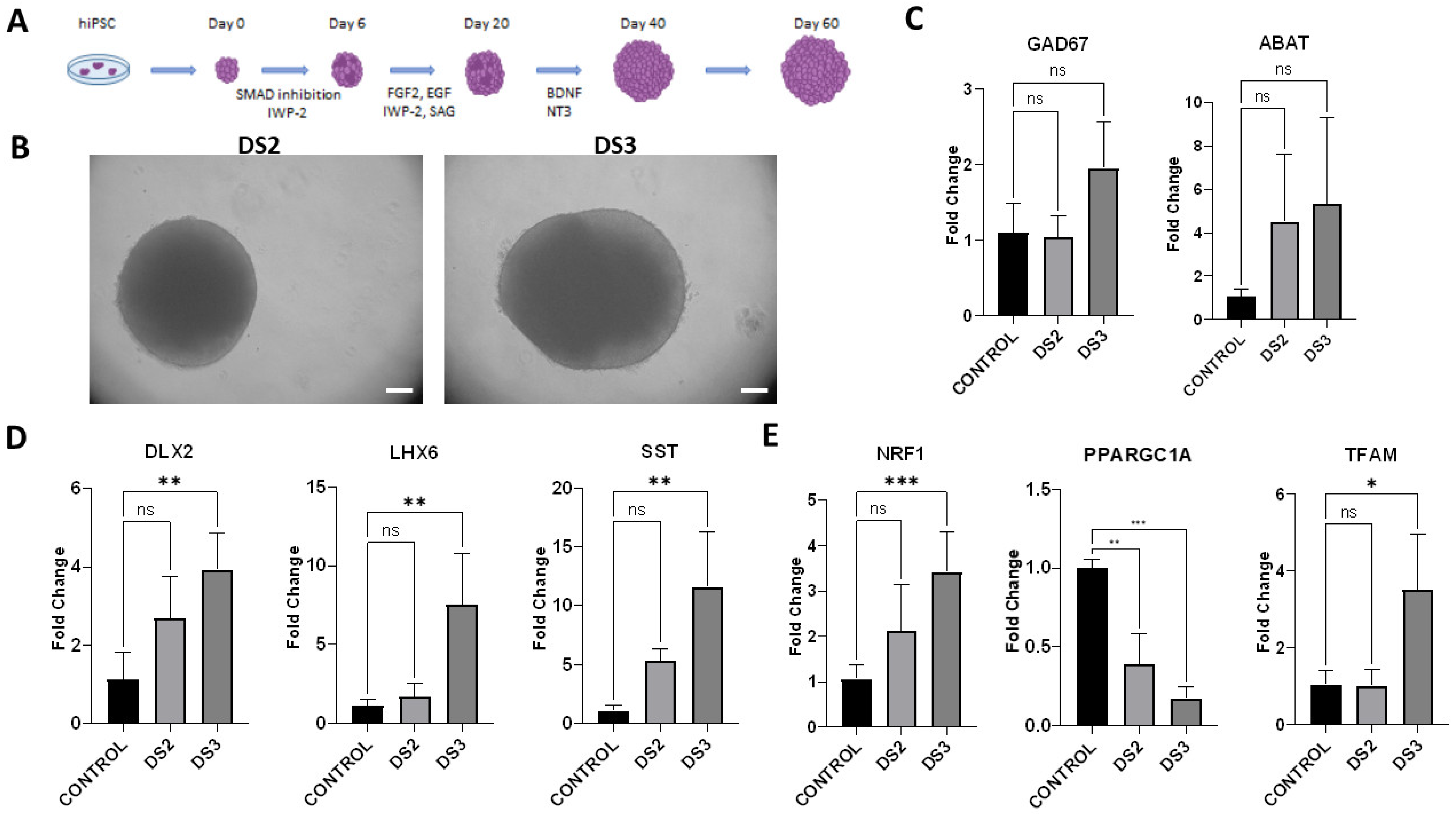
3.3. Generation of Ventral Forebrain Organoids
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Scheffer, I.E.; Liao, J. Deciphering the Concepts behind “Epileptic Encephalopathy” and “Developmental and Epileptic Encephalopathy”. Eur. J. Paediatr. Neurol. EJPN Off. J. Eur. Paediatr. Neurol. Soc. 2020, 24, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Brunklaus, A.; Brünger, T.; Feng, T.; Fons, C.; Lehikoinen, A.; Panagiotakaki, E.; Vintan, M.-A.; Symonds, J.; Andrew, J.; Arzimanoglou, A.; et al. The Gain of Function SCN1A Disorder Spectrum: Novel Epilepsy Phenotypes and Therapeutic Implications. Brain J. Neurol. 2022, 145, 3816–3831. [Google Scholar] [CrossRef] [PubMed]
- Wang, H. Modeling Neurological Diseases With Human Brain Organoids. Front. Synaptic Neurosci. 2018, 10, 15. [Google Scholar] [CrossRef]
- Ben Jehuda, R.; Shemer, Y.; Binah, O. Genome Editing in Induced Pluripotent Stem Cells Using CRISPR/Cas9. Stem Cell Rev. Rep. 2018, 14, 323–336. [Google Scholar] [CrossRef]
- Nieto-Estévez, V.; Hsieh, J. Human Brain Organoid Models of Developmental Epilepsies. Epilepsy Curr. 2020, 20, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Schuster, J.; Laan, L.; Klar, J.; Jin, Z.; Huss, M.; Korol, S.; Noraddin, F.H.; Sobol, M.; Birnir, B.; Dahl, N. Transcriptomes of Dravet Syndrome IPSC Derived GABAergic Cells Reveal Dysregulated Pathways for Chromatin Remodeling and Neurodevelopment. Neurobiol. Dis. 2019, 132, 104583. [Google Scholar] [CrossRef]
- Higurashi, N.; Uchida, T.; Lossin, C.; Misumi, Y.; Okada, Y.; Akamatsu, W.; Imaizumi, Y.; Zhang, B.; Nabeshima, K.; Mori, M.X.; et al. A Human Dravet Syndrome Model from Patient Induced Pluripotent Stem Cells. Mol. Brain 2013, 6, 19. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.H.; Mantegazza, M.; Westenbroek, R.E.; Robbins, C.A.; Kalume, F.; Burton, K.A.; Spain, W.J.; McKnight, G.S.; Scheuer, T.; Catterall, W.A. Reduced Sodium Current in GABAergic Interneurons in a Mouse Model of Severe Myoclonic Epilepsy in Infancy. Nat. Neurosci. 2006, 9, 1142–1149. [Google Scholar] [CrossRef]
- Martin, M.S.; Dutt, K.; Papale, L.A.; Dubé, C.M.; Dutton, S.B.; de Haan, G.; Shankar, A.; Tufik, S.; Meisler, M.H.; Baram, T.Z.; et al. Altered Function of the SCN1A Voltage-Gated Sodium Channel Leads to γ-Aminobutyric Acid-Ergic (GABAergic) Interneuron Abnormalities*. J. Biol. Chem. 2010, 285, 9823–9834. [Google Scholar] [CrossRef] [Green Version]
- Scalise, S.; Scaramuzzino, L.; Lucchino, V.; Esposito, C.; Malatesta, P.; Grillone, K.; Perrotti, N.; Cuda, G.; Parrotta, E.I. Generation of IPSC Lines from Two Patients Affected by Febrile Seizure Due to Inherited Missense Mutation in SCN1A Gene. Stem Cell Res. 2020, 49, 102083. [Google Scholar] [CrossRef]
- Schuster, J.; Fatima, A.; Sobol, M.; Norradin, F.H.; Laan, L.; Dahl, N. Generation of Three Human Induced Pluripotent Stem Cell (IPSC) Lines from Three Patients with Dravet Syndrome Carrying Distinct SCN1A Gene Mutations. Stem Cell Res. 2019, 39, 101523. [Google Scholar] [CrossRef]
- Kaminska, A.; Wedzinska, A.; Kot, M.; Sarnowska, A. Effect of Long-Term 3D Spheroid Culture on WJ-MSC. Cells 2021, 10, 719. [Google Scholar] [CrossRef]
- Birey, F.; Andersen, J.; Makinson, C.D.; Islam, S.; Wei, W.; Huber, N.; Fan, H.C.; Metzler, K.R.C.; Panagiotakos, G.; Thom, N.; et al. Assembly of Functionally Integrated Human Forebrain Spheroids. Nature 2017, 545, 54–59. [Google Scholar] [CrossRef] [Green Version]
- Paşca, A.M.; Sloan, S.A.; Clarke, L.E.; Tian, Y.; Makinson, C.D.; Huber, N.; Kim, C.H.; Park, J.-Y.; O’Rourke, N.A.; Nguyen, K.D.; et al. Functional Cortical Neurons and Astrocytes from Human Pluripotent Stem Cells in 3D Culture. Nat. Methods 2015, 12, 671–678. [Google Scholar] [CrossRef] [Green Version]
- Sloan, S.A.; Andersen, J.; Pașca, A.M.; Birey, F.; Pașca, S.P. Generation and Assembly of Human Brain Region–Specific Three-Dimensional Cultures. Nat. Protoc. 2018, 13, 2062–2085. [Google Scholar] [CrossRef]
- Sobol, M.; Raykova, D.; Cavelier, L.; Khalfallah, A.; Schuster, J.; Dahl, N. Methods of Reprogramming to Induced Pluripotent Stem Cell Associated with Chromosomal Integrity and Delineation of a Chromosome 5q Candidate Region for Growth Advantage. Stem Cells Dev. 2015, 24, 2032–2040. [Google Scholar] [CrossRef]
- Harkin, L.A.; McMahon, J.M.; Iona, X.; Dibbens, L.; Pelekanos, J.T.; Zuberi, S.M.; Sadleir, L.G.; Andermann, E.; Gill, D.; Farrell, K.; et al. The Spectrum of SCN1A-Related Infantile Epileptic Encephalopathies. Brain J. Neurol. 2007, 130, 843–852. [Google Scholar] [CrossRef]
- Hoffman-Zacharska, D.; Szczepanik, E.; Terczynska, I.; Goszczanska-Ciuchta, A.; Zalewska-Miszkurka, Z.; Tataj, R.; Bal, J. From Focal Epilepsy to Dravet Syndrome--Heterogeneity of the Phenotype Due to SCN1A Mutations of the p.Arg1596 Amino Acid Residue in the Nav1.1 Subunit. Neurol. Neurochir. Pol. 2015, 49, 258–266. [Google Scholar] [CrossRef]
- Kim, H.W.; Quan, Z.; Kim, Y.-B.; Cheong, E.; Kim, H.D.; Cho, M.; Jang, J.; Yoo, Y.R.; Lee, J.S.; Kim, J.H.; et al. Differential Effects on Sodium Current Impairments by Distinct SCN1A Mutations in GABAergic Neurons Derived from Dravet Syndrome Patients. Brain Dev. 2018, 40, 287–298. [Google Scholar] [CrossRef]
- Xie, Y.; Ng, N.N.; Safrina, O.S.; Ramos, C.M.; Ess, K.C.; Schwartz, P.H.; Smith, M.A.; O’Dowd, D.K. Comparisons of Dual Isogenic Human IPSC Pairs Identify Functional Alterations Directly Caused by an Epilepsy Associated SCN1A Mutation. Neurobiol. Dis. 2020, 134, 104627. [Google Scholar] [CrossRef]
- Zayat, V.; Szlendak, R.; Hoffman-Zacharska, D. Concise Review: Stem Cell Models of SCN1A-Related Encephalopathies—Current Perspective and Future Therapies. Cells 2022, 11, 3119. [Google Scholar] [CrossRef] [PubMed]
- Tai, C.; Abe, Y.; Westenbroek, R.E.; Scheuer, T.; Catterall, W.A. Impaired Excitability of Somatostatin- and Parvalbumin-Expressing Cortical Interneurons in a Mouse Model of Dravet Syndrome. Proc. Natl. Acad. Sci. USA 2014, 111, E3139–E3148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheah, C.S.; Yu, F.H.; Westenbroek, R.E.; Kalume, F.K.; Oakley, J.C.; Potter, G.B.; Rubenstein, J.L.; Catterall, W.A. Specific Deletion of NaV1.1 Sodium Channels in Inhibitory Interneurons Causes Seizures and Premature Death in a Mouse Model of Dravet Syndrome. Proc. Natl. Acad. Sci. USA 2012, 109, 14646–14651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marini, C.; Scheffer, I.E.; Nabbout, R.; Suls, A.; De Jonghe, P.; Zara, F.; Guerrini, R. The Genetics of Dravet Syndrome. Epilepsia 2011, 52 (Suppl. 2), 24–29. [Google Scholar] [CrossRef]
- Kumar, M.G.; Rowley, S.; Fulton, R.; Dinday, M.T.; Baraban, S.C.; Patel, M. Altered Glycolysis and Mitochondrial Respiration in a Zebrafish Model of Dravet Syndrome. eNeuro 2016, 3, ENEURO.0008-16.2016. [Google Scholar] [CrossRef] [Green Version]
- Craig, A.K.; de Menezes, M.S.; Saneto, R.P. Dravet Syndrome: Patients with Co-Morbid SCN1A Gene Mutations and Mitochondrial Electron Transport Chain Defects. Seizure 2012, 21, 17–20. [Google Scholar] [CrossRef] [Green Version]
- McMeekin, L.J.; Fox, S.N.; Boas, S.M.; Cowell, R.M. Dysregulation of PGC-1α-Dependent Transcriptional Programs in Neurological and Developmental Disorders: Therapeutic Challenges and Opportunities. Cells 2021, 10, 352. [Google Scholar] [CrossRef]
- Kuczynska, Z.; Metin, E.; Liput, M.; Buzanska, L. Covering the Role of PGC-1α in the Nervous System. Cells 2021, 11, 111. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| RT-PCR and Mutation Validation Primers | Target | Forward/Reverse Sequences (5′-3′) |
|---|---|---|
| Pluripotency markers | OCT4 [12] | F: CCTGAAGCAGAAGAGGATCACC |
| R: AAAGCGGCAGATGGTCGTTTGG | ||
| Pluripotency markers | NANOG [12] | F: GAACCTCAGCTACAAACAGG |
| R: CGTCACACCATTGCTATTCT | ||
| Pluripotency markers | SOX2 [12] | F: GTGGAAACTTTTGTCGGAGA |
| R: TTATAATCCGGGTGCTCCTT | ||
| GABAergic neuronal markers | GAD67 1 | F: TAGCGAGAACGAGGAAGCAG |
| R: GCAGAAACAGGCTCGGCT | ||
| GABAergic neuronal markers | ABAT [13] | F: TGAAATACCCTCTGGAAGAG |
| R: CAATCAGATCCTCCACCTC | ||
| Ventral forebrain interneuronal markers | DLX2 [13] | F: ACGTCCCTTACTCCGCCAAG |
| R: AGTAGATGGTGCGGGGTTTCC | ||
| Ventral forebrain interneuronal markers | LHX6 [13] | F: CCGTCTGCAGGCAAGAACAT |
| R: GACACACGGAGCACTCGAG | ||
| Ventral forebrain interneuronal markers | SST 1 | F: AGTTTGACCAGCCACTCTCC |
| R: GTACTTGGCCAGTTCCTGCTT | ||
| Mitochondrial biogenesis markers | NRF1 1 | F: CACTTATCCAGGTTGGTACG |
| R: CAGCCACGGCAGAATAAT | ||
| Mitochondrial biogenesis markers | PPARGC1A 1 | F: CCCAGAGTCACCAAATGAC |
| R: TCCAGAGAGTTCCACACTTA | ||
| Mitochondrial biogenesis markers | TFAM 1 | F: CTCAGAACCCAGATGCAAA |
| R: GCCACTCCGCCCTATAA | ||
| Housekeeping genes | GAPDH [14] | F: GAACGGGAAGCTTGTCATCAA |
| R: ATCGCCCCACTTGATTTTGG | ||
| Mutation analysis/sequencing | SCN1A | F: TACTTCGCGTTTCCACAAGG |
| c.2589+2dupT | R: GCTATGCAAGAACCCTGATTG | |
| SCN1A | F: CAAAAATCAGGGCCAATGAC | |
| p.Arg1596His | R: TGATTGCTGGGATGATCTTG | |
| SCN1A | F: GAGATTTGGGGGTGTTTGTC | |
| p.Ser1516* | R: GGATTGTAATGGGGTGCTTC |
| Immunocytochemical Markers | Antibody | Dilution | Source |
|---|---|---|---|
| Pluripotency markers | Mouse anti- | 0.1111111 | Santa Cruz Biotechnology, Dallas, TX, USA, Cat# sc-5279, RRID: AB_628051 |
| OCT3/4 | Cell Signaling Technology, Danvers, MA, USA, Cat# D73G4), RRID: AB_10559205 | ||
| Pluripotency markers | Rabbit anti- | 0.1111111 | Thermo Fisher Scientific, Waltham, MA USA, Cat# MA1-014, RRID: AB_2536667 |
| NANOG | Millipore, Burlington, MA, USA, Cat# MAB4304, RRID: AB_177629 | ||
| Pluripotency markers | Mouse anti- | 0.1111111 | R&D Systems, Minneapolis, MN, USA, Cat# NL557, RRID: AB 2157172 |
| SOX2 | R&D Systems, Minneapolis, MN, USA, Cat# NL493, RRID: AB_2239879 | ||
| Pluripotency markers | Mouse anti- | 0.1805556 | R&D Systems, Minneapolis, MN, USA, Cat# NL557, RRID: AB_2303012 |
| SSEA4 | R&D Systems, Minneapolis, MN, USA, Cat# NL637, RRID: AB_2115853 | ||
| Differentiation markers | Goat anti- | 01:10 | R&D Systems, Minneapolis, MN, USA, Cat# NL493, RRID: AB_1964595 |
| Otx2 | R&D Systems, Minneapolis, MN, USA, Cat# NL637, RRID: AB_2195645 | ||
| Differentiation markers | Goat anti- | 01:10 | Millipore, Burlington, MA, USA, Cat# AB9260, RRID: AB_2142366 |
| SOX1 | Santa Cruz, Biotechnology, Dallas, TX, USA, Cat# sc-32299, RRID: AB_2033967 | ||
| Differentiation markers | Goat anti- | 01:10 | Thermo Fisher Scientific, Waltham, MA USA, Cat# 14-6494-82, RRID: AB_2848274 |
| Brachyury | Thermo Fisher Scientific, Waltham, MA USA, Cat# A-21131, RRID: AB_2535771 | ||
| Differentiation markers | Goat anti- | 01:10 | Thermo Fisher Scientific, Waltham, MA USA, Cat# A-21141, RRID: AB_2535778 |
| HAND1 | Thermo Fisher Scientific, Waltham, MA USA, Cat# A-21151, RRID: AB_2535784 | ||
| Differentiation markers | Goat anti- | 01:10 | Thermo Fisher Scientific, Waltham, MA USA, Cat# A-11035, RRID: AB_2534093 |
| GATA-4 | Thermo Fisher Scientific, Waltham, MA USA, Cat# A-21123, RRID: AB_2535765 | ||
| Differentiation markers | Goat anti- | 01:10 | |
| SOX17 | |||
| Proliferation marker | Rabbit anti- | 0.7361111 | |
| Ki-67 | |||
| Mesenchymal marker | Mouse anti- | 0.1805556 | |
| CD73 | |||
| Sendai virus marker | Mouse anti- | 0.7361111 | |
| Sendai virus | |||
| Secondary antibody | Goat anti- | 0.3888889 | |
| Mouse IgG2a | |||
| Secondary antibody | (Alexa Fluor®® 488) | 0.3888889 | |
| Goat anti-Mouse IgG2b | |||
| Secondary antibody | (Alexa Fluor®® 488) | 0.3888889 | |
| Goat anti-Mouse IgG3 | |||
| Secondary antibody | (Alexa Fluor®® 488) | 0.3888889 | |
| Goat anti-Rabbit IgG H+L (Alexa Fluor®® 546) | |||
| Secondary antibody | Goat anti-Mouse IgG1 | 0.3888889 | |
| (Alexa Fluor®® 546) |
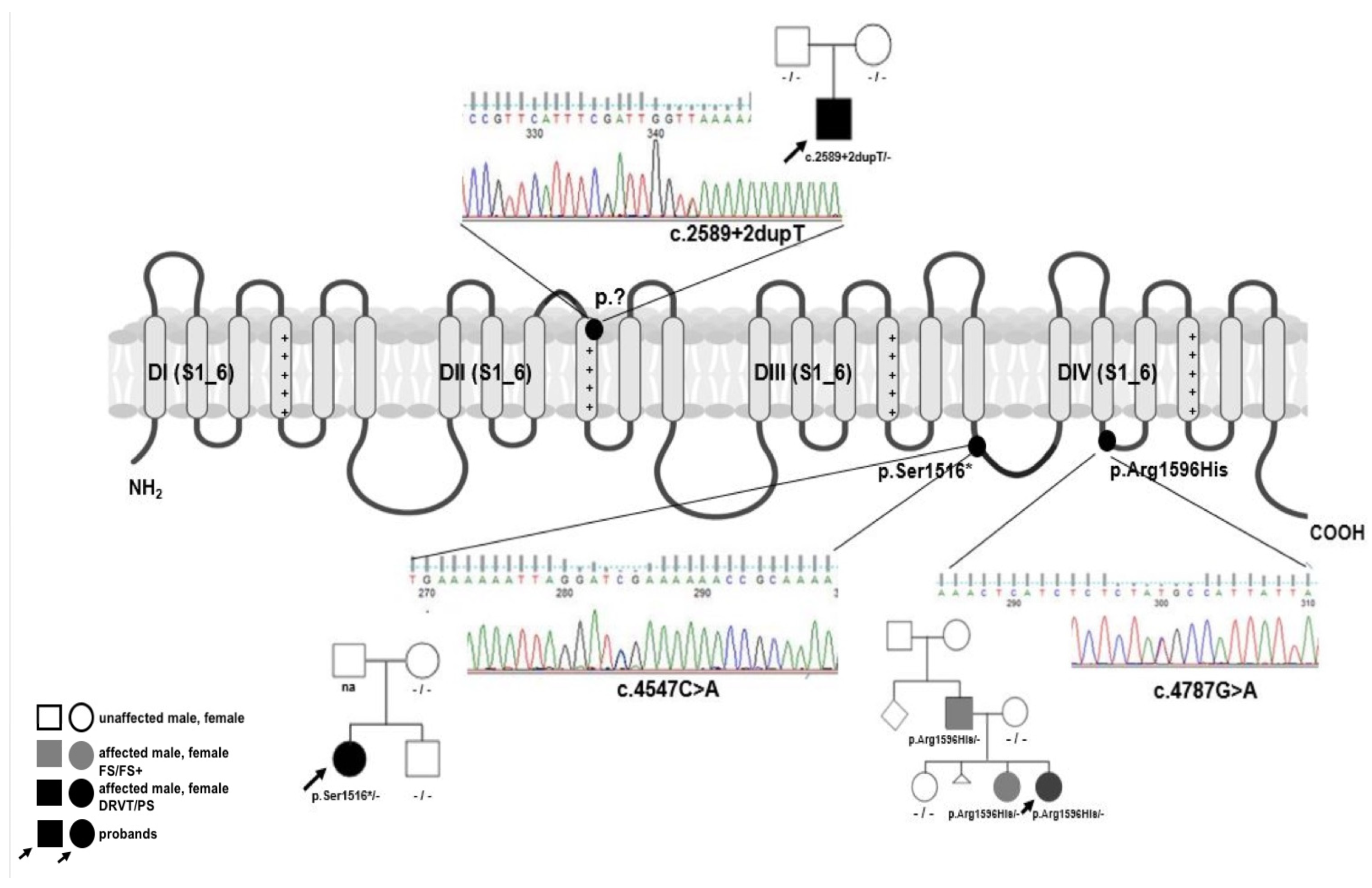
| iPSC Line | SCN1A Mutation (NM_001165963.4) | Disease | Gender | Ethnic Background |
|---|---|---|---|---|
| DS1 | c.2589+2dupT | Dravet syndrome | Female | Caucasian |
| DS2 | p.Arg1596His | FS/FS+/Panayiotopoulos syndrome | Female | Caucasian |
| DS3 | p.Ser1516* | Dravet syndrome | Male | Caucasian |
| Classification | Test | Results | Data |
|---|---|---|---|
| Mutation analysis | Sequencing | DS1: confirmation of splicing c.2589+2dupT/p.? mutation in the SCN1A gene; | Figure 1 |
| DS2: confirmation of missense c.4787G>A/p.Arg1596His mutation in the SCN1A gene; | |||
| DS3: confirmation of nonsense c.4547C>A/p.Ser156* mutation in the SCN1A gene | |||
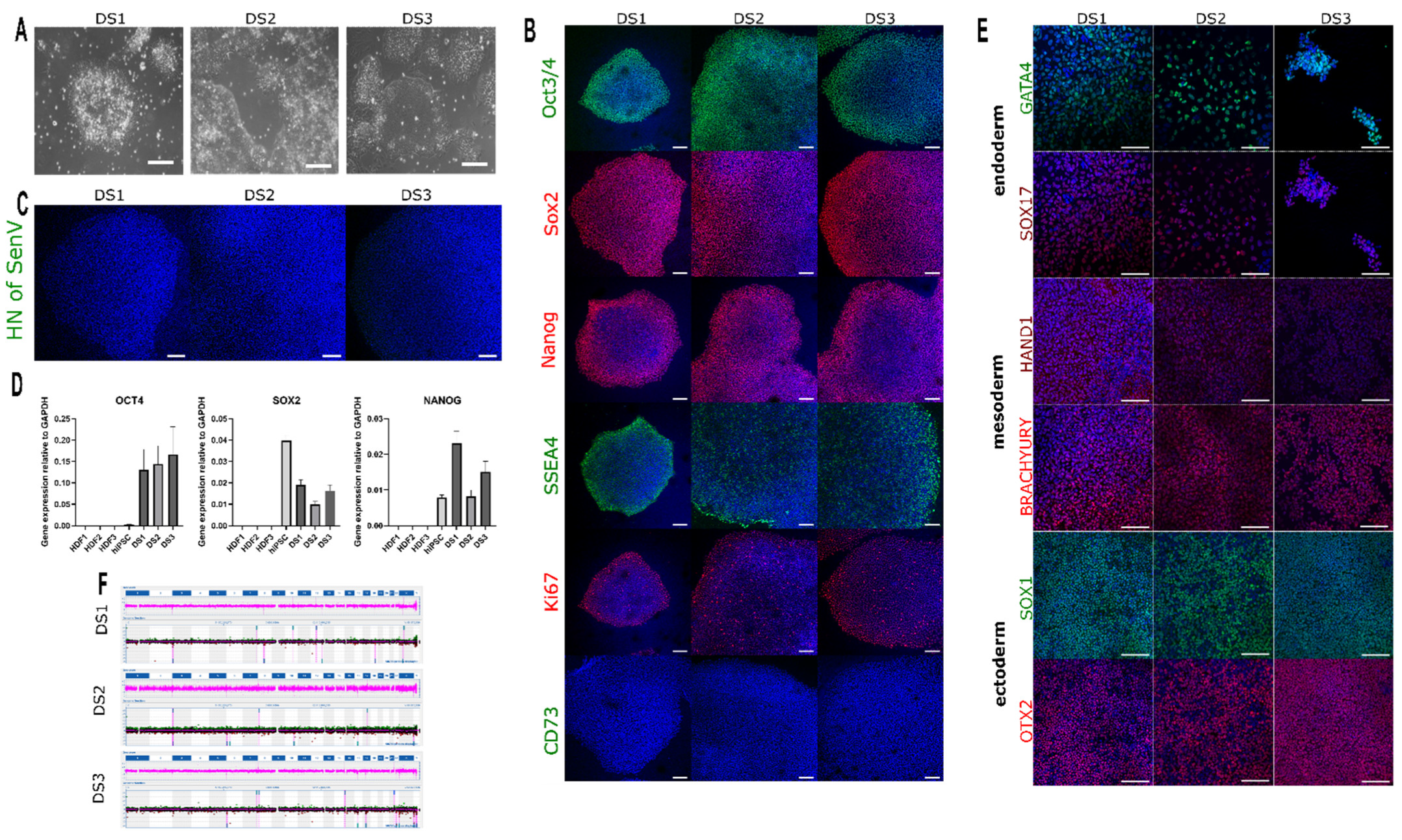
| Morphology | Microscope | All iPSC lines expressed the pluripotency markers: OCT4, SOX2, NANOG, and SSEA4; no presence of Sendai virus. Scale bars: 100 μm | Figure 2A |
| Phenotype | Immunocytochemistry | All iPSC lines have normal morphology. Scale bars: 100 μm | Figure 2B,C |
| RT-PCR | All iPSC lines expressed high levels of pluripotency markers such as OCT4, SOX2, and NANOG comparable to hiPSC positive control, whereas HDFs of origin showed no expression | Figure 2D | |
| Differentiation potential | Trilineage | All iPSC lines differentiated into the three germ layers including ectoderm (Otx2, SOX1), mesoderm (Brachyury, HAND1), and endoderm (GATA4, SOX-17). Scale bars: 100 μm | Figure 2E |
| differentiation | |||
| Genotype | Karyotype (G-banding) | DS1: 46, XX; | Figure 2F |
| DS2: 46, XX; | |||
| DS3: 46, XY | |||
| Microbiology/Virology | Mycoplasma testing | Negative |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zayat, V.; Kuczynska, Z.; Liput, M.; Metin, E.; Rzonca-Niewczas, S.; Smyk, M.; Mazurczak, T.; Goszczanska-Ciuchta, A.; Leszczynski, P.; Hoffman-Zacharska, D.; et al. The Generation of Human iPSC Lines from Three Individuals with Dravet Syndrome and Characterization of Neural Differentiation Markers in iPSC-Derived Ventral Forebrain Organoid Model. Cells 2023, 12, 339. https://doi.org/10.3390/cells12020339
Zayat V, Kuczynska Z, Liput M, Metin E, Rzonca-Niewczas S, Smyk M, Mazurczak T, Goszczanska-Ciuchta A, Leszczynski P, Hoffman-Zacharska D, et al. The Generation of Human iPSC Lines from Three Individuals with Dravet Syndrome and Characterization of Neural Differentiation Markers in iPSC-Derived Ventral Forebrain Organoid Model. Cells. 2023; 12(2):339. https://doi.org/10.3390/cells12020339
Chicago/Turabian StyleZayat, Valery, Zuzanna Kuczynska, Michal Liput, Erkan Metin, Sylwia Rzonca-Niewczas, Marta Smyk, Tomasz Mazurczak, Alicja Goszczanska-Ciuchta, Pawel Leszczynski, Dorota Hoffman-Zacharska, and et al. 2023. "The Generation of Human iPSC Lines from Three Individuals with Dravet Syndrome and Characterization of Neural Differentiation Markers in iPSC-Derived Ventral Forebrain Organoid Model" Cells 12, no. 2: 339. https://doi.org/10.3390/cells12020339