
Canonical and Noncanonical ER Stress-Mediated Autophagy Is a Bite the Bullet in View of Cancer Therapy


Abstract
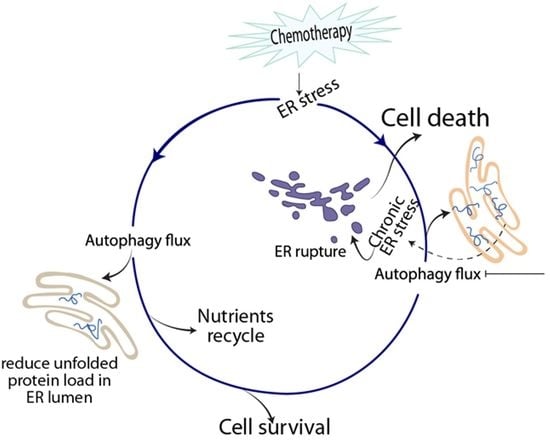
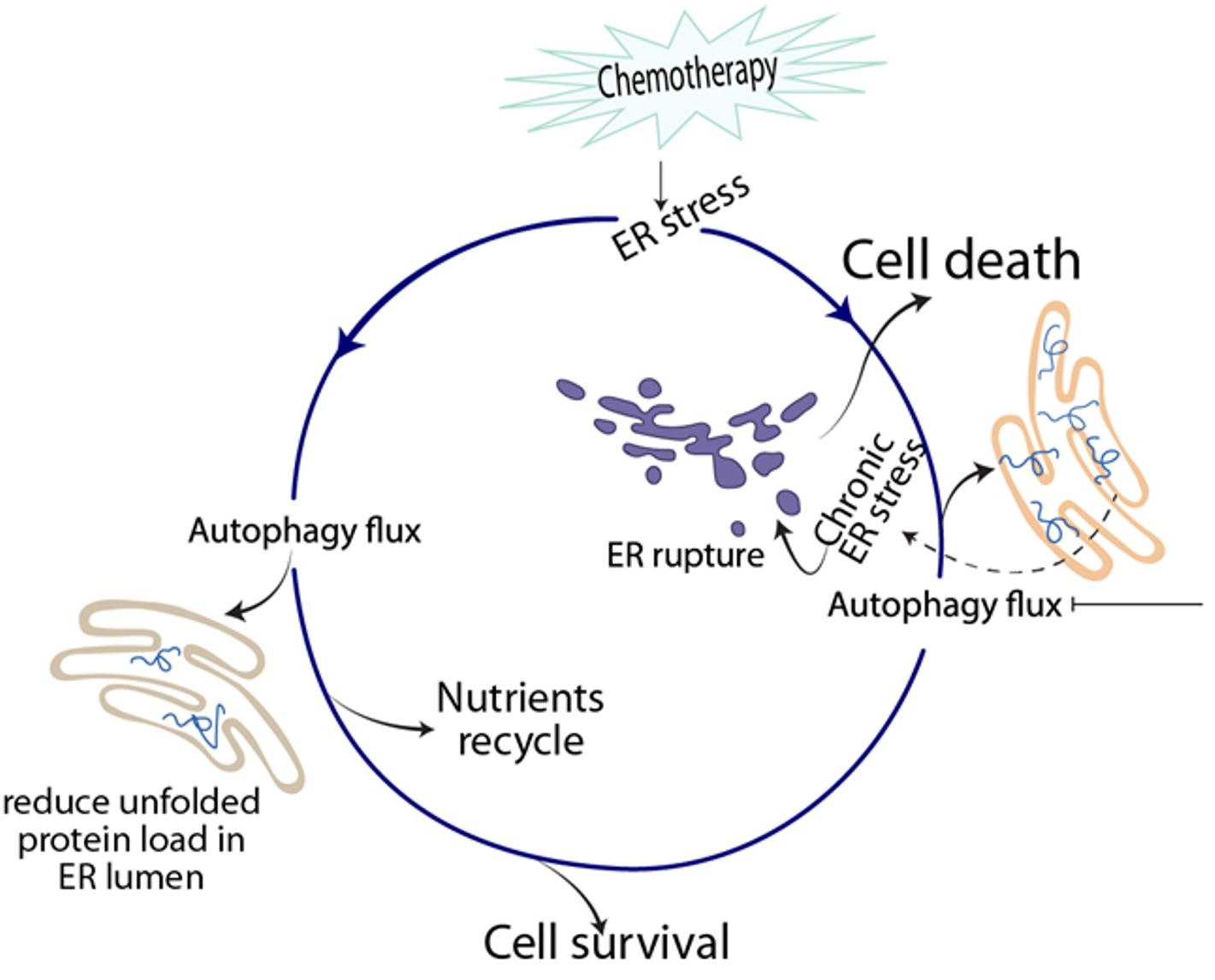
:
1. Introduction
2. Molecular Mechanism of Autophagy
3. ER Stress Assuage by Autophagy
4. Significance of Molecular Mechanisms Regulating ER-Phagy in Selection of Therapeutic Targets
5. Autophagic By-Product Effect on Cellular Stress Tolerance
6. Canonical Pathway Regulates ER-Stress Response
The Interplay between Canonical ER Stress and Autophagy in Cancer
7. Noncanonical Stress Response Induces Autophagy and Promotes Cancer Progression
8. ER-Specific Autophagy
9. ER Ca2+ Regulates Autophagy
10. DNA Damage Response (DDR)
11. Integrated Stress Response (ISR)
12. The Consequence of Unresolved ER Stress to Cell Death Mechanism
13. Maximize the Therapeutic Benefit by Manipulating ER Stress and Autophagy
14. A Comprehensive Patient’s Data Analysis for Accelerating Cancer Research and Precision Medicine
15. Concluding Remarks, Open Question, and Future Perspective
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Deng, J.; Bai, X.; Tang, H.; Pang, S. DNA damage promotes ER stress resistance through elevation of unsaturated phosphatidylcholine in Caenorhabditis elegans. J. Biol. Chem. 2021, 296, 100095. [Google Scholar] [CrossRef]
- Nakashima, A.; Cheng, S.B.; Kusabiraki, T.; Motomura, K.; Aoki, A.; Ushijima, A.; Ono, Y.; Tsuda, S.; Shima, T.; Yoshino, O.; et al. Endoplasmic reticulum stress disrupts lysosomal homeostasis and induces blockade of autophagic flux in human trophoblasts. Sci. Rep. 2019, 9, 11466. [Google Scholar] [CrossRef] [Green Version]
- Fregno, I.; Molinari, M. Endoplasmic reticulum turnover: ER-phagy and other flavors in selective and non-selective ER clearance. F1000Res 2018, 7, 454. [Google Scholar] [CrossRef]
- Zhao, G.; Blackstone, C. ER morphology: Sculpting with XendoU. Curr. Biol. 2014, 24, R1170–R1172. [Google Scholar] [CrossRef] [Green Version]
- Madden, E.; Logue, S.E.; Healy, S.J.; Manie, S.; Samali, A. The role of the unfolded protein response in cancer progression: From oncogenesis to chemoresistance. Biol. Cell 2019, 111, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zhang, L.; Zhou, L.; Lei, Y.; Zhang, Y.; Huang, C. Redox signaling and unfolded protein response coordinate cell fate decisions under ER stress. Redox. Biol. 2019, 25, 101047. [Google Scholar] [CrossRef]
- Andruska, N.; Zheng, X.; Yang, X.; Helferich, W.G.; Shapiro, D.J. Anticipatory estrogen activation of the unfolded protein response is linked to cell proliferation and poor survival in estrogen receptor alpha-positive breast cancer. Oncogene 2015, 34, 3760–3769. [Google Scholar] [CrossRef] [Green Version]
- Epple, L.M.; Dodd, R.D.; Merz, A.L.; Dechkovskaia, A.M.; Herring, M.; Winston, B.A.; Lencioni, A.M.; Russell, R.L.; Madsen, H.; Nega, M.; et al. Induction of the unfolded protein response drives enhanced metabolism and chemoresistance in glioma cells. PLoS ONE 2013, 8, e73267. [Google Scholar] [CrossRef]
- McGrath, E.P.; Logue, S.E.; Mnich, K.; Deegan, S.; Jager, R.; Gorman, A.M.; Samali, A. The Unfolded Protein Response in Breast Cancer. Cancers 2018, 10, 344. [Google Scholar] [CrossRef] [Green Version]
- Senft, D.; Ronai, Z.A. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 2015, 40, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Cregg, J.M.; Dunn, W.A., Jr.; Emr, S.D.; Sakai, Y.; Sandoval, I.V.; Sibirny, A.; Subramani, S.; Thumm, M.; Veenhuis, M.; et al. A unified nomenclature for yeast autophagy-related genes. Dev. Cell 2003, 5, 539–545. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Noda, T.; Yoshimori, T.; Tanaka, Y.; Ishii, T.; George, M.D.; Klionsky, D.J.; Ohsumi, M.; Ohsumi, Y. A protein conjugation system essential for autophagy. Nature 1998, 395, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Cicchini, M.; Karantza, V.; Xia, B. Molecular pathways: Autophagy in cancer--a matter of timing and context. Clin. Cancer Res. 2015, 21, 498–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farre, J.C.; Subramani, S. Mechanistic insights into selective autophagy pathways: Lessons from yeast. Nat. Rev. Mol. Cell Biol. 2016, 17, 537–552. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.Y. mTORC1 phosphorylates the ULK1-mAtg13-FIP200 autophagy regulatory complex. Sci. Signal 2009, 2, pe51. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Jun, C.B.; Ro, S.H.; Kim, Y.M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, S.; Bell, C.M.; Rothbart, S.B.; Moran, R.G. AMP-activated Protein Kinase (AMPK) Control of mTORC1 Is p53- and TSC2-independent in Pemetrexed-treated Carcinoma Cells. J. Biol. Chem. 2015, 290, 27473–27486. [Google Scholar] [CrossRef] [Green Version]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Yuan, H.X.; Russell, R.C.; Guan, K.L. Regulation of PIK3C3/VPS34 complexes by MTOR in nutrient stress-induced autophagy. Autophagy 2013, 9, 1983–1995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brier, L.W.; Ge, L.; Stjepanovic, G.; Thelen, A.M.; Hurley, J.H.; Schekman, R. Regulation of LC3 lipidation by the autophagy-specific class III phosphatidylinositol-3 kinase complex. Mol. Biol. Cell 2019, 30, 1098–1107. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T.; Tooze, S.A. Emerging roles of ATG proteins and membrane lipids in autophagosome formation. Cell Discov. 2020, 6, 32. [Google Scholar] [CrossRef]
- Reggiori, F.; Shintani, T.; Nair, U.; Klionsky, D.J. Atg9 cycles between mitochondria and the pre-autophagosomal structure in yeasts. Autophagy 2005, 1, 101–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tooze, S.A.; Yoshimori, T. The origin of the autophagosomal membrane. Nat. Cell Biol. 2010, 12, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zheng, L.; Zheng, X.; Ge, L. Autophagosomal Membrane Origin and Formation. Adv. Exp. Med. Biol. 2021, 1208, 17–42. [Google Scholar] [CrossRef]
- Walczak, M.; Martens, S. Dissecting the role of the Atg12-Atg5-Atg16 complex during autophagosome formation. Autophagy 2013, 9, 424–425. [Google Scholar] [CrossRef] [Green Version]
- Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Mizushima, N.; Tanida, I.; Kominami, E.; Ohsumi, M.; et al. A ubiquitin-like system mediates protein lipidation. Nature 2000, 408, 488–492. [Google Scholar] [CrossRef]
- Graef, M. Recent advances in the understanding of autophagosome biogenesis. F1000Res 2020, 9, 212. [Google Scholar] [CrossRef]
- Lee, Y.K.; Lee, J.A. Role of the mammalian ATG8/LC3 family in autophagy: Differential and compensatory roles in the spatiotemporal regulation of autophagy. BMB Rep. 2016, 49, 424–430. [Google Scholar] [CrossRef]
- Lippai, M.; Low, P. The role of the selective adaptor p62 and ubiquitin-like proteins in autophagy. Biomed. Res. Int. 2014, 2014, 832704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.N.; Padman, B.S.; Usher, J.; Oorschot, V.; Ramm, G.; Lazarou, M. Atg8 family LC3/GABARAP proteins are crucial for autophagosome-lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J. Cell Biol. 2016, 215, 857–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Sanchez, R.; Tooze, S.A.; Reggiori, F. Membrane supply and remodeling during autophagosome biogenesis. Curr. Opin. Cell Biol. 2021, 71, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Kishi-Itakura, C.; Mizushima, N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 2012, 151, 1256–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamb, C.A.; Yoshimori, T.; Tooze, S.A. The autophagosome: Origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 759–774. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.J.; Voeltz, G.K. Structure and function of ER membrane contact sites with other organelles. Nat. Rev. Mol. Cell Biol. 2016, 17, 69–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Brodsky, J.L. Protein quality control in the secretory pathway. J. Cell Biol. 2019, 218, 3171–3187. [Google Scholar] [CrossRef] [Green Version]
- Fregno, I.; Molinari, M. Proteasomal and lysosomal clearance of faulty secretory proteins: ER-associated degradation (ERAD) and ER-to-lysosome-associated degradation (ERLAD) pathways. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 153–163. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef]
- Yamasaki, A.; Alam, J.M.; Noshiro, D.; Hirata, E.; Fujioka, Y.; Suzuki, K.; Ohsumi, Y.; Noda, N.N. Liquidity Is a Critical Determinant for Selective Autophagy of Protein Condensates. Mol. Cell 2020, 77, 1163–1175e9. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.; Obara, C.J.; Szczesna, E.; Nixon-Abell, J.; Mahalingan, K.K.; Roll-Mecak, A.; Lippincott-Schwartz, J.; Blackstone, C. ER proteins decipher the tubulin code to regulate organelle distribution. Nature 2022, 601, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Desai, T.; McNew, J.A.; Gerard, P.; Novick, P.J.; Ferro-Novick, S. Lunapark stabilizes nascent three-way junctions in the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2015, 112, 418–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Craene, J.O.; Coleman, J.; Estrada de Martin, P.; Pypaert, M.; Anderson, S.; Yates, J.R., 3rd; Ferro-Novick, S.; Novick, P. Rtn1p is involved in structuring the cortical endoplasmic reticulum. Mol. Biol. Cell 2006, 17, 3009–3020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Shibata, Y.; Zhu, P.P.; Voss, C.; Rismanchi, N.; Prinz, W.A.; Rapoport, T.A.; Blackstone, C. A class of dynamin-like GTPases involved in the generation of the tubular ER network. Cell 2009, 138, 549–561. [Google Scholar] [CrossRef] [Green Version]
- Orso, G.; Pendin, D.; Liu, S.; Tosetto, J.; Moss, T.J.; Faust, J.E.; Micaroni, M.; Egorova, A.; Martinuzzi, A.; McNew, J.A.; et al. Homotypic fusion of ER membranes requires the dynamin-like GTPase atlastin. Nature 2009, 460, 978–983. [Google Scholar] [CrossRef]
- Voeltz, G.K.; Prinz, W.A.; Shibata, Y.; Rist, J.M.; Rapoport, T.A. A class of membrane proteins shaping the tubular endoplasmic reticulum. Cell 2006, 124, 573–586. [Google Scholar] [CrossRef] [Green Version]
- Hubner, C.A.; Dikic, I. ER-phagy and human diseases. Cell Death Differ. 2020, 27, 833–842. [Google Scholar] [CrossRef]
- Wilkinson, S. Picky Eating at the ER-phagy Buffet. Trends Biochem. Sci. 2019, 44, 731–733. [Google Scholar] [CrossRef]
- Chino, H.; Mizushima, N. ER-Phagy: Quality Control and Turnover of Endoplasmic Reticulum. Trends Cell Biol. 2020, 30, 384–398. [Google Scholar] [CrossRef]
- Ferro-Novick, S.; Reggiori, F.; Brodsky, J.L. ER-Phagy, ER Homeostasis, and ER Quality Control: Implications for Disease. Trends Biochem. Sci. 2021, 46, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, S. Emerging Principles of Selective ER Autophagy. J. Mol. Biol. 2020, 432, 185–205. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg-Lerner, A.; Ciechanover, A.; Merbl, Y. Post-translational modification profiling—A novel tool for mapping the protein modification landscape in cancer. Exp. Biol. Med. 2016, 241, 1475–1482. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Li, F.; Zhang, X.; Lin, H.K.; Xu, C. Insights into the post-translational modification and its emerging role in shaping the tumor microenvironment. Signal. Transduct. Target Ther. 2021, 6, 422. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.H.; Kim, G.R.; Seong, M.; Baek, S.H.; Seol, J.H.; Bang, O.S.; Ovaa, H.; Tatsumi, K.; Komatsu, M.; Tanaka, K.; et al. Two novel ubiquitin-fold modifier 1 (Ufm1)-specific proteases, UfSP1 and UfSP2. J. Biol. Chem. 2007, 282, 5256–5262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabinowitz, J.D.; White, E. Autophagy and metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.Y.; Chen, H.Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.; Karantza, V.; et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.Y.; Teng, X.; Laddha, S.V.; Ma, S.; Van Nostrand, S.C.; Yang, Y.; Khor, S.; Chan, C.S.; Rabinowitz, J.D.; White, E. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev. 2016, 30, 1704–1717. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Kumanova, M.; Hart, L.S.; Sloane, K.; Zhang, H.; De Panis, D.N.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Ron, D.; Koumenis, C. The GCN2-ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient deprivation. EMBO J. 2010, 29, 2082–2096. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Villani, R.M.; Wang, H.; Simpson, M.J.; Roberts, M.S.; Tang, M.; Liang, X. The role of cellular reactive oxygen species in cancer chemotherapy. J. Exp. Clin. Cancer Res. 2018, 37, 266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukagoshi, S.; Mikami, R.; Arai, K. Basic Amino Acid Conjugates of 1,2-Diselenan-4-amine with Protein Disulfide Isomerase-like Functions as a Manipulator of Protein Quality Control. Chem. Asian. J. 2020, 15, 2646–2652. [Google Scholar] [CrossRef]
- Fu, J.; Gao, J.; Liang, Z.; Yang, D. PDI-Regulated Disulfide Bond Formation in Protein Folding and Biomolecular Assembly. Molecules 2020, 26, 171. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J. Autophagy: From phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 2007, 8, 931–937. [Google Scholar] [CrossRef]
- Bosc, C.; Broin, N.; Fanjul, M.; Saland, E.; Farge, T.; Courdy, C.; Batut, A.; Masoud, R.; Larrue, C.; Skuli, S.; et al. Autophagy regulates fatty acid availability for oxidative phosphorylation through mitochondria-endoplasmic reticulum contact sites. Nat. Commun. 2020, 11, 4056. [Google Scholar] [CrossRef]
- Junjappa, R.P.; Patil, P.; Bhattarai, K.R.; Kim, H.R.; Chae, H.J. IRE1alpha Implications in Endoplasmic Reticulum Stress-Mediated Development and Pathogenesis of Autoimmune Diseases. Front. Immunol. 2018, 9, 1289. [Google Scholar] [CrossRef] [Green Version]
- Rashid, H.O.; Yadav, R.K.; Kim, H.R.; Chae, H.J. ER stress: Autophagy induction, inhibition and selection. Autophagy 2015, 11, 1956–1977. [Google Scholar] [CrossRef] [Green Version]
- Bhattarai, K.R.; Riaz, T.A.; Kim, H.R.; Chae, H.J. The aftermath of the interplay between the endoplasmic reticulum stress response and redox signaling. Exp. Mol. Med. 2021, 53, 151–167. [Google Scholar] [CrossRef]
- Maurel, M.; Chevet, E.; Tavernier, J.; Gerlo, S. Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem. Sci. 2014, 39, 245–254. [Google Scholar] [CrossRef]
- Le Thomas, A.; Ferri, E.; Marsters, S.; Harnoss, J.M.; Lawrence, D.A.; Zuazo-Gaztelu, I.; Modrusan, Z.; Chan, S.; Solon, M.; Chalouni, C.; et al. Decoding non-canonical mRNA decay by the endoplasmic-reticulum stress sensor IRE1alpha. Nat. Commun. 2021, 12, 7310. [Google Scholar] [CrossRef] [PubMed]
- Rainbolt, T.K.; Frydman, J. Dynamics and clustering of IRE1alpha during ER stress. Proc. Natl. Acad. Sci. USA 2020, 117, 3352–3354. [Google Scholar] [CrossRef] [PubMed]
- Bae, D.; Moore, K.A.; Mella, J.M.; Hayashi, S.Y.; Hollien, J. Degradation of Blos1 mRNA by IRE1 repositions lysosomes and protects cells from stress. J. Cell Biol. 2019, 218, 1118–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, T.K.; Lawrence, D.A.; Lu, M.; Tan, J.; Harnoss, J.M.; Marsters, S.A.; Liu, P.; Sandoval, W.; Martin, S.E.; Ashkenazi, A. Coordination between Two Branches of the Unfolded Protein Response Determines Apoptotic Cell Fate. Mol. Cell 2018, 71, 629–636.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dufey, E.; Bravo-San Pedro, J.M.; Eggers, C.; Gonzalez-Quiroz, M.; Urra, H.; Sagredo, A.I.; Sepulveda, D.; Pihan, P.; Carreras-Sureda, A.; Hazari, Y.; et al. Genotoxic stress triggers the activation of IRE1alpha-dependent RNA decay to modulate the DNA damage response. Nat. Commun. 2020, 11, 2401. [Google Scholar] [CrossRef]
- Lu, M.; Lawrence, D.A.; Marsters, S.; Acosta-Alvear, D.; Kimmig, P.; Mendez, A.S.; Paton, A.W.; Paton, J.C.; Walter, P.; Ashkenazi, A. Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science 2014, 345, 98–101. [Google Scholar] [CrossRef] [Green Version]
- So, J.S.; Hur, K.Y.; Tarrio, M.; Ruda, V.; Frank-Kamenetsky, M.; Fitzgerald, K.; Koteliansky, V.; Lichtman, A.H.; Iwawaki, T.; Glimcher, L.H.; et al. Silencing of lipid metabolism genes through IRE1alpha-mediated mRNA decay lowers plasma lipids in mice. Cell Metab. 2012, 16, 487–499. [Google Scholar] [CrossRef] [Green Version]
- Darling, N.J.; Cook, S.J. The role of MAPK signalling pathways in the response to endoplasmic reticulum stress. Biochim. Biophys. Acta. 2014, 1843, 2150–2163. [Google Scholar] [CrossRef] [Green Version]
- Siegmund, D.; Wagner, J.; Wajant, H. TNF Receptor Associated Factor 2 (TRAF2) Signaling in Cancer. Cancers 2022, 14, 4055. [Google Scholar] [CrossRef]
- Tournier, C.; Hess, P.; Yang, D.D.; Xu, J.; Turner, T.K.; Nimnual, A.; Bar-Sagi, D.; Jones, S.N.; Flavell, R.A.; Davis, R.J. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 2000, 288, 870–874. [Google Scholar] [CrossRef]
- Dang Do, A.N.; Kimball, S.R.; Cavener, D.R.; Jefferson, L.S. eIF2alpha kinases GCN2 and PERK modulate transcription and translation of distinct sets of mRNAs in mouse liver. Physiol. Genom. 2009, 38, 328–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Wang, M.; Cheng, A.; Yang, Q.; Wu, Y.; Jia, R.; Liu, M.; Zhu, D.; Chen, S.; Zhang, S.; et al. The role of host eIF2alpha in viral infection. Virol. J. 2020, 17, 112. [Google Scholar] [CrossRef]
- Asano, K.; Clayton, J.; Shalev, A.; Hinnebusch, A.G. A multifactor complex of eukaryotic initiation factors, eIF1, eIF2, eIF3, eIF5, and initiator tRNA(Met) is an important translation initiation intermediate in vivo. Genes Dev. 2000, 14, 2534–2546. [Google Scholar] [CrossRef] [Green Version]
- Sudhakar, A.; Ramachandran, A.; Ghosh, S.; Hasnain, S.E.; Kaufman, R.J.; Ramaiah, K.V. Phosphorylation of serine 51 in initiation factor 2 alpha (eIF2 alpha) promotes complex formation between eIF2 alpha(P) and eIF2B and causes inhibition in the guanine nucleotide exchange activity of eIF2B. Biochemistry 2000, 39, 12929–12938. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. ER stress activates immunosuppressive network: Implications for aging and Alzheimer’s disease. J. Mol. Med. 2020, 98, 633–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brush, M.H.; Weiser, D.C.; Shenolikar, S. Growth arrest and DNA damage-inducible protein GADD34 targets protein phosphatase 1 alpha to the endoplasmic reticulum and promotes dephosphorylation of the alpha subunit of eukaryotic translation initiation factor 2. Mol. Cell Biol. 2003, 23, 1292–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novoa, I.; Zeng, H.; Harding, H.P.; Ron, D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J. Cell Biol. 2001, 153, 1011–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Cubillos-Ruiz, J.R. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat. Rev. Cancer 2021, 21, 71–88. [Google Scholar] [CrossRef]
- Shen, J.; Chen, X.; Hendershot, L.; Prywes, R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell 2002, 3, 99–111. [Google Scholar] [CrossRef] [Green Version]
- Glembotski, C.C.; Arrieta, A.; Blackwood, E.A.; Stauffer, W.T. ATF6 as a Nodal Regulator of Proteostasis in the Heart. Front. Physiol. 2020, 11, 267. [Google Scholar] [CrossRef]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gelinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef] [Green Version]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Moenner, M.; Pluquet, O.; Bouchecareilh, M.; Chevet, E. Integrated endoplasmic reticulum stress responses in cancer. Cancer Res. 2007, 67, 10631–10634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Kumano, M.; Beraldi, E.; Fazli, L.; Du, C.; Moore, S.; Sorensen, P.; Zoubeidi, A.; Gleave, M.E. Clusterin facilitates stress-induced lipidation of LC3 and autophagosome biogenesis to enhance cancer cell survival. Nat. Commun. 2014, 5, 5775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Carbonero, N.; Li, W.; Cabeza-Morales, M.; Martinez-Useros, J.; Garcia-Foncillas, J. New Hope for Pancreatic Ductal Adenocarcinoma Treatment Targeting Endoplasmic Reticulum Stress Response: A Systematic Review. Int. J. Mol. Sci. 2018, 19, 2468. [Google Scholar] [CrossRef] [Green Version]
- Thakur, P.C.; Miller-Ocuin, J.L.; Nguyen, K.; Matsuda, R.; Singhi, A.D.; Zeh, H.J.; Bahary, N. Inhibition of endoplasmic-reticulum-stress-mediated autophagy enhances the effectiveness of chemotherapeutics on pancreatic cancer. J. Transl. Med. 2018, 16, 190. [Google Scholar] [CrossRef]
- Kishino, A.; Hayashi, K.; Hidai, C.; Masuda, T.; Nomura, Y.; Oshima, T. XBP1-FoxO1 interaction regulates ER stress-induced autophagy in auditory cells. Sci. Rep. 2017, 7, 4442. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Liu, H.; Jiang, C.C.; Fang, L.; Chen, C.; Zhang, X.D.; Jiang, Z.W. Connecting endoplasmic reticulum stress to autophagy through IRE1/JNK/beclin-1 in breast cancer cells. Int. J. Mol. Med. 2014, 34, 772–781. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Wang, Q.; Michalak, M. Inositol Requiring Enzyme (IRE), a multiplayer in sensing endoplasmic reticulum stress. Anim. Cells. Syst. 2021, 25, 347–357. [Google Scholar] [CrossRef]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef] [Green Version]
- Ogata, M.; Hino, S.; Saito, A.; Morikawa, K.; Kondo, S.; Kanemoto, S.; Murakami, T.; Taniguchi, M.; Tanii, I.; Yoshinaga, K.; et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell Biol. 2006, 26, 9220–9231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chipurupalli, S.; Ganesan, R.; Martini, G.; Mele, L.; Reggio, A.; Esposito, M.; Kannan, E.; Namasivayam, V.; Grumati, P.; Desiderio, V.; et al. Cancer cells adapt FAM134B/BiP mediated ER-phagy to survive hypoxic stress. Cell. Death. Dis. 2022, 13, 357. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.X.; Ni, H.M.; Gao, W.; Hou, Y.F.; Melan, M.A.; Chen, X.; Stolz, D.B.; Shao, Z.M.; Yin, X.M. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J. Biol. Chem. 2007, 282, 4702–4710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox. Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [Green Version]
- Leitman, J.; Ulrich Hartl, F.; Lederkremer, G.Z. Soluble forms of polyQ-expanded huntingtin rather than large aggregates cause endoplasmic reticulum stress. Nat. Commun. 2013, 4, 2753. [Google Scholar] [CrossRef] [Green Version]
- Remondelli, P.; Renna, M. The Endoplasmic Reticulum Unfolded Protein Response in Neurodegenerative Disorders and Its Potential Therapeutic Significance. Front. Mol. Neurosci. 2017, 10, 187. [Google Scholar] [CrossRef] [Green Version]
- Tashiro, E.; Zako, T.; Muto, H.; Itoo, Y.; Sorgjerd, K.; Terada, N.; Abe, A.; Miyazawa, M.; Kitamura, A.; Kitaura, H.; et al. Prefoldin protects neuronal cells from polyglutamine toxicity by preventing aggregation formation. J. Biol. Chem. 2013, 288, 19958–19972. [Google Scholar] [CrossRef] [Green Version]
- Balsa, E.; Soustek, M.S.; Thomas, A.; Cogliati, S.; Garcia-Poyatos, C.; Martin-Garcia, E.; Jedrychowski, M.; Gygi, S.P.; Enriquez, J.A.; Puigserver, P. ER and Nutrient Stress Promote Assembly of Respiratory Chain Supercomplexes through the PERK-eIF2alpha Axis. Mol. Cell 2019, 74, 877–890.e876. [Google Scholar] [CrossRef]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability--an evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef]
- Robert, T.; Vanoli, F.; Chiolo, I.; Shubassi, G.; Bernstein, K.A.; Rothstein, R.; Botrugno, O.A.; Parazzoli, D.; Oldani, A.; Minucci, S.; et al. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature 2011, 471, 74–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouschop, K.M.; Dubois, L.J.; Keulers, T.G.; van den Beucken, T.; Lambin, P.; Bussink, J.; van der Kogel, A.J.; Koritzinsky, M.; Wouters, B.G. PERK/eIF2alpha signaling protects therapy resistant hypoxic cells through induction of glutathione synthesis and protection against ROS. Proc. Natl. Acad. Sci. USA 2013, 110, 4622–4627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verfaillie, T.; Salazar, M.; Velasco, G.; Agostinis, P. Linking ER Stress to Autophagy: Potential Implications for Cancer Therapy. Int. J. Cell Biol. 2010, 2010, 930509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.H.; Piao, S.F.; Dey, S.; McAfee, Q.; Karakousis, G.; Villanueva, J.; Hart, L.S.; Levi, S.; Hu, J.; Zhang, G.; et al. Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma. J. Clin. Investig. 2014, 124, 1406–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, W.; Xie, W.; Yin, D.; Luo, R.; Liu, M.; Guo, F. ATG5 and ATG7 induced autophagy interplays with UPR via PERK signaling. Cell. Commun. Signal. 2019, 17, 42. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Wu, Y.; Lian, X. Targeted inhibition of GRP78 by HA15 promotes apoptosis of lung cancer cells accompanied by ER stress and autophagy. Biol. Open 2020, 9, bio053298. [Google Scholar] [CrossRef]
- Huang, H.; Kang, R.; Wang, J.; Luo, G.; Yang, W.; Zhao, Z. Hepatitis C virus inhibits AKT-tuberous sclerosis complex (TSC), the mechanistic target of rapamycin (MTOR) pathway, through endoplasmic reticulum stress to induce autophagy. Autophagy 2013, 9, 175–195. [Google Scholar] [CrossRef] [Green Version]
- Chang, K.C.; Chen, P.C.; Chen, Y.P.; Chang, Y.; Su, I.J. Dominant expression of survival signals of endoplasmic reticulum stress response in Hodgkin lymphoma. Cancer Sci. 2011, 102, 275–281. [Google Scholar] [CrossRef]
- Shuda, M.; Kondoh, N.; Imazeki, N.; Tanaka, K.; Okada, T.; Mori, K.; Hada, A.; Arai, M.; Wakatsuki, T.; Matsubara, O.; et al. Activation of the ATF6, XBP1 and grp78 genes in human hepatocellular carcinoma: A possible involvement of the ER stress pathway in hepatocarcinogenesis. J. Hepatol. 2003, 38, 605–614. [Google Scholar] [CrossRef]
- Yarapureddy, S.; Abril, J.; Foote, J.; Kumar, S.; Asad, O.; Sharath, V.; Faraj, J.; Daniel, D.; Dickman, P.; White-Collins, A.; et al. ATF6alpha Activation Enhances Survival against Chemotherapy and Serves as a Prognostic Indicator in Osteosarcoma. Neoplasia 2019, 21, 516–532. [Google Scholar] [CrossRef]
- Kalvakolanu, D.V.; Gade, P. IFNG and autophagy: A critical role for the ER-stress mediator ATF6 in controlling bacterial infections. Autophagy 2012, 8, 1673–1674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schewe, D.M.; Aguirre-Ghiso, J.A. ATF6alpha-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 10519–10524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrkamp, A.; Herrmann, C.; Stoll, R.; Heumann, R. Ras and rheb signaling in survival and cell death. Cancers 2013, 5, 639–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usman, R.M.; Razzaq, F.; Akbar, A.; Farooqui, A.A.; Iftikhar, A.; Latif, A.; Hassan, H.; Zhao, J.; Carew, J.S.; Nawrocki, S.T.; et al. Role and mechanism of autophagy-regulating factors in tumorigenesis and drug resistance. Asia Pac. J. Clin. Oncol. 2021, 17, 193–208. [Google Scholar] [CrossRef]
- Yarapureddy, S.; Hingorani, P.; Kumar, S.; Asad, O.; Abril, J.; Sharath, V.; Dickman, P.; Sertil, A.C. Abstract 4505: Activating transcription factor-6α dependent regulation of Rheb-mTOR and Notch signaling contributes to chemoresistance in osteosarcoma. Cancer Res. 2017, 77, 4505. [Google Scholar] [CrossRef]
- Dooley, H.C.; Wilson, M.I.; Tooze, S.A. WIPI2B links PtdIns3P to LC3 lipidation through binding ATG16L1. Autophagy 2015, 11, 190–191. [Google Scholar] [CrossRef]
- Bhardwaj, M.; Leli, N.M.; Koumenis, C.; Amaravadi, R.K. Regulation of autophagy by canonical and non-canonical ER stress responses. Semin. Cancer Biol. 2020, 66, 116–128. [Google Scholar] [CrossRef]
- Yao, Y.; Lu, Q.; Hu, Z.; Yu, Y.; Chen, Q.; Wang, Q.K. A non-canonical pathway regulates ER stress signaling and blocks ER stress-induced apoptosis and heart failure. Nat. Commun. 2017, 8, 133. [Google Scholar] [CrossRef] [Green Version]
- León-Annicchiarico, C.L.; Ramírez-Peinado, S.; Domínguez-Villanueva, D.; Gonsberg, A.; Lampidis, T.J.; Muñoz-Pinedo, C. ATF4 mediates necrosis induced by glucose deprivation and apoptosis induced by 2-deoxyglucose in the same cells. FEBS J. 2015, 282, 3647–3658. [Google Scholar] [CrossRef] [Green Version]
- Jiang, C.C.; Lucas, K.; Avery-Kiejda, K.A.; Wade, M.; deBock, C.E.; Thorne, R.F.; Allen, J.; Hersey, P.; Zhang, X.D. Up-regulation of Mcl-1 is critical for survival of human melanoma cells upon endoplasmic reticulum stress. Cancer Res. 2008, 68, 6708–6717. [Google Scholar] [CrossRef]
- Martin-Perez, R.; Palacios, C.; Yerbes, R.; Cano-Gonzalez, A.; Iglesias-Serret, D.; Gil, J.; Reginato, M.J.; Lopez-Rivas, A. Activated ERBB2/HER2 licenses sensitivity to apoptosis upon endoplasmic reticulum stress through a PERK-dependent pathway. Cancer Res. 2014, 74, 1766–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verfaillie, T.; van Vliet, A.; Garg, A.D.; Dewaele, M.; Rubio, N.; Gupta, S.; de Witte, P.; Samali, A.; Agostinis, P. Pro-apoptotic signaling induced by photo-oxidative ER stress is amplified by Noxa, not Bim. Biochem. Biophys. Res. Commun. 2013, 438, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, J.L.; Flockhart, R.; Veal, G.J.; Lovat, P.E.; Redfern, C.P. Regulation of endoplasmic reticulum stress-induced cell death by ATF4 in neuroectodermal tumor cells. J. Biol. Chem. 2010, 285, 6091–6100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, J.A.; Ullman, E.; Dou, Z.; Zong, W.X. Inhibition of protein degradation induces apoptosis through a microtubule-associated protein 1 light chain 3-mediated activation of caspase-8 at intracellular membranes. Mol. Cell Biol. 2011, 31, 3158–3170. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Duan, B.; Zhang, Y.; Zhang, X.; Xia, B. Excessive ER-phagy mediated by the autophagy receptor FAM134B results in ER stress, the unfolded protein response, and cell death in HeLa cells. J. Biol. Chem. 2019, 294, 20009–20023. [Google Scholar] [CrossRef]
- Kim, C.; Kim, B. Anti-Cancer Natural Products and Their Bioactive Compounds Inducing ER Stress-Mediated Apoptosis: A Review. Nutrients 2018, 10, 1021. [Google Scholar] [CrossRef] [Green Version]
- Wei, M.C.; Zong, W.X.; Cheng, E.H.; Lindsten, T.; Panoutsakopoulou, V.; Ross, A.J.; Roth, K.A.; MacGregor, G.R.; Thompson, C.B.; Korsmeyer, S.J. Proapoptotic BAX and BAK: A requisite gateway to mitochondrial dysfunction and death. Science 2001, 292, 727–730. [Google Scholar] [CrossRef] [Green Version]
- Bernales, S.; McDonald, K.L.; Walter, P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 2006, 4, e423. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Gao, W.; Zhou, L.; Chen, Y.; Qin, S.; Zhang, L.; Liu, J.; He, Y.; Lei, Y.; Chen, H.N.; et al. Repurposing Brigatinib for the Treatment of Colorectal Cancer Based on Inhibition of ER-phagy. Theranostics 2019, 9, 4878–4892. [Google Scholar] [CrossRef]
- Kasem, K.; Gopalan, V.; Salajegheh, A.; Lu, C.T.; Smith, R.A.; Lam, A.K. The roles of JK-1 (FAM134B) expressions in colorectal cancer. Exp. Cell Res. 2014, 326, 166–173. [Google Scholar] [CrossRef]
- Tang, W.K.; Chui, C.H.; Fatima, S.; Kok, S.H.; Pak, K.C.; Ou, T.M.; Hui, K.S.; Wong, M.M.; Wong, J.; Law, S.; et al. Oncogenic properties of a novel gene JK-1 located in chromosome 5p and its overexpression in human esophageal squamous cell carcinoma. Int. J. Mol. Med. 2007, 19, 915–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fumagalli, F.; Noack, J.; Bergmann, T.J.; Cebollero, E.; Pisoni, G.B.; Fasana, E.; Fregno, I.; Galli, C.; Loi, M.; Soldà, T.; et al. Translocon component Sec62 acts in endoplasmic reticulum turnover during stress recovery. Nat. Cell Biol. 2016, 18, 1173–1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linxweiler, M.; Schick, B.; Zimmermann, R. Let’s talk about Secs: Sec61, Sec62 and Sec63 in signal transduction, oncology and personalized medicine. Signal Transduct. Target. Ther. 2017, 2, 17002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergmann, T.J.; Fumagalli, F.; Loi, M.; Molinari, M. Role of SEC62 in ER maintenance: A link with ER stress tolerance in SEC62-overexpressing tumors? Mol. Cell Oncol. 2017, 4, e1264351. [Google Scholar] [CrossRef] [Green Version]
- Muller, C.S.L.; Pfohler, C.; Wahl, M.; Bochen, F.; Korner, S.; Kuhn, J.P.; Bozzato, A.; Schick, B.; Linxweiler, M. Expression of SEC62 Oncogene in Benign, Malignant and Borderline Melanocytic Tumors-Unmasking the Wolf in Sheep’s Clothing? Cancers 2021, 13, 1645. [Google Scholar] [CrossRef]
- Lee, K.T.; Islam, F.; Vider, J.; Martin, J.; Chruscik, A.; Lu, C.T.; Gopalan, V.; Lam, A.K. Overexpression of family with sequence similarity 134, member B (FAM134B) in colon cancers and its tumor suppressive properties in vitro. Cancer Biol. Ther. 2020, 21, 954–962. [Google Scholar] [CrossRef]
- Zielke, S.; Kardo, S.; Zein, L.; Mari, M.; Covarrubias-Pinto, A.; Kinzler, M.N.; Meyer, N.; Stolz, A.; Fulda, S.; Reggiori, F.; et al. ATF4 links ER stress with reticulophagy in glioblastoma cells. Autophagy 2021, 17, 2432–2448. [Google Scholar] [CrossRef]
- Smith, M.D.; Harley, M.E.; Kemp, A.J.; Wills, J.; Lee, M.; Arends, M.; von Kriegsheim, A.; Behrends, C.; Wilkinson, S. CCPG1 Is a Non-canonical Autophagy Cargo Receptor Essential for ER-Phagy and Pancreatic ER Proteostasis. Dev. Cell 2018, 44, 217–232.e211. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.T.; Lee, T.J.; Kim, C.H.; Kim, N.S.; Kwon, T.K. Over-expression of Reticulon 3 (RTN3) enhances TRAIL-mediated apoptosis via up-regulation of death receptor 5 (DR5) and down-regulation of c-FLIP. Cancer Lett. 2009, 279, 185–192. [Google Scholar] [CrossRef]
- Sannino, S.; Yates, M.E.; Schurdak, M.E.; Oesterreich, S.; Lee, A.V.; Wipf, P.; Brodsky, J.L. Unique integrated stress response sensors regulate cancer cell susceptibility when Hsp70 activity is compromised. Elife 2021, 10, e64977. [Google Scholar] [CrossRef]
- Chang, S.H.; Huang, S.W.; Wang, S.T.; Chung, K.C.; Hsieh, C.W.; Kao, J.K.; Chen, Y.J.; Wu, C.Y.; Shieh, J.J. Imiquimod-induced autophagy is regulated by ER stress-mediated PKR activation in cancer cells. J. Dermatol. Sci. 2017, 87, 138–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouyang, D.; Zhang, Y.; Xu, L.; Li, J.; Zha, Q.; He, X. Histone deacetylase inhibitor valproic acid sensitizes B16F10 melanoma cells to cucurbitacin B treatment. Acta Biochim. Biophys. Sin. 2011, 43, 487–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, B.; Lu, Q.; Liu, J.; Fan, L.; Wang, Y.; Wei, W.; Wang, H.; Sun, G. Melatonin Increases the Sensitivity of Hepatocellular Carcinoma to Sorafenib through the PERK-ATF4-Beclin1 Pathway. Int. J. Biol. Sci. 2019, 15, 1905–1920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Tang, B.; Yu, P.W.; Tang, B.; Hao, Y.X.; Lei, X.; Luo, H.X.; Zeng, D.Z. Autophagy protects against oxaliplatin-induced cell death via ER stress and ROS in Caco-2 cells. PLoS ONE 2012, 7, e51076. [Google Scholar] [CrossRef]
- Huang, J.; Pan, H.; Wang, J.; Wang, T.; Huo, X.; Ma, Y.; Lu, Z.; Sun, B.; Jiang, H. Unfolded protein response in colorectal cancer. Cell Biosci. 2021, 11, 26. [Google Scholar] [CrossRef]
- Park, J.M.; Huang, S.; Wu, T.T.; Foster, N.R.; Sinicrope, F.A. Prognostic impact of Beclin 1, p62/sequestosome 1 and LC3 protein expression in colon carcinomas from patients receiving 5-fluorouracil as adjuvant chemotherapy. Cancer Biol. Ther. 2013, 14, 100–107. [Google Scholar] [CrossRef] [Green Version]
- Yao, X.; Tu, Y.; Xu, Y.; Guo, Y.; Yao, F.; Zhang, X. Endoplasmic reticulum stress confers 5-fluorouracil resistance in breast cancer cell via the GRP78/OCT4/lncRNA MIAT/AKT pathway. Am. J. Cancer Res. 2020, 10, 838–855. [Google Scholar]
- Ji, G.R.; Yu, N.C.; Xue, X.; Li, Z.G. PERK-mediated Autophagy in Osteosarcoma Cells Resists ER Stress-induced Cell Apoptosis. Int. J. Biol. Sci. 2015, 11, 803–812. [Google Scholar] [CrossRef] [Green Version]
- Li, L.Y.; Chen, X.S.; Wang, K.S.; Guan, Y.D.; Ren, X.C.; Cao, D.S.; Sun, X.Y.; Li, A.X.; Tao, Y.G.; Zhang, Y.; et al. RSK2 protects human breast cancer cells under endoplasmic reticulum stress through activating AMPKalpha2-mediated autophagy. Oncogene 2020, 39, 6704–6718. [Google Scholar] [CrossRef]
- He, Y.; Su, J.; Lan, B.; Gao, Y.; Zhao, J. Targeting off-target effects: Endoplasmic reticulum stress and autophagy as effective strategies to enhance temozolomide treatment. Onco-Targets Ther. 2019, 12, 1857–1865. [Google Scholar] [CrossRef] [Green Version]
- Debernardi, J.; Pioche-Durieu, C.; Cam, E.L.; Wiels, J.; Robert, A. Verotoxin-1-Induced ER Stress Triggers Apoptotic or Survival Pathways in Burkitt Lymphoma Cells. Toxins 2020, 12, 316. [Google Scholar] [CrossRef] [PubMed]
- Salazar, M.; Carracedo, A.; Salanueva, I.J.; Hernandez-Tiedra, S.; Lorente, M.; Egia, A.; Vazquez, P.; Blazquez, C.; Torres, S.; Garcia, S.; et al. Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J. Clin. Investig. 2009, 119, 1359–1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.W. Cinnamaldehyde induces autophagy-mediated cell death through ER stress and epigenetic modification in gastric cancer cells. Acta Pharmacol. Sin. 2022, 43, 712–723. [Google Scholar] [CrossRef] [PubMed]
- Roderick, H.L.; Cook, S.J. Ca2+ signalling checkpoints in cancer: Remodelling Ca2+ for cancer cell proliferation and survival. Nat. Rev. Cancer 2008, 8, 361–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapovalov, G.; Ritaine, A.; Skryma, R.; Prevarskaya, N. Role of TRP ion channels in cancer and tumorigenesis. Semin Immunopathol. 2016, 38, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Xu, X.; Wang, X.; Zhang, B. Regulation of autophagy by Ca2+. Tumour Biol. 2016, 37, 15467–15476. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.H.; Shih, Y.L.; Ko, W.C.; Wei, Y.H.; Shih, C.M. Cadmium-induced autophagy and apoptosis are mediated by a calcium signaling pathway. Cell Mol. Life Sci. 2008, 65, 3640–3652. [Google Scholar] [CrossRef]
- Bialik, S.; Kimchi, A. The death-associated protein kinases: Structure, function, and beyond. Annu. Rev. Biochem. 2006, 75, 189–210. [Google Scholar] [CrossRef] [Green Version]
- Hoyer-Hansen, M.; Bastholm, L.; Szyniarowski, P.; Campanella, M.; Szabadkai, G.; Farkas, T.; Bianchi, K.; Fehrenbacher, N.; Elling, F.; Rizzuto, R.; et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol. Cell 2007, 25, 193–205. [Google Scholar] [CrossRef]
- Luo, Z.; Zang, M.; Guo, W. AMPK as a metabolic tumor suppressor: Control of metabolism and cell growth. Future Oncol. 2010, 6, 457–470. [Google Scholar] [CrossRef] [Green Version]
- Laski, J.; Singha, B.; Wang, X.; Valdes, Y.R.; Collins, O.; Shepherd, T.G. Activated CAMKKbeta-AMPK signaling promotes autophagy in a spheroid model of ovarian tumour metastasis. J. Ovarian Res. 2020, 13, 58. [Google Scholar] [CrossRef] [PubMed]
- Gong, G.; Waris, G.; Tanveer, R.; Siddiqui, A. Human hepatitis C virus NS5A protein alters intracellular calcium levels, induces oxidative stress, and activates STAT-3 and NF-kappa B. Proc. Natl. Acad. Sci. USA 2001, 98, 9599–9604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maycotte, P.; Gearheart, C.M.; Barnard, R.; Aryal, S.; Mulcahy Levy, J.M.; Fosmire, S.P.; Hansen, R.J.; Morgan, M.J.; Porter, C.C.; Gustafson, D.L.; et al. STAT3-mediated autophagy dependence identifies subtypes of breast cancer where autophagy inhibition can be efficacious. Cancer Res. 2014, 74, 2579–2590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinsey, C.G.; Camolotto, S.A.; Boespflug, A.M.; Guillen, K.P.; Foth, M.; Truong, A.; Schuman, S.S.; Shea, J.E.; Seipp, M.T.; Yap, J.T.; et al. Protective autophagy elicited by RAF-->MEK-->ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med. 2019, 25, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.J.; Wang, S.H.; Chen, K.C.; Kuei, H.P.; Shih, Y.L.; Hou, S.Y.; Chiu, W.T.; Hsiao, S.H.; Shih, C.M. Evodiamine, a plant alkaloid, induces calcium/JNK-mediated autophagy and calcium/mitochondria-mediated apoptosis in human glioblastoma cells. Chem. Biol. Interact. 2013, 205, 20–28. [Google Scholar] [CrossRef]
- Doycheva, D.; Xu, N.; Kaur, H.; Malaguit, J.; McBride, D.W.; Tang, J.; Zhang, J.H. Adenoviral TMBIM6 vector attenuates ER-stress-induced apoptosis in a neonatal hypoxic-ischemic rat model. Dis. Model. Mech. 2019, 12, dmm040352. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.K.; Bhattarai, K.R.; Junjappa, R.P.; Ahn, J.H.; Pagire, S.H.; Yoo, H.J.; Han, J.; Lee, D.; Kim, K.W.; Kim, H.R.; et al. TMBIM6/BI-1 contributes to cancer progression through assembly with mTORC2 and AKT activation. Nat. Commun. 2020, 11, 4012. [Google Scholar] [CrossRef]
- Kim, H.K.; Lee, G.H.; Bhattarai, K.R.; Lee, M.S.; Back, S.H.; Kim, H.R.; Chae, H.J. TMBIM6 (transmembrane BAX inhibitor motif containing 6) enhances autophagy through regulation of lysosomal calcium. Autophagy 2021, 17, 761–778. [Google Scholar] [CrossRef] [Green Version]
- Yadav, R.K.; Lee, G.H.; Lee, H.Y.; Li, B.; Jung, H.E.; Rashid, H.O.; Choi, M.K.; Yadav, B.K.; Kim, W.H.; Kim, K.W.; et al. TMBIM6 (transmembrane BAX inhibitor motif containing 6) enhances autophagy and reduces renal dysfunction in a cyclosporine A-induced nephrotoxicity model. Autophagy 2015, 11, 1760–1774. [Google Scholar] [CrossRef] [Green Version]
- Sano, R.; Hou, Y.C.; Hedvat, M.; Correa, R.G.; Shu, C.W.; Krajewska, M.; Diaz, P.W.; Tamble, C.M.; Quarato, G.; Gottlieb, R.A.; et al. Endoplasmic reticulum protein BI-1 regulates Ca2+-mediated bioenergetics to promote autophagy. Genes Dev. 2012, 26, 1041–1054. [Google Scholar] [CrossRef] [Green Version]
- Florea, A.M.; Varghese, E.; McCallum, J.E.; Mahgoub, S.; Helmy, I.; Varghese, S.; Gopinath, N.; Sass, S.; Theis, F.J.; Reifenberger, G.; et al. Calcium-regulatory proteins as modulators of chemotherapy in human neuroblastoma. Oncotarget 2017, 8, 22876–22893. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 2016, 16, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Galati, S.; Boni, C.; Gerra, M.C.; Lazzaretti, M.; Buschini, A. Autophagy: A Player in response to Oxidative Stress and DNA Damage. Oxid. Med. Cell Longev. 2019, 2019, 5692958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eliopoulos, A.G.; Havaki, S.; Gorgoulis, V.G. DNA Damage Response and Autophagy: A Meaningful Partnership. Front. Genet. 2016, 7, 204. [Google Scholar] [CrossRef] [Green Version]
- Cremona, C.A.; Behrens, A. ATM signalling and cancer. Oncogene 2014, 33, 3351–3360. [Google Scholar] [CrossRef] [Green Version]
- Edifizi, D.; Nolte, H.; Babu, V.; Castells-Roca, L.; Mueller, M.M.; Brodesser, S.; Kruger, M.; Schumacher, B. Multilayered Reprogramming in Response to Persistent DNA Damage in C. elegans. Cell Rep. 2017, 20, 2026–2043. [Google Scholar] [CrossRef] [Green Version]
- Moslemi, M.; Vafaei, M.; Khani, P.; Soheili, M.; Nedaeinia, R.; Manian, M.; Moradi, Y.; Sohrabi, E. The prevalence of ataxia telangiectasia mutated (ATM) variants in patients with breast cancer patients: A systematic review and meta-analysis. Cancer Cell Int. 2021, 21, 474. [Google Scholar] [CrossRef]
- Tran, H.; Brunet, A.; Grenier, J.M.; Datta, S.R.; Fornace, A.J., Jr.; DiStefano, P.S.; Chiang, L.W.; Greenberg, M.E. DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science 2002, 296, 530–534. [Google Scholar] [CrossRef] [Green Version]
- Tsai, W.B.; Chung, Y.M.; Takahashi, Y.; Xu, Z.; Hu, M.C. Functional interaction between FOXO3a and ATM regulates DNA damage response. Nat. Cell Biol. 2008, 10, 460–467. [Google Scholar] [CrossRef] [Green Version]
- Alasiri, G.; Fan, L.Y.; Zona, S.; Goldsbrough, I.G.; Ke, H.L.; Auner, H.W.; Lam, E.W. ER stress and cancer: The FOXO forkhead transcription factor link. Mol. Cell Endocrinol. 2018, 462, 67–81. [Google Scholar] [CrossRef]
- Rupp, M.; Hagenbuchner, J.; Rass, B.; Fiegl, H.; Kiechl-Kohlendorfer, U.; Obexer, P.; Ausserlechner, M.J. FOXO3-mediated chemo-protection in high-stage neuroblastoma depends on wild-type TP53 and SESN3. Oncogene 2017, 36, 6190–6203. [Google Scholar] [CrossRef] [PubMed]
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006, 126, 121–134. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Liang, Q.Y.; Chen, H.K.; Wu, P.F.; Feng, Z.Y.; Ma, X.M.; Wu, H.R.; Zhou, G.Q. DRAM1 regulates the migration and invasion of hepatoblastoma cells via autophagy-EMT pathway. Oncol. Lett. 2018, 16, 2427–2433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, C.; Zhao, Y. Autophagy and DNA damage repair. Genome Instab. Dis. 2020, 1, 172–183. [Google Scholar] [CrossRef]
- Abedin, M.J.; Wang, D.; McDonnell, M.A.; Lehmann, U.; Kelekar, A. Autophagy delays apoptotic death in breast cancer cells following DNA damage. Cell Death Differ. 2007, 14, 500–510. [Google Scholar] [CrossRef]
- Liang, D.H.; Choi, D.S.; Ensor, J.E.; Kaipparettu, B.A.; Bass, B.L.; Chang, J.C. The autophagy inhibitor chloroquine targets cancer stem cells in triple negative breast cancer by inducing mitochondrial damage and impairing DNA break repair. Cancer Lett. 2016, 376, 249–258. [Google Scholar] [CrossRef] [Green Version]
- Arnould, T.; Michel, S.; Renard, P. Mitochondria Retrograde Signaling and the UPR mt: Where Are We in Mammals? Int. J. Mol. Sci. 2015, 16, 18224–18251. [Google Scholar] [CrossRef] [Green Version]
- McConkey, D.J. The integrated stress response and proteotoxicity in cancer therapy. Biochem. Biophys. Res. Commun. 2017, 482, 450–453. [Google Scholar] [CrossRef] [Green Version]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [Green Version]
- Talloczy, Z.; Jiang, W.; Virgin, H.W.t.; Leib, D.A.; Scheuner, D.; Kaufman, R.J.; Eskelinen, E.L.; Levine, B. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc. Natl. Acad. Sci. USA 2002, 99, 190–195. [Google Scholar] [CrossRef] [Green Version]
- Lu, P.D.; Harding, H.P.; Ron, D. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J. Cell Biol. 2004, 167, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Humeau, J.; Leduc, M.; Cerrato, G.; Loos, F.; Kepp, O.; Kroemer, G. Phosphorylation of eukaryotic initiation factor-2alpha (eIF2alpha) in autophagy. Cell Death. Dis. 2020, 11, 433. [Google Scholar] [CrossRef] [PubMed]
- Corazzari, M.; Gagliardi, M.; Fimia, G.M.; Piacentini, M. Endoplasmic Reticulum Stress, Unfolded Protein Response, and Cancer Cell Fate. Front. Oncol. 2017, 7, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giampietri, C.; Petrungaro, S.; Conti, S.; Facchiano, A.; Filippini, A.; Ziparo, E. Cancer Microenvironment and Endoplasmic Reticulum Stress Response. Mediat. Inflamm. 2015, 2015, 417281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazova, R.; Camp, R.L.; Klump, V.; Siddiqui, S.F.; Amaravadi, R.K.; Pawelek, J.M. Punctate LC3B expression is a common feature of solid tumors and associated with proliferation, metastasis, and poor outcome. Clin. Cancer Res. 2012, 18, 370–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margariti, A.; Li, H.; Chen, T.; Martin, D.; Vizcay-Barrena, G.; Alam, S.; Karamariti, E.; Xiao, Q.; Zampetaki, A.; Zhang, Z.; et al. XBP1 mRNA splicing triggers an autophagic response in endothelial cells through BECLIN-1 transcriptional activation. J. Biol. Chem. 2013, 288, 859–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, M.M.; Ni, J.D.; Song, D.; Ding, M.; Huang, J. Interplay between unfolded protein response and autophagy promotes tumor drug resistance. Oncol. Lett. 2015, 10, 1959–1969. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Yang, M.; Zhao, J.; Wang, J.; Zhang, Y.; Zhang, Q. High expression of LC3B is associated with progression and poor outcome in triple-negative breast cancer. Med. Oncol. 2013, 30, 475. [Google Scholar] [CrossRef]
- Hart, L.S.; Cunningham, J.T.; Datta, T.; Dey, S.; Tameire, F.; Lehman, S.L.; Qiu, B.; Zhang, H.; Cerniglia, G.; Bi, M.; et al. ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. J. Clin. Investig. 2012, 122, 4621–4634. [Google Scholar] [CrossRef] [Green Version]
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [Green Version]
- Albert, A.E.; Adua, S.J.; Cai, W.L.; Arnal-Estape, A.; Cline, G.W.; Liu, Z.; Zhao, M.; Cao, P.D.; Mariappan, M.; Nguyen, D.X. Adaptive Protein Translation by the Integrated Stress Response Maintains the Proliferative and Migratory Capacity of Lung Adenocarcinoma Cells. Mol. Cancer Res. 2019, 17, 2343–2355. [Google Scholar] [CrossRef] [PubMed]
- Pataer, A.; Ozpolat, B.; Shao, R.; Cashman, N.R.; Plotkin, S.S.; Samuel, C.E.; Lin, S.H.; Kabil, N.N.; Wang, J.; Majidi, M.; et al. Therapeutic targeting of the PI4K2A/PKR lysosome network is critical for misfolded protein clearance and survival in cancer cells. Oncogene 2020, 39, 801–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pataer, A.; Swisher, S.G.; Roth, J.A.; Logothetis, C.J.; Corn, P.G. Inhibition of RNA-dependent protein kinase (PKR) leads to cancer cell death and increases chemosensitivity. Cancer Biol. Ther. 2009, 8, 245–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iurlaro, R.; Munoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.-H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef] [Green Version]
- McCullough, K.D.; Martindale, J.L.; Klotz, L.O.; Aw, T.Y.; Holbrook, N.J. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell Biol. 2001, 21, 1249–1259. [Google Scholar] [CrossRef] [Green Version]
- Logue, S.E.; Cleary, P.; Saveljeva, S.; Samali, A. New directions in ER stress-induced cell death. Apoptosis 2013, 18, 537–546. [Google Scholar] [CrossRef]
- Shore, G.C.; Papa, F.R.; Oakes, S.A. Signaling cell death from the endoplasmic reticulum stress response. Curr. Opin. Cell Biol. 2011, 23, 143–149. [Google Scholar] [CrossRef] [Green Version]
- Button, R.W.; Roberts, S.L.; Willis, T.L.; Hanemann, C.O.; Luo, S. Accumulation of autophagosomes confers cytotoxicity. J. Biol. Chem. 2017, 292, 13599–13614. [Google Scholar] [CrossRef] [Green Version]
- Deegan, S.; Saveljeva, S.; Logue, S.E.; Pakos-Zebrucka, K.; Gupta, S.; Vandenabeele, P.; Bertrand, M.J.; Samali, A. Deficiency in the mitochondrial apoptotic pathway reveals the toxic potential of autophagy under ER stress conditions. Autophagy 2014, 10, 1921–1936. [Google Scholar] [CrossRef] [PubMed]
- Tomar, D.; Prajapati, P.; Sripada, L.; Singh, K.; Singh, R.; Singh, A.K.; Singh, R. TRIM13 regulates caspase-8 ubiquitination, translocation to autophagosomes and activation during ER stress induced cell death. Biochim. Biophys. Acta 2013, 1833, 3134–3144. [Google Scholar] [CrossRef] [Green Version]
- Laussmann, M.A.; Passante, E.; Dussmann, H.; Rauen, J.A.; Wurstle, M.L.; Delgado, M.E.; Devocelle, M.; Prehn, J.H.; Rehm, M. Proteasome inhibition can induce an autophagy-dependent apical activation of caspase-8. Cell Death Differ. 2011, 18, 1584–1597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, M.M.; Takahashi, Y.; Khan, O.; Park, S.; Hori, T.; Yun, J.; Sharma, A.K.; Amin, S.; Hu, C.D.; Zhang, J.; et al. Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated caspase-8 activation and apoptosis. J. Biol. Chem. 2012, 287, 12455–12468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, E.; Mehnert, J.M.; Chan, C.S. Autophagy, Metabolism, and Cancer. Clin. Cancer Res. 2015, 21, 5037–5046. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Zhang, J.; Li, K.; Deng, L.; Wang, H. Combination of an Autophagy Inducer and an Autophagy Inhibitor: A Smarter Strategy Emerging in Cancer Therapy. Front. Pharmacol. 2020, 11, 408. [Google Scholar] [CrossRef] [Green Version]
- Vu, H.T.; Kobayashi, M.; Hegazy, A.M.; Tadokoro, Y.; Ueno, M.; Kasahara, A.; Takase, Y.; Nomura, N.; Peng, H.; Ito, C.; et al. Autophagy inhibition synergizes with calcium mobilization to achieve efficient therapy of malignant gliomas. Cancer Sci. 2018, 109, 2497–2508. [Google Scholar] [CrossRef]
- Nakano, K.; Masui, T.; Yogo, A.; Uchida, Y.; Sato, A.; Kasai, Y.; Nagai, K.; Anazawa, T.; Kawaguchi, Y.; Uemoto, S. Chloroquine induces apoptosis in pancreatic neuroendocrine neoplasms via endoplasmic reticulum stress. Endocr. Relat. Cancer 2020, 27, 431–439. [Google Scholar] [CrossRef]
- Wu, J.; Chen, S.; Liu, H.; Zhang, Z.; Ni, Z.; Chen, J.; Yang, Z.; Nie, Y.; Fan, D. Tunicamycin specifically aggravates ER stress and overcomes chemoresistance in multidrug-resistant gastric cancer cells by inhibiting N-glycosylation. J. Exp. Clin. Cancer Res. 2018, 37, 272. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Li, Q.; Pang, J.; Jin, H.; Li, H.; Yang, X. Blocking autophagy enhances the pro-apoptotic effect of bufalin on human gastric cancer cells through endoplasmic reticulum stress. Biol. Open 2017, 6, 1416–1422. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Zhou, P.; Chen, X.; Zhao, L.; Tan, J.; Yang, Y.; Fang, Y.; Zhou, J. The novel autophagy inhibitor elaiophylin exerts antitumor activity against multiple myeloma with mutant TP53 in part through endoplasmic reticulum stress-induced apoptosis. Cancer Biol. Ther. 2017, 18, 584–595. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Bamodu, O.A.; Huang, W.C.; Zucha, M.A.; Lin, Y.K.; Wu, A.T.H.; Huang, C.C.; Lee, W.H.; Yuan, C.C.; Hsiao, M.; et al. 4-Acetylantroquinonol B suppresses autophagic flux and improves cisplatin sensitivity in highly aggressive epithelial cancer through the PI3K/Akt/mTOR/p70S6K signaling pathway. Toxicol. Appl. Pharmacol. 2017, 325, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Du, H.; Huang, Z.; Zhang, P.; Yue, Y.; Wang, W.; Liu, W.; Zeng, J.; Ma, J.; Chen, G.; et al. Thymoquinone induces apoptosis in bladder cancer cell via endoplasmic reticulum stress-dependent mitochondrial pathway. Chem. Biol. Interact. 2018, 292, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Hong, L.; Xu, J.; Zhong, G.; Gu, Q.; Gu, Q.; Guan, Y.; Zheng, X.; Dai, Q.; Luo, X.; et al. Discovery of a small molecule targeting autophagy via ATG4B inhibition and cell death of colorectal cancer cells in vitro and in vivo. Autophagy 2019, 15, 295–311. [Google Scholar] [CrossRef] [Green Version]
- DeVorkin, L.; Hattersley, M.; Kim, P.; Ries, J.; Spowart, J.; Anglesio, M.S.; Levi, S.M.; Huntsman, D.G.; Amaravadi, R.K.; Winkler, J.D.; et al. Autophagy Inhibition Enhances Sunitinib Efficacy in Clear Cell Ovarian Carcinoma. Mol. Cancer. Res. 2017, 15, 250–258. [Google Scholar] [CrossRef] [Green Version]
- Tang, F.; Hu, P.; Yang, Z.; Xue, C.; Gong, J.; Sun, S.; Shi, L.; Zhang, S.; Li, Z.; Yang, C.; et al. SBI0206965, a novel inhibitor of Ulk1, suppresses non-small cell lung cancer cell growth by modulating both autophagy and apoptosis pathways. Oncol. Rep. 2017, 37, 3449–3458. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Gao, Y.; Wang, D.; Xu, Z.; Sun, W.; Ren, P. Autophagy Inhibitor (LY294002) and 5-fluorouracil (5-FU) Combination-Based Nanoliposome for Enhanced Efficacy Against Esophageal Squamous Cell Carcinoma. Nanoscale Res. Lett. 2018, 13, 325. [Google Scholar] [CrossRef] [Green Version]
- Giorgi, C.; Bonora, M.; Missiroli, S.; Poletti, F.; Ramirez, F.G.; Morciano, G.; Morganti, C.; Pandolfi, P.P.; Mammano, F.; Pinton, P. Intravital imaging reveals p53-dependent cancer cell death induced by phototherapy via calcium signaling. Oncotarget 2015, 6, 1435–1445. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Zhong, F.; Yi, X.; Shi, Z.; Ou, F.; Xu, Z.; Zuo, Y. Application of an Autophagy-Related Gene Prognostic Risk Model Based on TCGA Database in Cervical Cancer. Front. Genet. 2020, 11, 616998. [Google Scholar] [CrossRef]
- Chen, K.; Yang, D.; Zhao, F.; Wang, S.; Ye, Y.; Sun, W.; Lu, H.; Ruan, Z.; Xu, J.; Wang, T.; et al. Autophagy and Tumor Database: ATdb, a novel database connecting autophagy and tumor. Database 2020, 2020, baaa052. [Google Scholar] [CrossRef]
- Lindstrom, S.; Finucane, H.; Bulik-Sullivan, B.; Schumacher, F.R.; Amos, C.I.; Hung, R.J.; Rand, K.; Gruber, S.B.; Conti, D.; Permuth, J.B.; et al. Quantifying the Genetic Correlation between Multiple Cancer Types. Cancer Epidemiol. Biomark. Prev. 2017, 26, 1427–1435. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
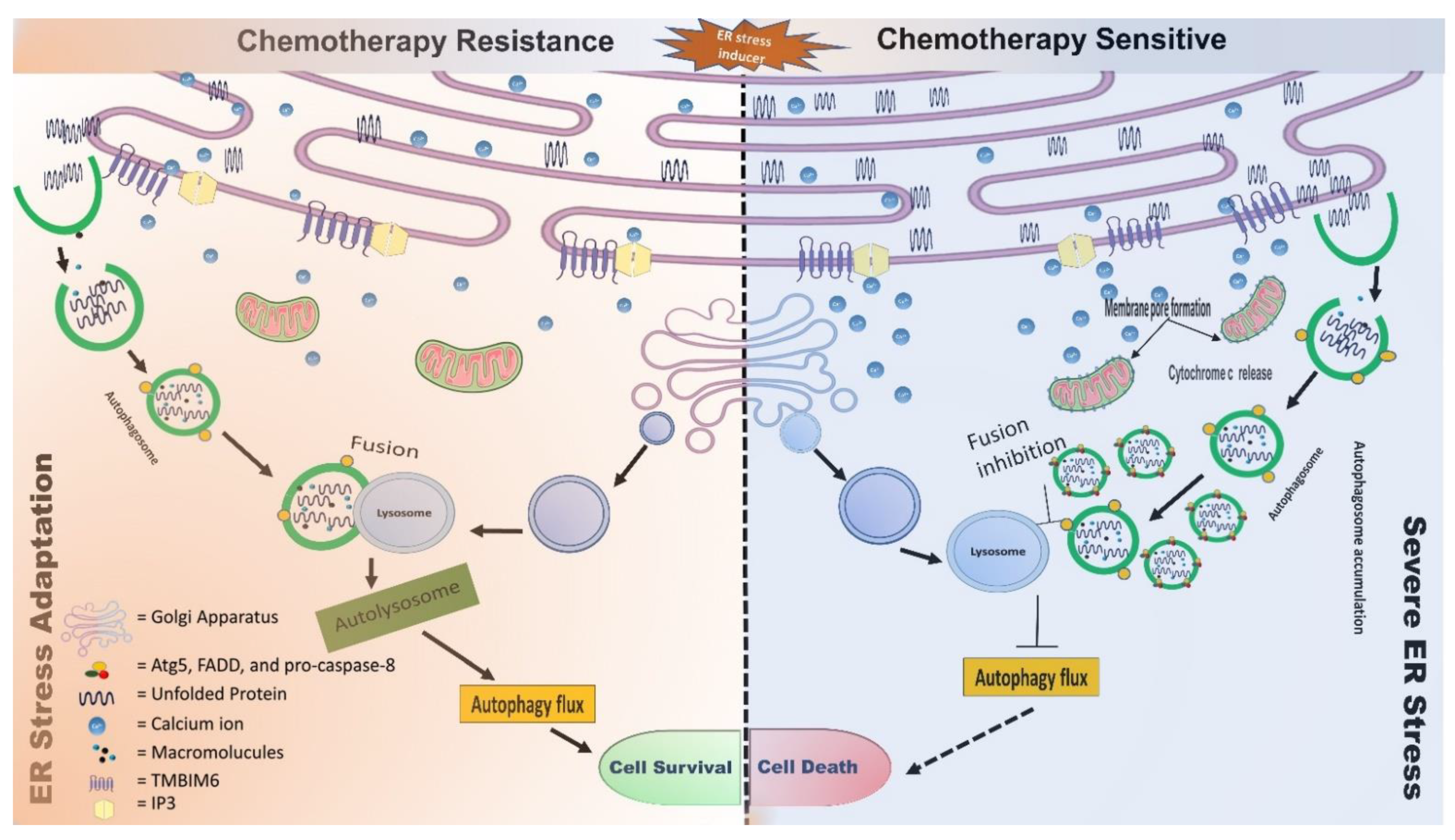{kind=link}
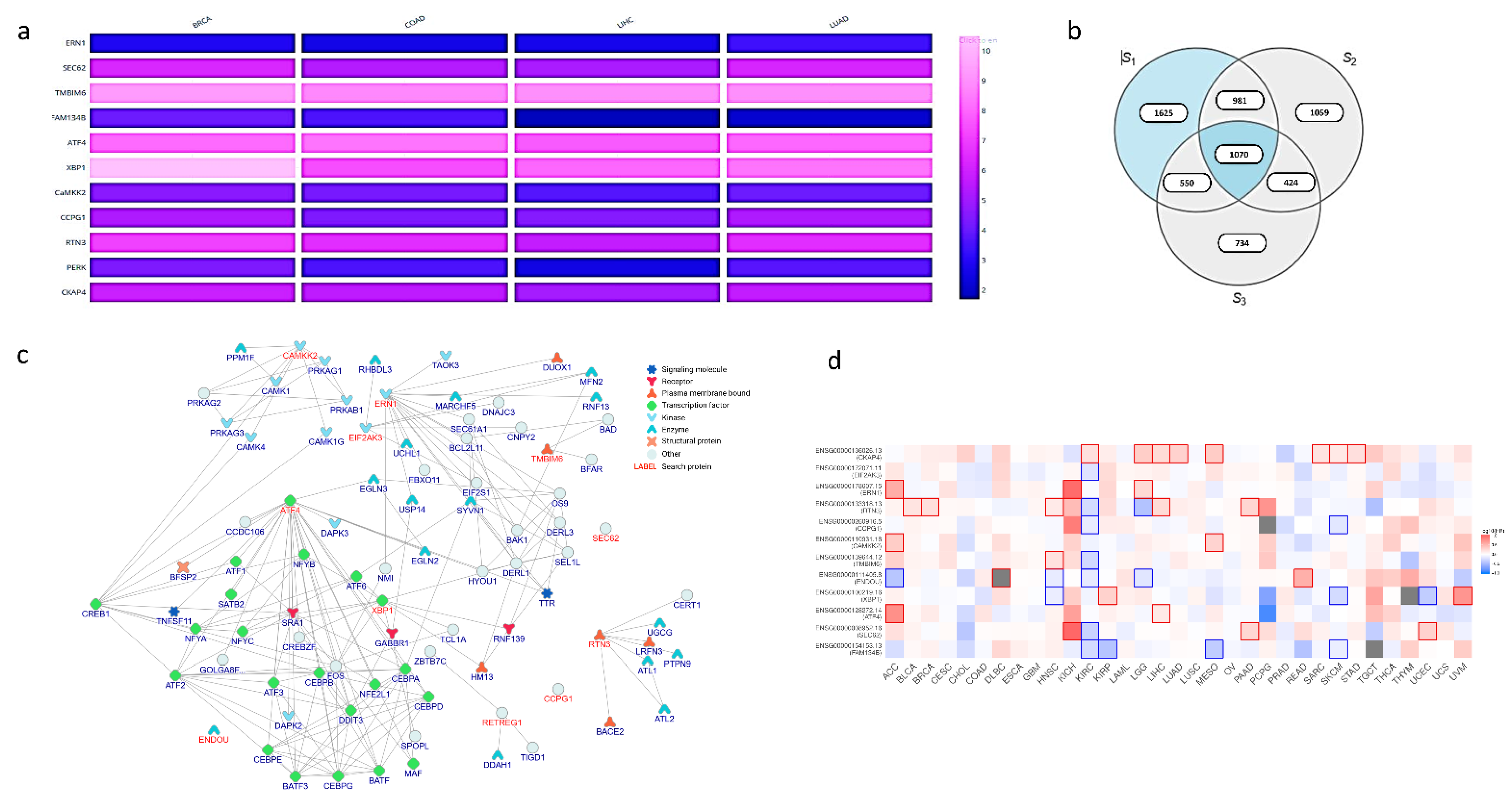{kind=link}
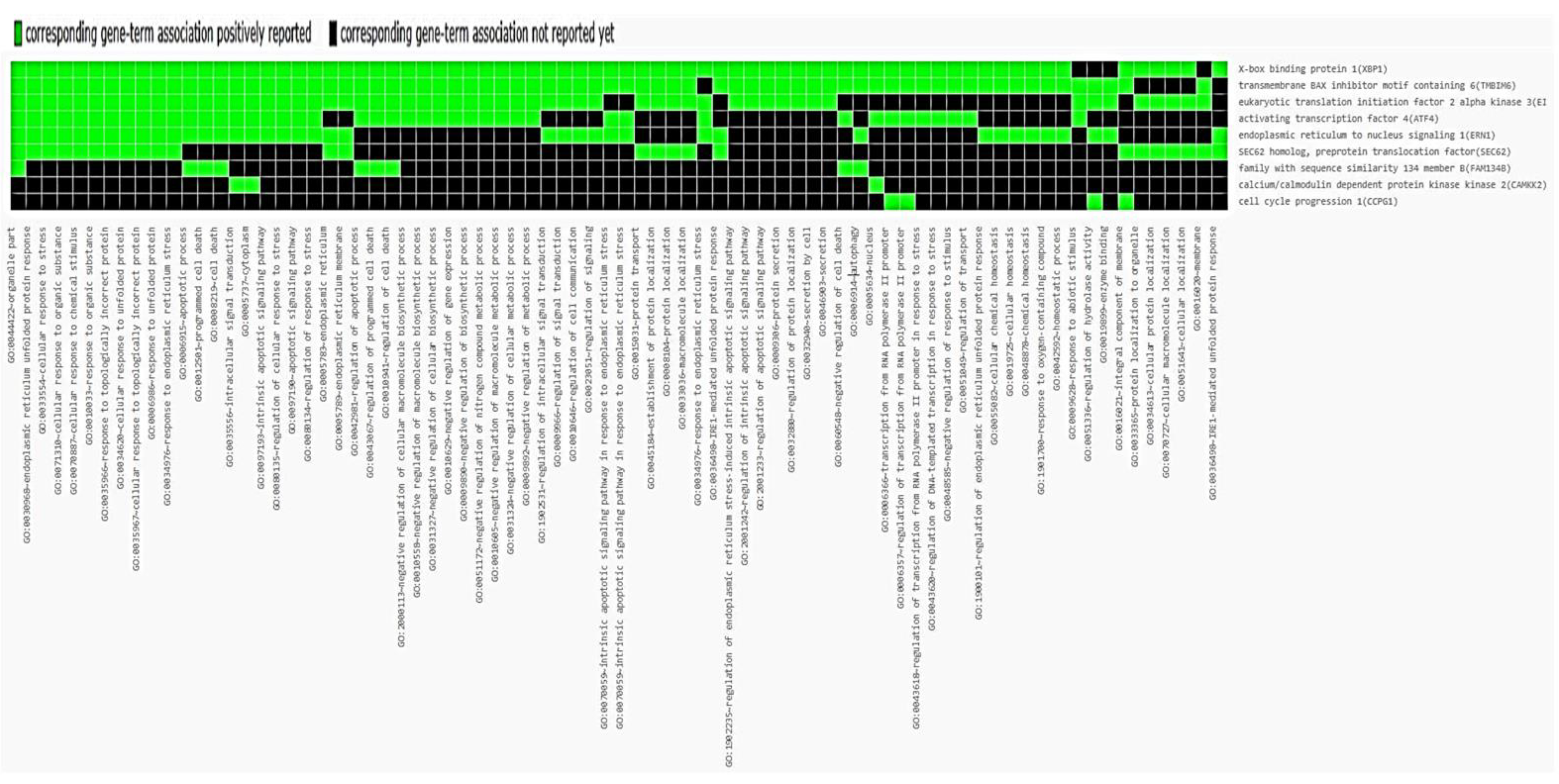{kind=link}
| Cell Death Mode | Apoptosis Marker | Stress Inducer | Canonical and Noncanonical Stress Response | Cell Type | Reference |
|---|---|---|---|---|---|
| Apoptosis | Mcl-1 (anti), Noxa | 2-Deoxyglucose (2-DG) | ATF4 | Rhabdomyosarcoma | [129] |
| Apoptosis | Puma, Noxa, | Tunicamycin, thapsigargin | IRE1α, ATF6 Protective effect | Melanoma cells | [130] |
| Apoptosis | Death receptor 5 | Thapsigargin | PERK, ATF4-CHOP | MDA-MB231 | [131] |
| Apoptosis | Noxa | Hypericin-based photodynamic therapy | PERK | T24 bladder carcinoma | [132] |
| Apoptosis | Noxa | Bortezomib | ATF4 | Neuroectodermal tumor cells | [133] |
| Apoptosis | caspase-8 activation | Bortezomib/MG132 | HEK293, MDAMB231, MCF7 | [134] | |
| Apoptosis | CHOP | Z36 | p-PERK | HeLa cells | [135] |
| Apoptosis | Cleaved PARP | Brefeldin A (BFA) | C/EBP homologous protein (CHOP) | A549 cells | [136] |
| Apoptosis | Bax, Bak | Tunicamycin, Thapsigargin, Brefeldin A | MEF | [137] |
| Drug Name | Target | Types of Tumor | ER Stress | Autophagy | Reference |
|---|---|---|---|---|---|
| Bortezomib | Molecular chaperones Hsp70 | Breast cancers | Integrated stress response (ISR) and UPR activation | eIF2α mediated induction of autophagy | [150] |
| Imiquimod (IMQ) | Toll-like receptor (TLR) 7 ligand | Basal cell carcinoma | PKR is activated to phosphorylate eIF2α | PKR markedly enhanced IMQ-induced conversion of LC3-I to LC3-II | [151] |
| Cucurbitacin B (CuB) | cell cycle in G2/M phases | Melanoma cells | Phosphorylation of eIF2α also mediates the conversion of LC3-I | CuB-induced autophagy was associated with c-Jun N-terminal kinase (JNK) activation | [152] |
| Sorafenib | Tyrosine kinase inhibitor | Hepatocellular carcinoma | PERK-ATF4 pathway correlation with drug resistance | Beclin 1 plays a role in ER stress-related autophagy | [153] |
| Oxaliplatin | DNA damage | Colon Cancer | Activation of UPR components | Enhanced autophagy genes (ATG5 or Beclin 1). | [154] |
| 5-fluorouracil | Inhibition of thymidylate synthase (TS) and incorporation of its metabolites into RNA and DNA | Colon cancer | Activation of different signal branches in UPR | Protective autophagy is induced by Beclin-1 expression conversion of LC3I to LC3II. | [155,156,157] |
| Thapsigargin | Oxidative DNA damage | Osteosarcoma | PERK mediated cytoprotection | Inhibits mTORC1 activity and induces autophagy | [158] |
| Paclitaxel | ER stress-inducing agents against cancer | Breast cancer | IRE1α-ERK1/2 mediates the activation of RSK2 | RSK2 enhanced autophagy | [159] |
| Sunitinib and Gemcitabine | Vascular endothelial growth factor receptor | Pancreatic ductal adenocarcinoma (PDAC) | GRP78 and XBP1 splicing mediated ER homeostasis. | Increased lysosomal enzymatic activity and autophagy | [106] |
| Brigatinib | Anaplastic lymphoma kinase (ALK) inhibitor | Non-small-cell lung cancer | IRE1α/JNK signaling | Enhanced ER-Phagy | [139] |
| Tunicamycin | Target calcium | Breast cancer | IRE1-TRAF2 complex formation | Autophagy regulated by IRE1/JNK/Beclin 1 | [99] |
| Temozolomide (TMZ) | These agents act directly on DNA | Glioblastoma multiforme (GBM) | Activated PERK, XBP1 | Autophagy genes (e.g., ATG5, ATG7, BECN1) | [160] |
| Verotoxin-1 | ER stress inducer | Lymphoma cancer | ER stress response by IRE1 and ATF6—two ER stress sensors | Protective role through ER-phagy, depending on the cell line | [161] |
| Tetrahydrocannabinol (THC) | Stimulation of ER stress | Glioma cell | Eukaryotic translation initiation factor 2α (eIF2α) phosphorylation | ER stress response promotes autophagy | [162] |
| Cinnamomum cassia | ER stress inducer | Gastric cancer | ER stress-induced eIF2α/ATF4 axis induces AMP-activated protein kinase (AMPK) phosphorylation | Ca2+ release induced autophagy by Beclin 1, ATG5, and LC3B expression | [163] |
| Name of Drug | Target | ER Stress | Class | Cancer Types | Reference |
|---|---|---|---|---|---|
| Chloroquine (CQ) | Fusion process of autophagosome and lysosome | Increase apoptosis via PERK-eIF2α-ATF4 pathway | Lysosomotropic agents | Pancreatic neuroendocrine neoplasms (PanNENs) | [228] |
| 3 methyladenine (3-MA) | PI3K | Activated IRE1α-ER stress sensor | PI3K inhibitor | Colon & breast cancer | [99] |
| Hydroxychloroquine (HCQ) | Lysosomal cathepsin D | Not observed | Lysosome inhibitor | Gastric cancer | [229] |
| Bafilomycin A1 | V-ATPase | ER stress via the IRE1 α -JNK pathway | Lysosomal H+-ATPase inhibitor | Gastric cancer cells | [230] |
| Elaiophylin | Inhibition of autophagy flux | Fetal ER stress-induced apoptosis | Autophagy flux inhibitor | Multiple myeloma (MM) | [231] |
| 4-Acetylantroquinonol B | Inhibition of autophagy flux | Not observed | Autophagy flux inhibitors | Epithelial cancer cells | [232] |
| Thymoquinone | Permeabilization of the lysosome membrane | ER stress markers (GRP78, CHOP) | Lysosomotropic agents | Bladder cancer | [233] |
| S130 | Target ATG4B | Not observed | Autophagy inhibitor | Colorectal cancer | [234] |
| Single Drug | Cancer Types | Dose | Mechanism | Combination | Target | Final Outcome | Ref |
|---|---|---|---|---|---|---|---|
| Vitexin | Breast cancer | Vitexin concentration 20 µM | Disrupts FAM134B-BiP complex inhibits ER-phagy and suppresses breast cancer progression | Tunicamycin | ER stress inducer | When used in combination with ER stress inducer, synergistically stunted cell proliferation | [103] |
| Temozolomide (TMZ) | Malignant Glioma | 10–15 μmol/mL active concentration | calcium-mobilizing compound activated autophagy-related gene BECN1, ATG7 | Chloroquine | Autophagy Inhibitor | CQ treatment-enhanced TMZ can synergize with the activation of Ca2+ signaling and reactive oxygen species (ROS) | [227] |
| Sunitinib | Ovarian Carcinoma | 10 μm/mL sunitinib | Hypoxia–induce autophagy | Lys05 Active concentration 10 μmol/L | Autophagy Inhibitor | Autophagy inhibition as a strategy to overcome resistance to RTK inhibitors such as sunitinib | [235] |
| Brigatinib | Colorectal Cancer | 2 μM Brigatinib | Activates ER-Phagy via ER stress-signaling pathway | Chloroquine | Autolysosome Inhibitor | Inhibition of ER-phagy enhances the susceptibility of CRC cells to brigatinib in vitro and in vivo | [139] |
| Carboplatin | Triple-negative breast cancer | 24 mg/kg | DNA damage induces autophagy | Chloroquine | Autophagy Inhibitor | Autophagy inhibitor CQ causes sustained oxidative DNA damage | [196] |
| Cisplatin and Daunorubicin | Non-small cell lung cancer (NSCLC) | 10 µM effective concentration | DNA alkylating agents | 0.1 µM effective concentration of SBI0206965 | Inhibition of Ulk1 | Inhibition of Ulk1 suppresses NSCLC cell growth and sensitizes NSCLC cells to cisplatin by modulating both autophagy and apoptosis pathways | [236] |
| Fluorouracil (5FU) | Esophageal squamous cell carcinoma | 5 mg/kg | Inhibits the nucleotide synthesis | LY294002 Effective concentration 2 µM | Autophagy Inhibitor | Autophagy inhibitor (LY) will disrupt the protective mechanism of cancer cells | [237] |
| Photodynamic therapy (PDT) aluminum phthalocyanine chloride | MEF cells | Active concentration 15 µM | photosensitizing agents target Ca2+ signaling | Not use | Generating oxidative stress capable of causing damage to cell membranes, proteins, or DNA. Increasing intracellular Ca2+concentration and activating the apoptotic pathway | [238] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alam, R.; Kabir, M.F.; Kim, H.-R.; Chae, H.-J. Canonical and Noncanonical ER Stress-Mediated Autophagy Is a Bite the Bullet in View of Cancer Therapy. Cells 2022, 11, 3773. https://doi.org/10.3390/cells11233773
Alam R, Kabir MF, Kim H-R, Chae H-J. Canonical and Noncanonical ER Stress-Mediated Autophagy Is a Bite the Bullet in View of Cancer Therapy. Cells. 2022; 11(23):3773. https://doi.org/10.3390/cells11233773
Chicago/Turabian StyleAlam, Rashedul, Mohammad Fazlul Kabir, Hyung-Ryong Kim, and Han-Jung Chae. 2022. "Canonical and Noncanonical ER Stress-Mediated Autophagy Is a Bite the Bullet in View of Cancer Therapy" Cells 11, no. 23: 3773. https://doi.org/10.3390/cells11233773