1. Introduction
Up-regulation of the mechanistic Target of Rapamycin (mTOR) in glioblastoma multiforme (GBM) is established in human patients and experimental models [
1,
2,
3,
4,
5]. An excess of mTOR activity leads to detrimental effects by acting on a number of intracellular pathways [
6,
7,
8,
9,
10]. Among these, suppression of autophagy and impairment of mitochondrial turn-over prevail at large [
11]. In fact, the loss of autophagy activity correlates with disease severity, tumor relapse, and resistance to therapy, while it stimulates the shift towards a stem cell phenotype. All these effects are responsible for a poor prognosis in GBM patients. In contrast, when autophagy is re-established via mTOR inhibition, an improvement of mitochondrial status and differentiated cell phenotype occur in GBM cells [
6,
7,
12]. Thus, suppression of the autophagy pathway is sufficient to produce cell pathology induced by mTOR overexpression. In fact, when autophagy is stimulated cell pathology is greatly reduced [
6]. Despite its prominent role, autophagy suppression implicates a number of downstream phenomena. Thus, the role played by autophagy in the biology of GBM cells is multifaceted. The role of autophagy in the clearance of misfolded proteins may extend to clear the cells from proteins which are relevant for cancerogenesis. In fact, in previous manuscripts we described that autophagy promotes the suppression of cell proliferation [
6], it occludes the expression of stem cell markers such as nestin [
7] and clears GBM cells from misfolded proteins [
13,
14].
A bulk of proteins owing influence on neighboring or distant cells may be overproduced or under-metabolized during autophagy suppression. This is the case of the cellular prion protein (PrPc). In line with this, recent evidence indicates that PrPc plays a key role in promoting GBM malignancy [
15,
16]. It is remarkable that, in prion disorders, elevated PrPc was shown to induce a defect in mitochondrial removal. This is likely to be caused by PrPc-mediated suppression of PTEN-induced putative kinase1 (Pink1)/Parkin (PARK) proteins at mitochondrial level [
17]. Thus, one may hypothesize that an impairment of the autophagy machinery may induce an increase in PrPc [
18], which in turn suppresses mitochondria turn-over, thus altering the mitochondrial compartment. This would lead to a vicious circle, since mitochondrial aberrations promote cell pathology in GBM [
12,
19].
Therefore, the present research study is designed to assess whether activation of the autophagy flux following mTOR inhibition leads to a significant clearance of PrPc along with removal of altered mitochondria concomitantly with persistent inhibition of cell proliferation.
In detail, the present study investigates whether PrPc accumulation along with mitochondrial impairment may all be reverted along with the expression of specific proteins promoting mitochondrial turn-over, when the autophagy flux is rescued through mTOR inhibition. The present study investigates whether all these effects, following a brief exposure to the mTOR inhibitor rapamycin, are long-lasting when administered to different GBM cell lines. This includes a persistent removal of PrPc and a long-lasting correction of the aberrant mitochondrial status.
2. Materials and Methods
2.1. Experimental Design
Experiments were carried out in human U87MG and A172 GBM cell lines. U87MG cells were obtained from Cell Bank (IRCCS San Martino-Institute, Genova, Italy). U87MG cells were maintained in DMEM growth medium (Sigma-Aldrich, Saint Louis, MO, USA) containing 10% Fetal Bovine Serum (FBS, Sigma-Aldrich, Saint Louis, MO, USA), 1% of MEM Non-Essential Amino-Acid (MEM-NEAA, Sigma-Aldrich, Saint Louis, MO, USA), penicillin, and streptomycin (50 IU/mL and 100 μg respectively, Sigma-Aldrich, Saint Louis, MO, USA). A172 cells were obtained from the European Collection of Authenticated Cell Cultures (ECACC) and from Cell Bank (IRCCS San Martino-IST, Genova) and they were maintained in Modified Eagle’s Medium (Euroclone, Milan, Italy) supplemented with 10% FBS, 2 mM L-glutamine, 100 IU/mL penicillin and 100 μg streptomycin. Both GBM cell lines were kept at 37 °C in a humidified atmosphere containing 5% CO2.
Based on previous studies [
12,
19], we selected a protocol where rapamycin is administered for a short time interval (24 h) and measurement of autophagy flux, cell cycle, analysis of the mitochondria status (mitochondrial alterations, mitophagy and fission), and the tumorigenic protein, i.e., prion protein, are carried out at various time intervals following rapamycin withdrawal, ranging from 1 day up to 14 days for morphology and 4 days up 14 days for autophagy flux and cell cycle. The timing and dosing of rapamycin (10 nM) were selected based on pilot experiments and previous studies [
12,
19]. In detail, the dose of rapamycin (10 nM) was selected according to the average of its therapeutic range (from 3 nM up to15 nM [
20]). After rapamycin exposure (24 h), cell cultures were washed to remove rapamycin and kept in the culture medium, for 1 d, 4 d, 7 d, or 14 d. To keep cells alive for long time intervals (up to 14d), the culture medium was removed and replaced with fresh medium every three days.
The treatment solution of rapamycin (Sigma-Aldrich, Saint Louis, MO, USA) was prepared starting from a stock solution of 1 mM, which was dissolved in 1.41 M DMSO (Sigma-Aldrich, Saint Louis, MO, USA), and further diluted in the culture medium before being administered to the cell cultures. In this way, the final concentration of DMSO was 0.01%. Control cells were grown in the same culture medium containing 0.01% DMSO for the same time intervals and with the same washing procedure.
In experiments aimed at assessing the autophagy flux, the autophagy inhibitor bafilomycin A1 (100 nM, Sigma-Aldrich, Saint Louis, MO, USA) was added to the culture medium 3 h before the end of the treatments.
2.2. Light Microscopy for Concomitant MitoTracker Green and Immunohistochemistry for LC3 or BNIP3
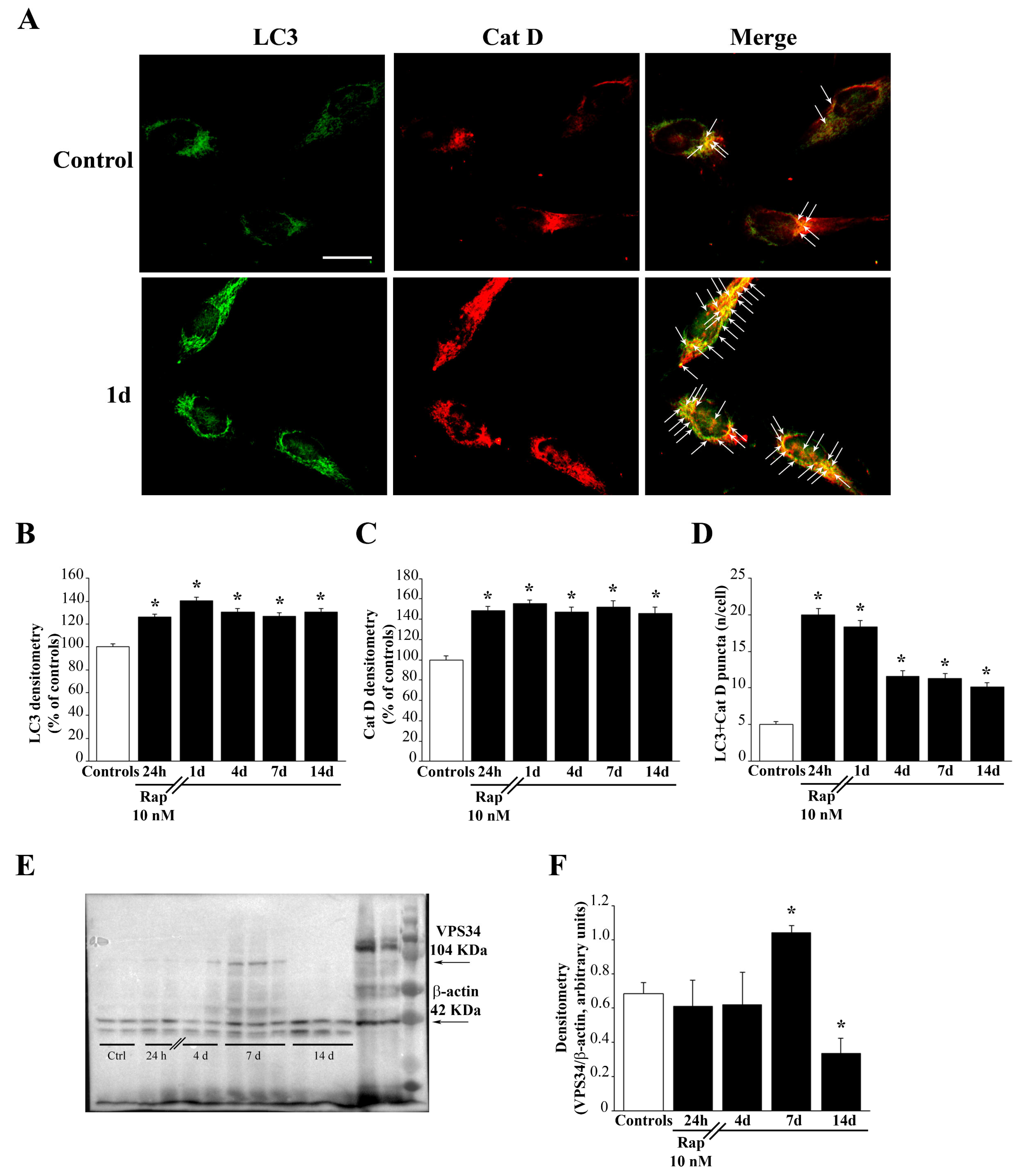
To label mitochondria in living cells, MitoTracker-Green (MTR-G) (Thermo-Fisher Scientific, Waltham, MA, USA) dye was used, which labels total mitochondria [
21,
22]. Briefly, 5 × 10
4 GBM cells were grown in 24-well plates containing 1 mL/well of culture medium. At the end of each experiment, the medium was removed, and cells were incubated in a serum free culture medium containing MTR-G at 500 nM for 45 min, at 37 °C and 5% CO
2. At the end of incubation, MTR-G solution was removed, and GBM cells were washed in fresh pre-warmed medium before being fixed in 4% paraformaldehyde for 10 min. After fixation, GBM cells were permeabilized with 0.1% Triton-X 100 (Sigma-Aldrich, Saint Louis, MO, USA) for 15 min, and then incubated in 10% normal goat serum in PBS at 21 °C for 1h, followed by incubation with primary antibodies. In detail, microtubule-associated protein I/II light chain 3 (LC3, Abcam, Cambridge, UK) or BCL2/adenovirus E1B interacting protein 3 (BNIP3, Thermo-Fisher Scientific, Waltham, MA, USA) primary antibodies were used. GBM cells were incubated with rabbit anti-LC3 (diluted 1:75) or rabbit anti-BNIP3 (diluted 1:50), at 4 °C overnight. Primary antibodies’ solutions were prepared in PBS containing 2% normal goat serum. After washing in PBS, cells were incubated at 21 °C in the dark for 90 min with fluorophore-conjugated secondary antibody Alexa Fluor 546 (anti-rabbit, 1:200, Life Technologies, Carlsabad, CA, USA) in PBS. Cells were observed under light microscope Nikon Eclipse 80i (Nikon, Tokyo, Japan) equipped with a fluorescent lamp and a digital camera connected to NIS Element Software for image analysis (Nikon, Tokyo, Japan). Negative control cells were incubated with secondary antibodies only. Stained pictures were acquired independently and then they were merged. The number of double MTR-G + LC3 or MTR-G+BNIP3 puncta per cell was counted. Values are given as the mean number ± SEM per cell from N = 100 cells/group. Optical density of BNIP3 fluorescent cells was measured using Image J software (NIH, Version 1.8.0_172, Bethesda, MD, USA). Values are given as the mean percentage ± SEM from N = 100 cells/group.
2.3. Immunohistochemistry
GBM cells (N = 5 × 104) were grown on poly-lysine slides placed in 24-well plates containing 1 mL/well of culture medium. At the end of the treatments, cells were washed in PBS and fixed with 4% paraformaldehyde in PBS for 15 min, incubated in 0.1% TritonX-100 for 15 min in PBS, and then blocked in PBS + 10% normal goat serum for 1h at 21 °C. Cells were then incubated overnight at 4 °C in 1% normal goat serum in PBS containing the primary antibodies (diluted 1:50) according to the following combinations: (i) anti-Phospho-S6 Ribosomal Protein (PS6RP, Cell Signaling, Milan, Italy); (ii) anti-Phospho p70 S6 Kinase (P70S6K) (Cell Signaling); (iii) anti-LC3 (Abcam) + Cathepsin D (Cat D) (Sigma-Aldrich, Saint Louis, MO, USA), (iv) anti-Pink1 (Abcam) + anti-PARK (Millipore, Burlington, MA, 808 USA); (v) anti-mitochondrial fission 1 protein (Fis1, GeneTex, Irvine, CA, USA) + anti-dynamin-related protein 1 (DRP1, Abcam) antibodies. After rinsing in PBS, GBM cells were incubated at 21 °C for 1h with the appropriate fluorophore-conjugated secondary antibodies (i.e., Alexa 488, Life Technologies, or Alexa 594 Life Technologies, Carlsabad, CA, USA) diluted 1:200. After washing in PBS, cells were transferred on coverslip and were mounted with the mounting medium Fluoroshield (Sigma-Aldrich, Saint Louis, MO, USA). GBM cells were observed under the Nikon Eclipse 80i light microscope (Nikon) equipped with a fluorescent lamp and a digital camera connected to the NIS Elements Software for image analysis (Nikon). Negative control cells were incubated with secondary antibodies only. Double stained pictures were acquired independently and then they were merged. Single fluorescent pictures were used to measure the optical density, using Image J software (NIH, Version 1.8.0_172, Version 1.8.0_172, Bethesda, MD, USA, Version 1.8.0_172). Values are given as the mean percentage ± SEM from N = 100 cells/group. Merged pictures were used to count the number of double fluorescent puncta per cell. Values are given as the mean number ± SEM per cell from N = 100 cells/group.
2.4. Confocal Microscopy
For confocal microscopy, 5 × 102 GBM cells were grown on poly-lysine slides placed in 24-well plates containing 1 mL/well of culture medium.
Cells were washed in PBS and fixed with methanol at 21 °C for 5 min, followed by incubation in 100 mM Tris-HCl, 5% urea at 95 °C for 10 min. Then, GBM cells were permeabilized in 0.2% Triton X-100 for 10 min and were blocked in PBS containing 0.1% Tween-20 (PBST), 1% bovine serum albumin (BSA), and 22.52 mg/mL of glycine for 30 min. Cells were then incubated in the primary antibodies’ solution overnight at 4 °C. Primary antibodies were diluted 1:50 in in PBST with 1% BSA. After washing in PBST, GBM cells were incubated at 21 °C for 1h with the appropriate fluorophore-conjugated secondary antibodies (i.e., Alexa 488, Life Technologies, or Alexa 594 Life Technologies, Carlsabad, CA, USA) diluted 1:200 in PBST with 1% BSA. After incubation with secondary antibody, GBM cells were washed in PBS, mounted in Prolong Diamond Antifade Mountant (Life Technologies, Carlsabad, CA, USA) and observed under a Leica TCSSP5 confocal laser-scanning microscope (Leica Microsystems, Wetzlar, Germany) using a sequential scan procedure. Confocal images were collected every 400 nm intervals through the z-axis by means of 63-oil lenses. Z-stacks of serial optical planes were analyzed using the Multicolor Packages (Leica Microsystems, Wetzlar, Germany). Negative control GBM cells were incubated with secondary antibodies only. The optical density of each single fluorescent antigen was measured using Image J software (NIH, Bethesda, MD, USA, Version 1.8.0_172) and values are given as the mean percentage ± SEM from N = 100 cells/group. The number of double fluorescent puncta per cell was counted and values are given as the mean number ± SEM per cell from N = 100 cells/group.
2.5. Transmission Electron Microscopy (TEM)
GBM cells (1 × 106) were seeded in 10 mm diameter culture dishes with 5 mL of culture medium. After removing culture medium, cells were fixed with the first fixing solution (2.0% paraformaldehyde/0.1% glutaraldehyde in 0.1M PBS pH 7.4) for 90 min at 4 °C. Then cells were gently scraped from the plate, centrifuged at 10,000× g rpm for 10 min and cell pellet was collected, washed in PBS, and fixed with the second fixing solution (1% osmium tetroxide) for 1 h at 4 °C. After washing, cell pellet was dehydrated in increasing ethanol solutions and embedded in epoxy resin.
Either plain electron microscopy or immuno-electron microscopy was carried out in ultra-thin sections, which were obtained at ultra-microtome (Leica Microsystems, Wetzlar, Germany) and were observed at Jeol JEM SX100 Transmission Electron Microscope (TEM, Jeol, Tokyo, Japan).
2.6. Ultra-Structural Analysis of Mitochondria
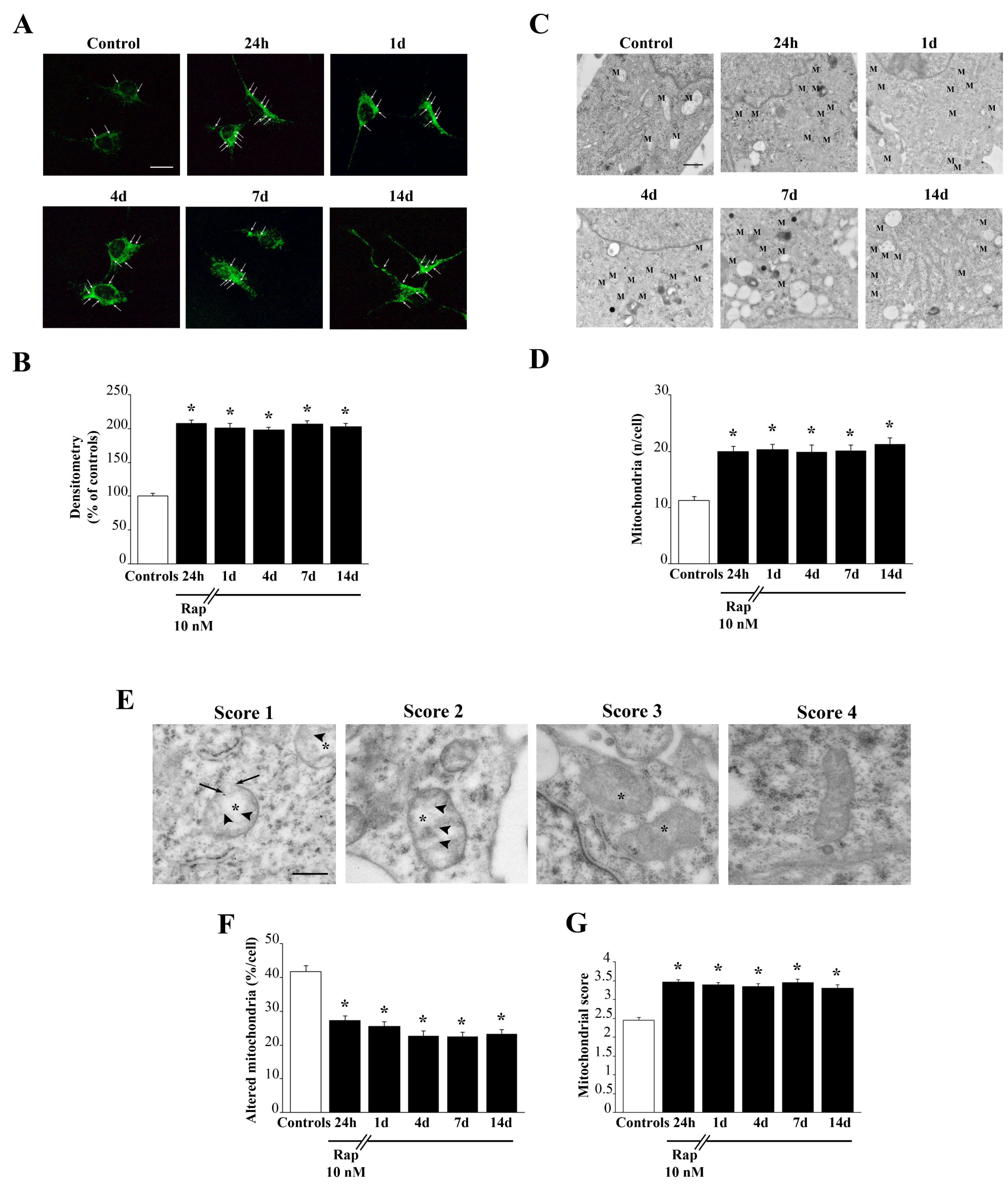
To analyze mitochondria status, non-serial ultra-thin sections (90 nm thick) were examined at TEM at 6000× magnification. Several grids were observed in order to analyse a total number of at least 50 cells for each experimental group. In detail, in order to examine the whole sectioned cell pellet, the grid was scanned in equally spaced parallel sweeps starting from a grid square corner.
Mitochondria were identified based on the presence of an inner and an outer membrane, an internal matrix, and a system of crests. Although the morphology of mitochondria may vary also in physiological conditions, altered mitochondria are defined according to ultrastructural criteria, which were validated in previous studies [
12,
19]. The number of mitochondrial alterations was used to provide a novel scoring system (modified from Flameng and coll. [
23]), which was extensively reported in the results section. For the purpose of clarity, it may be summarized here as follows: score 1, mitochondria with spread matrix dilution and membrane alterations concerning both crests and inner/outer membranes; score 2, mitochondria with spots of matrix dilution with some membrane alterations in the form of broken crests; score 3, mitochondria with intact membranes but owing spots of matrix dilution; score 4, intact mitochondria.
2.7. Post-Embedding Immuno-Electron Microscopy
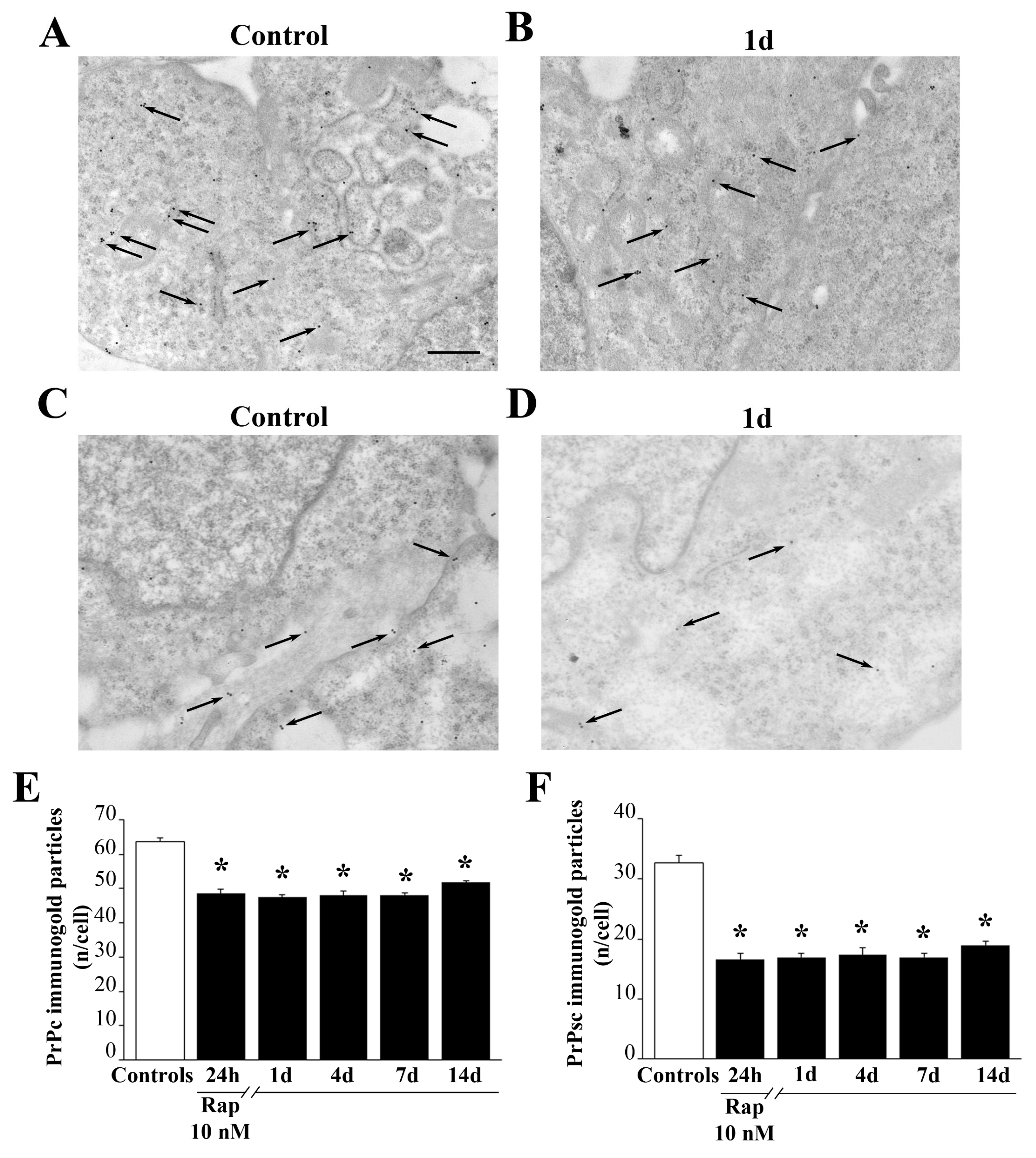
Ultra-thin sections were collected on nickel grids, they were de-osmicated in aqueous saturated solution of sodium metaperiodate (NaIO4) for 15 min, washed three times in ice cold filtered PBS, (pH 7.4) for 10 min, and then treated with ice-cold PBS containing 10% goat serum and 0.2% saponin to block non-specific antigens for 20 min at 21 °C.
Primary antibodies were incubated in ice-cold PBS containing 1% goat serum and 0.2% saponin in a humidified chamber overnight, at 4 °C. The following primary antibodies were used: anti-LC3 (1:50, Abcam); anti-Cat D (1:10, Sigma-Aldrich, Saint Louis, MO, USA); anti-Prion Protein (PrP, 1:10, Chemicon, Temecula CA, USA); anti-PARK (1:20, Millipore); anti-Pink1 (1:20, Abcam). Solutions containing two primary antibodies were used in order to detect co-localization of LC3 and Cat D proteins.
Pre-treatment of the sample with proteinase K (PK, Sigma-Aldrich, Saint Louis, MO, USA, 50 mg/mL) allows to digest all properly folded, non-aggregated proteins, including native PrPc. Based on our previous study [
24], PK tratment does not alter the ultrastructure of the cells. In this way, primary antibody against PrPc, when staining PK-resistant protein, documents the presence of scrapie-like prion protein (PrPsc-like). The treatment of the sample with PK was carried out with the specific purpose to carry out a quantitative ultrastructural stoichiometry in situ, which allowed optimizing detection and localization of either PrPc or PrPsc-like proteins within GBM cells. Therefore, sample grids were combined with drops of PK solution (50 mg/mL) in 0.1M PBS for 1 h at 37 °C. Proteinase K digests.
All primary antibodies were revealed through a solution containing gold-conjugated secondary antibodies (gold particle diameter, 10 nm or 20 nm, BB International, Cardiff UK) diluted 1:10, in PBS containing 1% goat serum and 0.2% saponin for 1 h, at room temperature. After rinsing in PBS, grids were incubated in 1% glutaraldehyde for 3 min, washed in distilled water, and further stained with uranyl acetate and lead citrate. Ultra-thin sections were finally observed at Jeol JEM SX100 TEM (Jeol). Control sections were incubated with secondary antibodies only.
2.8. Western Blotting
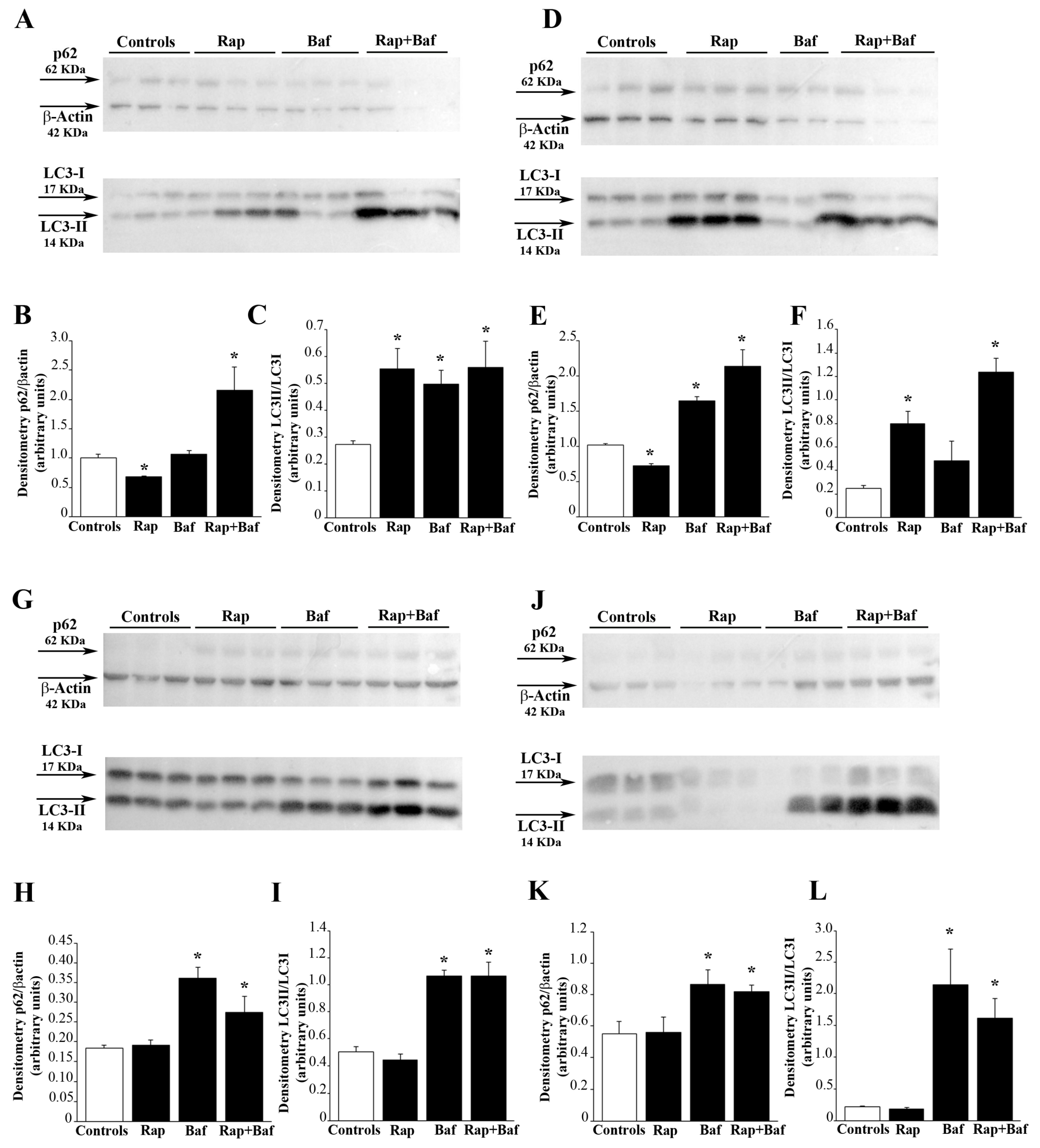
To analyze autophagy flux U87MG and A172 cells were treated with 10 nM rapamycin or vehicle (control) for 24 h immediately followed by cell culture washed out. Cells were lysed immediately after treatment or following 4, 7, or 14 days of rapamycin withdrawal. Bafilomycin A1 (100 nM; Sigma-Aldrich, Saint Louis, MO, USA) was added to the GBM cells alone or in combination with rapamycin during the last 3h before cell lysis. At the end of each time interval, cells were lysed for western blotting and electrophoretically resolved as previously reported [
25]. These proteins were electro-transferred onto PVDF membranes (BioRad Laboratories, Hercules, CA, USA) by a semi-dry system (BioRad Laboratories, Hercules, CA, USA). Membranes were blocked with 3% non-fat milk in PBS containing 0.1% Tween-20 (TBST) and then incubated (overnight at 4 °C) with the following antibodies: anti-LC3-I and LC3-II (MBL International, Woburn, MA, USA), anti-p62/SQSTM1 (Sigma-Aldrich, Saint Louis, MO, USA). After extensive washing with TBST, blots were incubated with a 1:3000 dilution of HRP-conjugated secondary antibody (Amersham Biosciences, Amersham, UK) for 1 h, at room temperature. Immunostained bands were detected with naked eye, and they were measured by using the classic non-quantitative, non-necessarily linear quantification through chemiluminescence (GE Healthcare Biosciences, Little Chalfont, Buckinghamshire, UK). Membranes were probed with the housekeeping mouse anti-β-actin (1:25,000, Sigma-Aldrich, Saint Louis, MO, USA). Densitometric analysis of p62/β-actin and LC3-II/LC3-I ratio was performed with ImageJ software (NIH, Bethesda, MD, USA, Version 1.8.0_172).
To analyze mTOR inhibition under the effect of rapamycin the downstream PS6RP was evaluated. Autophagy activation was evaluated by measuring the levels of the early autophagy stimulating protein VPS34. Finally, Fis1 and DRP1 were measured as markers of mitochondrial fission and mitophagy. U87MG cell pellet was placed in an Eppendorf tube containing 20 μL of ice-cold lysis buffer with phosphatase and protease inhibitors to be homogenized. An aliquot of the homogenate was used for Bradford protein assay. Proteins (20 μg) were separated on SDS-polyacrylamide gels (Mini Protean TGX precast gel 4–20% gradient, BioRad Laboratories, Hercules, CA, USA) on Trans-blot Turbo Transfer System Pack (for mixed molecular weight; 1.3 A-25 V-10 min). Membranes were blocked for 2 h in Tween-20 Tris-buffered saline (TTBS) (100 mM Tris-HCl, 0.9% NaCl, 1% Tween 20, pH 7.4) containing 5% non-fat dry milk (BioRad Laboratories, Hercules, CA, USA). We used the following primary antibodies: (i) anti-PS6RP (1:1000, Cell Signaling); (ii) anti-VPS34 (1:1000, Thermo-Fisher Scientific, Waltham, MA, USA); (iii) anti-Fis1 (1:1000, GeneTex); (iv) anti-DRP1 (1:2000, Abcam). Rabbit anti-β-actin (1:50,000; Sigma-Aldrich, Saint Louis, MO, USA) was used as an internal standard for semi-quantitative protein measurement.
Membranes were incubated overnight at 4 °C with primary antibodies diluted in TTBS containing 2.5% non-fat dry milk, and then they were washed in TTBS and incubated for 1 h with peroxidase-labeled secondary antibodies (anti-rabbit/anti-mouse, 1:3000; Calbiochem, Milan, Italy). Bands were visualized with enhanced chemiluminescence reagents (GE Healthcare, Milan, Italy). Image analysis was carried out by ChemiDoc System (BioRad Laboratories, Hercules, CA, USA). Optical density was normalized for relative β-actin using Image J software (NIH, Bethesda, MD, USA, Version 1.8.0_172).
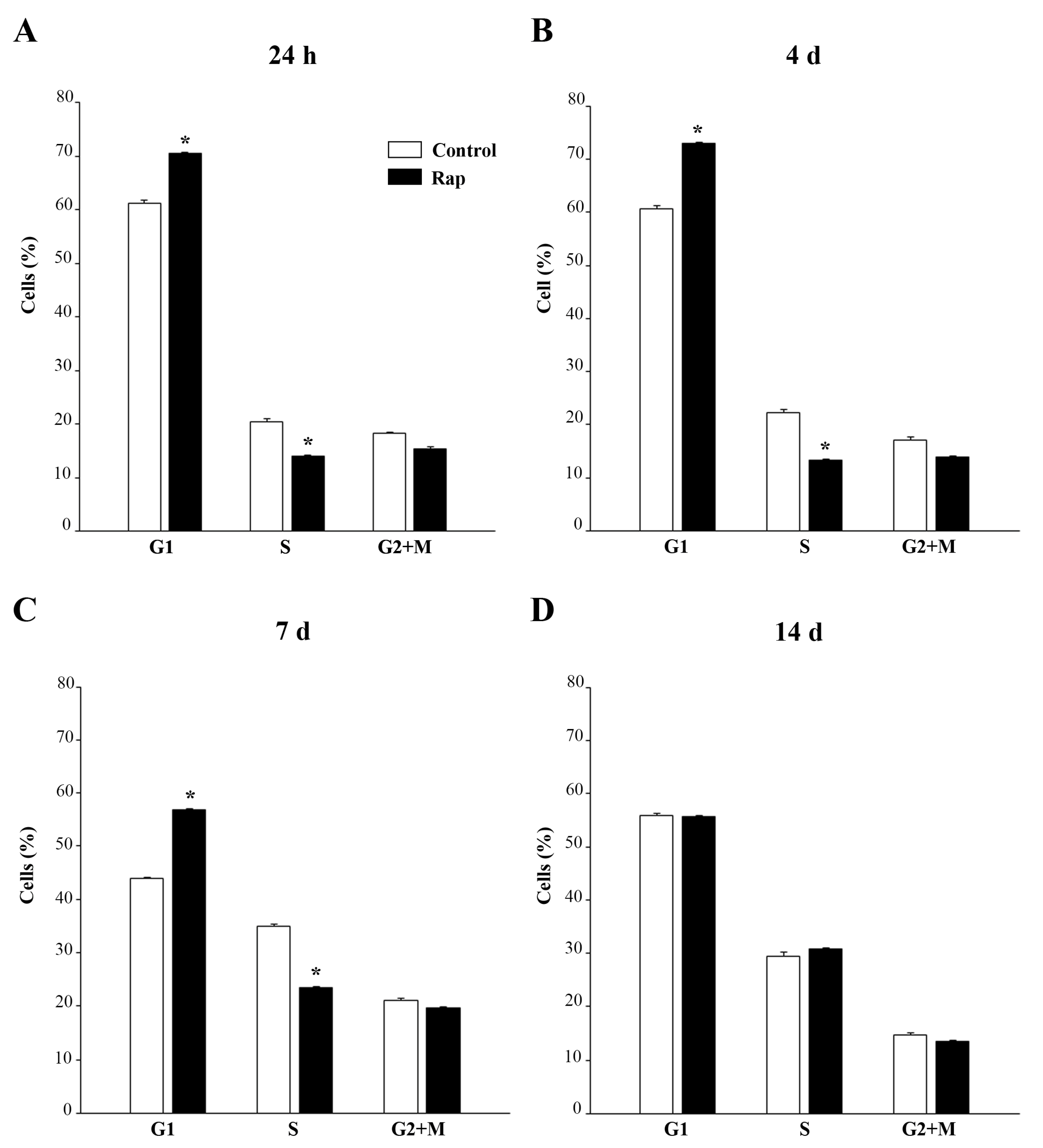
2.9. Cell Cycle Analysis
U87MG cells in logarithmic growth were treated with 10 nM rapamycin or vehicle. After 24 h a part of the sample was immediately prepared for flow cytometry, while the rest was maintained in culture for 4 days, 7 days, and 14 days without any further treatment. At the end of each incubation time, samples were fixed with 70% ethanol, stained with 20 μg/mL of propidium iodide and 100 μg/mL of RNase A, and then incubated at 37 °C for 30 min. Flow cytometric analysis was performed with appropriate gating on a FACScan (Becton Dickinson, Milan, Italy).
2.10. Statistical Analyses
The optical density of MTR-G histo-fluorescence and each protein immuno-fluorescence (PS6RP, LC3, Cat D, Pink1, PARK, BNIP3, Fis1 and DRP1) was given as mean percentage ± SEM per cell from N = 100 cells/group (assuming controls as 100%).
The merging of immuno-fluorescent puncta was counted and expressed as the mean ± SEM of puncta per cell in each experimental group (each group being representative of a specific dose and timing of rapamycin/vehicle saline) from N = 100 cells.
The optical density of immuno-blotting was given as mean ± SEM from 2 ≤ N ≤ 6 samples per experimental group.
Flow cytometry values were expressed as mean percentage ± SEM of cells in each cell cycle phase (G1, S, or G2+M).
For ultrastructural morphometry, values were expressed as following: (i) total number of mitochondria per cell; (ii) percentage of altered mitochondria per cell; (iii) number of Pink1- or PARK-positive mitochondria per cell; (iv) number of LC3- and/or Cat D-positive vacuoles per cell, (v) number of Pink1 or PARK immuno-gold particles per cell; (vi) number of Pink1 or PARK immuno-gold particles within mitochondria; (vii) the ratio between the number of Pink1 or PARK immuno-gold particles within mitochondria and the number of Pink1 or PARK immuno-gold particles within cytosol; (viii) number of PrPc or PrPsc-like immuno-gold particles per cell.
Values are given as the mean number or the mean percentage ± SEM per cell from 50 cells per group in all counts.
Mitochondrial score was calculated by averaging mitochondrial scores (as established from the first block of Results, in the Results section) from at least 150 mitochondria from each experimental group. Values are given as the mean ± SEM.
Data are compared using ANOVA with the Scheffe’s post-hoc test or Bonferroni’s corrected t test (for cell cycle data). Differences between the various groups are considered to be significant when the null hypothesis H0 is less than 5%.
4. Discussion
The present study, which is validate in different cell lines, indicates that, mTOR inhibition, which is evidenced by suppressed PS6RP and P70S6K expression, induced by rapamycin in the therapeutic range (10 nM, [
20]) produces a sudden and concomitant activation of the autophagy flux as measured by the classic assay with bafilomycin, the merging of LC3 with Cat D immunofluorescence, and the merging of autophagosomes with lysosomes by gold standard TEM. Most of these effects persist unmodified at least 14 days following rapamycin withdrawal and they are concomitant with a shift of a significant amount of GBM cells from S towards G1 phase, as measured by flow cytometry. These effects are long-lasting since they extend long beyond rapamycin withdrawal, for at least 14 days. Remarkably, this is concomitant with a marked suppression in the amount of the cellular prion protein, which is detected at molecular level considering the stoichiometry of both native PrPc and the protease resistant isotype PrPsc-like. These effects are tightened with an increase in the total number of mitochondria detected using both light (MTR-G fluorescence) and electron microscopy (direct count of the organelles). The increase in mitochondria occurs although the number of altered mitochondria and the number of mitochondrial alterations are steadily suppressed, at least for 14 days. This indicates a robust improvement in mitochondrial integrity. The autophagy-related increase in mitochondria is confirmed by the merging of LC3 with MTR-G, which increases following rapamycin administration. Similarly, the significance of increased LC3 is validated concerning the autophagy flux within different GBM cell lines, by using bafilomycin assay and the gold standard assessment of autophagy status at TEM. In fact, TEM also indicates that rapamycin stimulates the merging of autophagosomes with lysosomes, which is backed up by increased merging of LC3 with Cat D immuno-fluorescence. A marked effect of rapamycin in situ on mitochondrial proteins is confirmed by the merging of fluorescence for the mitophagy protein BNIP3 with MTR-G. This is further witnessed by AN increased amount and augmented merging of the mitophagy-related proteins Pink1 and PARK which is shown here at immunofluorescence and electron microscopy. This latter evidence confirms in GBM cells the effects of the prion proteins on suppressing Pink1/PARK interactions which were recently evidence in prion disorders [
17]. This is concomitant with an increase in the mitochondrial fission/mitophagy-related proteins Fis1 and DRP1, which persists at least for 14 days following rapamycin withdrawal. This is in line with the well-established concept that when promoting global autophagy, mitochondrial fission and mitophagy concomitantly increase [
26]. These data confirm a long-lasting effect following rapamycin withdrawal [
19] and indicate a powerful and persistent activation of autophagy and mitophagy under the effects of rapamycin. In detail, the long-lasting effects of rapamycin on the autophagy flux in GBM cells were not described previously. The stimulation of the autophagy flux, which is estimated by counting the amount of LC3-II and p62 with or without bafilomycin, persists during rapamycin withdrawal. This is concomitant with the merging of LC3 with Cat D immunofluorescence and the fusion of autophagosomes with lysosomes. The increase in proteins, which promote mitochondrial biogenesis explains the increase in healthy mitochondria reported here, which confirms previous recent data [
12]. In this recent report, evidence was provided that rapamycin increases the expression of those genes promoting mitochondrial biogenesis at 14 days following rapamycin withdrawal. In line with this, the present study indicates that rapamycin promotes a persistent clearance of PrPc, which takes place at least for 14 days following rapamycin withdrawal. Again, PrPc removal is consistent with enhanced autophagy flux, which efficiently clears such an aggregate prone protein mostly concerning its PK-resistant isoform.
Long-lasting improved mitochondrial turnover joined with persistent suppression of prion protein levels in GBM is likely to explain the shift of cell cycle from S towards G1 phase. Accordingly, a shift in the balance between mitochondrial fission and fusion can modulate the progression of the cell cycle [
27] and different phases of the cell cycle are associated with changes in mitochondrial status [
28]. In detail, the S to G1 transition is concomitant to increased mitochondrial fission [
28], while the opposite change from G1 to S phase is accompanied by increased mitochondrial fusion [
29].
In line with concomitant autophagy activation, the increase in DRP1 (fission protein) produces increased mitochondrial turnover with fragmentation of altered mitochondria [
30] and mitochondria biogenesis, which is evidenced by the presence of elongated mitochondria owing to densely packed crests, affecting the efficiency of ATP production [
31,
32,
33].
Early studies provided evidence that PrPc is highly expressed within human GBM cell lines [
34,
35], while recent evidence indicates that PrPc enhances the expression of a number of genes which are key in the onset and progression of GBM including a profound impact on mitochondrial turnover [
17]. The role of PrPc in GBM is remarkable and it is likely to operate upstream in controlling the mitochondrial status.
The dual effects of mTOR-dependent autophagy activation provided in the present study (mitochondria status and prion protein) in two GBM cell lines are indeed very much connected. In fact, the occurrence of mitochondrial alterations is dramatic in the presence of high levels of prion protein and abnormally high prion protein produces a defect in mitochondrial fission/mitophagy, as recently shown by Li and coll. [
17].
This is likely to explain why accumulation of altered mitochondria is considered a key factor in prion disease pathogenesis [
17]. Remarkably, this is specifically induced by a defect in Pink1/PARK-induced mitochondrial removal. In the present study we extended the evidence connecting PrPc and defective mitochondria to GBM cells. The study shows that such a mitochondrial defect can be corrected by reverting the Pink1/PARK defect. In fact, rapamycin improved mitochondrial status and increased Pink1/PARK levels, while occluding PrPc accumulation. This was concomitant with mTOR inhibition and autophagy activation. In fact, PARK overexpression was already shown to mitigate defective mitophagy when PrPc is overexpressed [
17]. Thus, an autophagy-dependent defect in Pink1/PARK-mediated mitophagy is induced by PrPc [
17].
The role of PrPc in the onset and progression of disease were already evidenced in recent studies as the effects of multiple mechanisms concerning the growth of various tumors, differentiation, and resistance to radio- and chemo- therapy [
15,
36,
37,
38,
39,
40,
41]. The occurrence of PrPc in GBM is more and more evident and it may contribute to cancer invasion and poor prognosis. It is remarkable that, according to Corsaro and coll. [
41], PrPc expression in GBM may be responsible for promoting stemness, which in turn relates to tumor aggression and relapse. In fact, within GBM glioblastoma cancer stem cells express the highest level of PrPc in the tumor. These cells form typical neurospheres and possess remarkable proliferation rate and a loss of differentiation [
15,
41,
42]. Conversely, suppression of PrPc expression produces a slower rate of proliferation along with cell differentiation and suppression of stem cell markers. All this evidence indicates that the expression of PrPc is key in the neurobiology of GBM and it seems to impact disease prognosis. The present study demonstrates the tighten relationship between autophagy, PrPc expression and mitochondrial status. The dual effects (mitochondrial status and PrPc expression) being scrutinized in the present research work using different GBM cell lines are likely to operate in symbiosis to transfer the impact of autophagy in the neurobiology of GBM. In fact, PrPc and mitochondria strongly impact cell proliferation. Thus, the autophagy dependent expression of proteins which promote mitochondrial fission is likely to strongly impact the course of GBM.

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






