
Potential Therapeutic Use of Stem Cells for Prion Diseases


Abstract
:1. Introduction
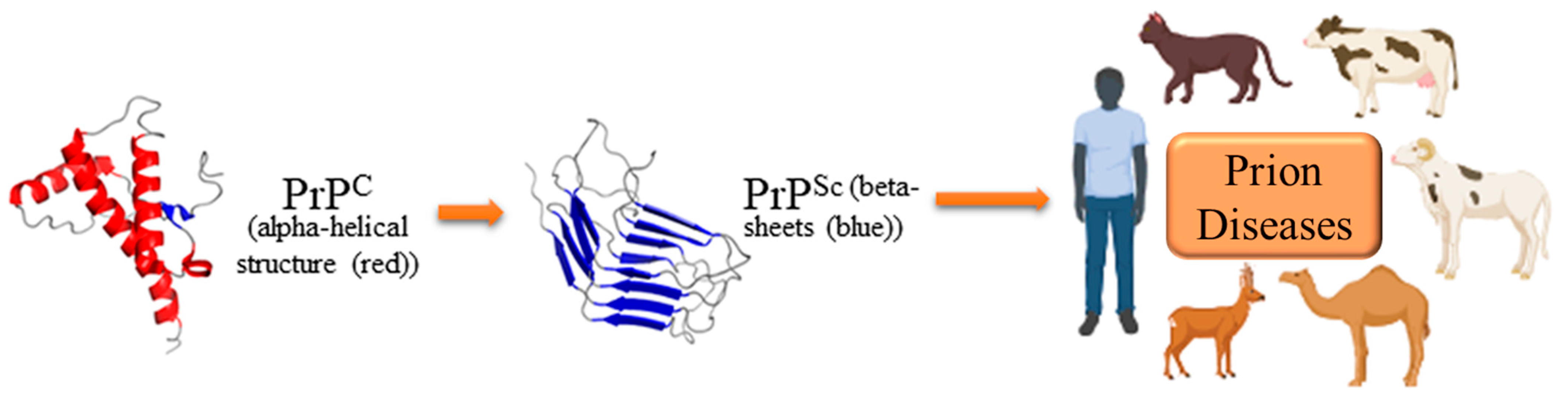
2. Molecular Biology and Pathogenesis of Prion Diseases
3. Current Therapeutic Strategies for Prion Diseases
3.1. Target PrPC
3.2. Inhibit the Conversion of PrPC to PrPSc
3.3. Clearance of PrPSc
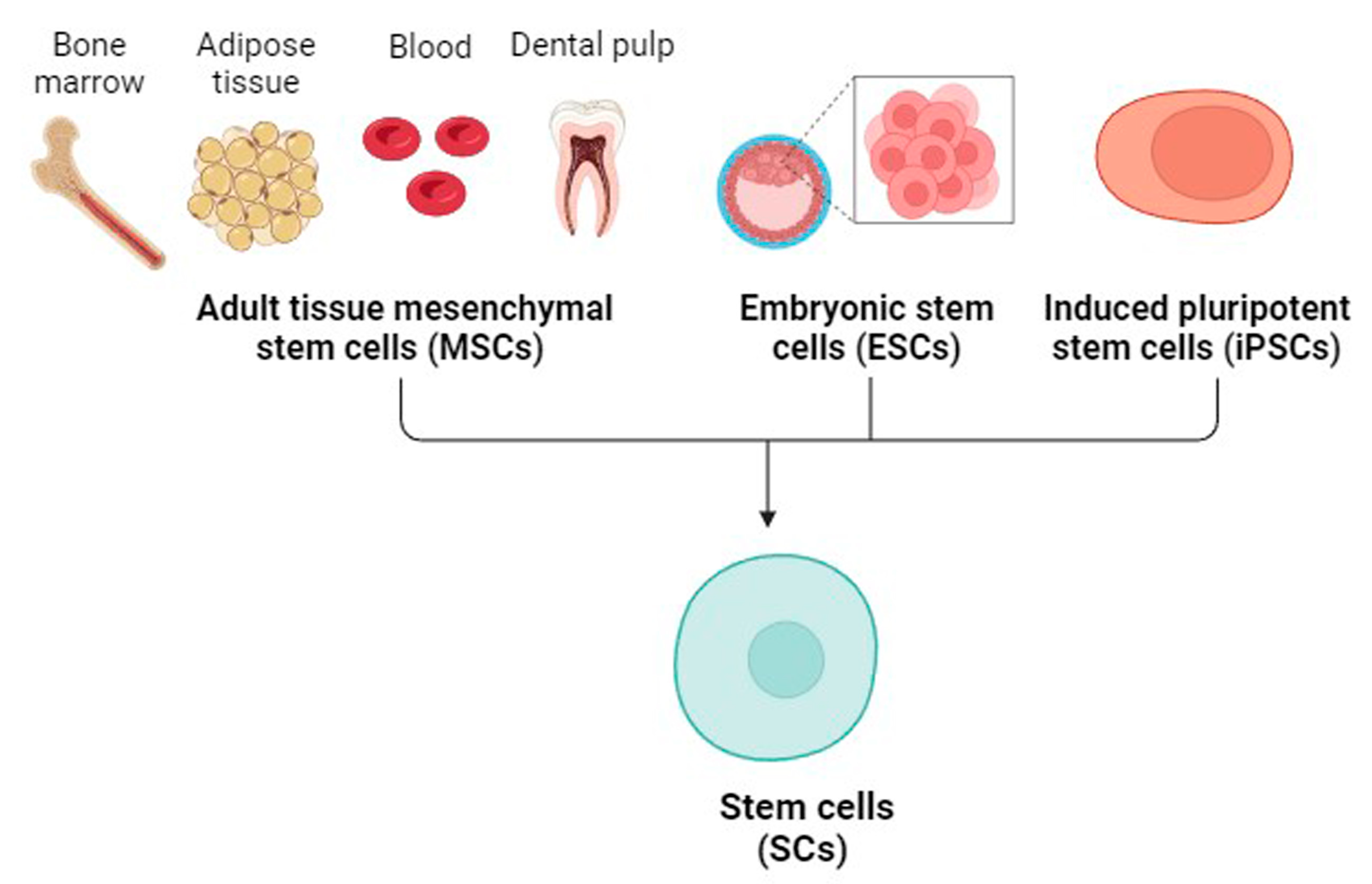
4. Mesenchymal SCs (MSCs)
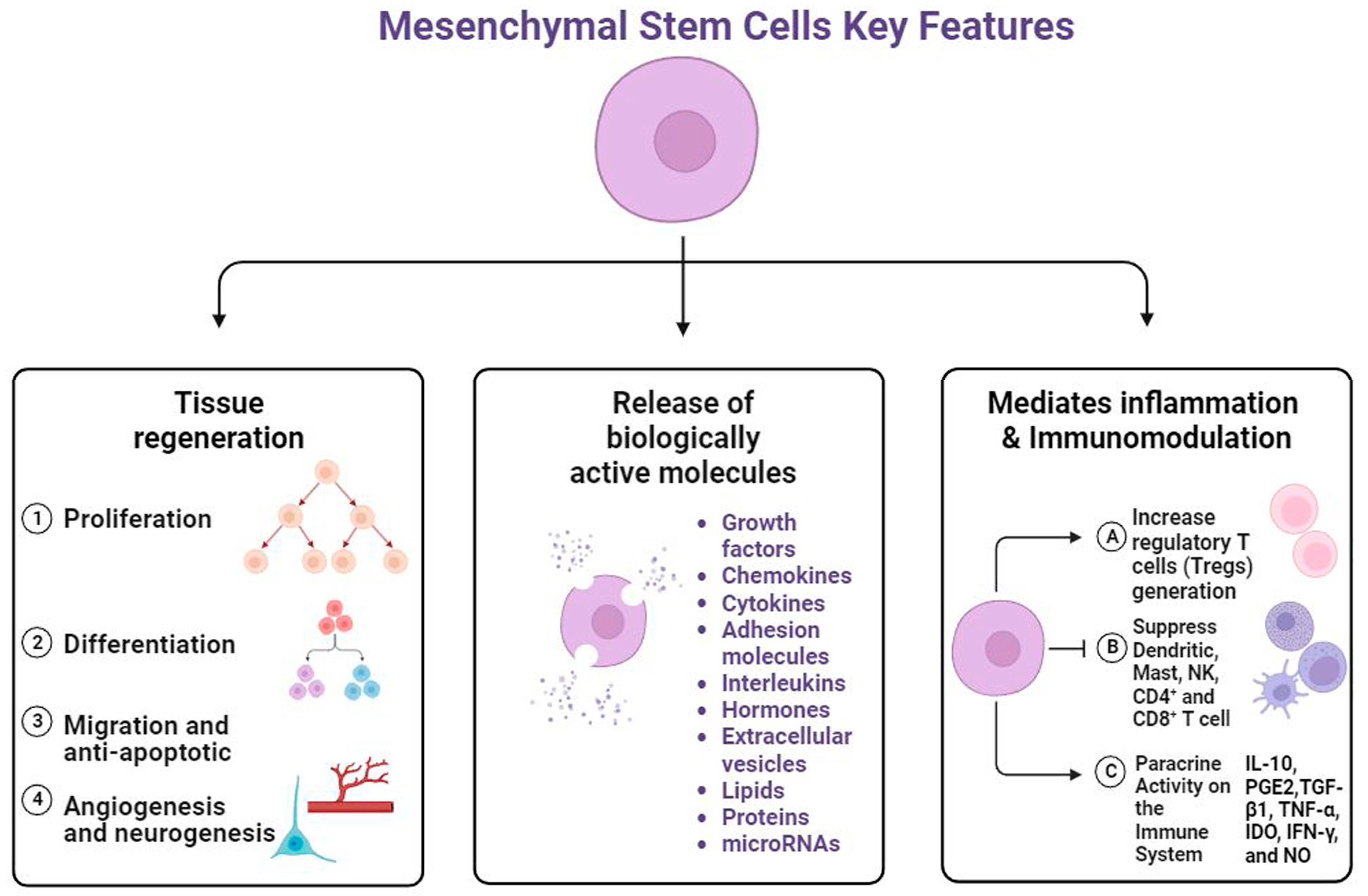
5. Regenerative Potential of MSCs
6. MSCs as a Cell Model for Prion Diseases
- Because SCs are derived from healthy tissues, they constantly exhibit normal physiological conditions.
- The genomes of SCs are devoid of aberrations and can be exceptionally durable [112].
- SCs can differentiate into a variety of cell types.
- SCs can produce organoids, which allow cellular processes to be investigated in the context of differentiated tissue.
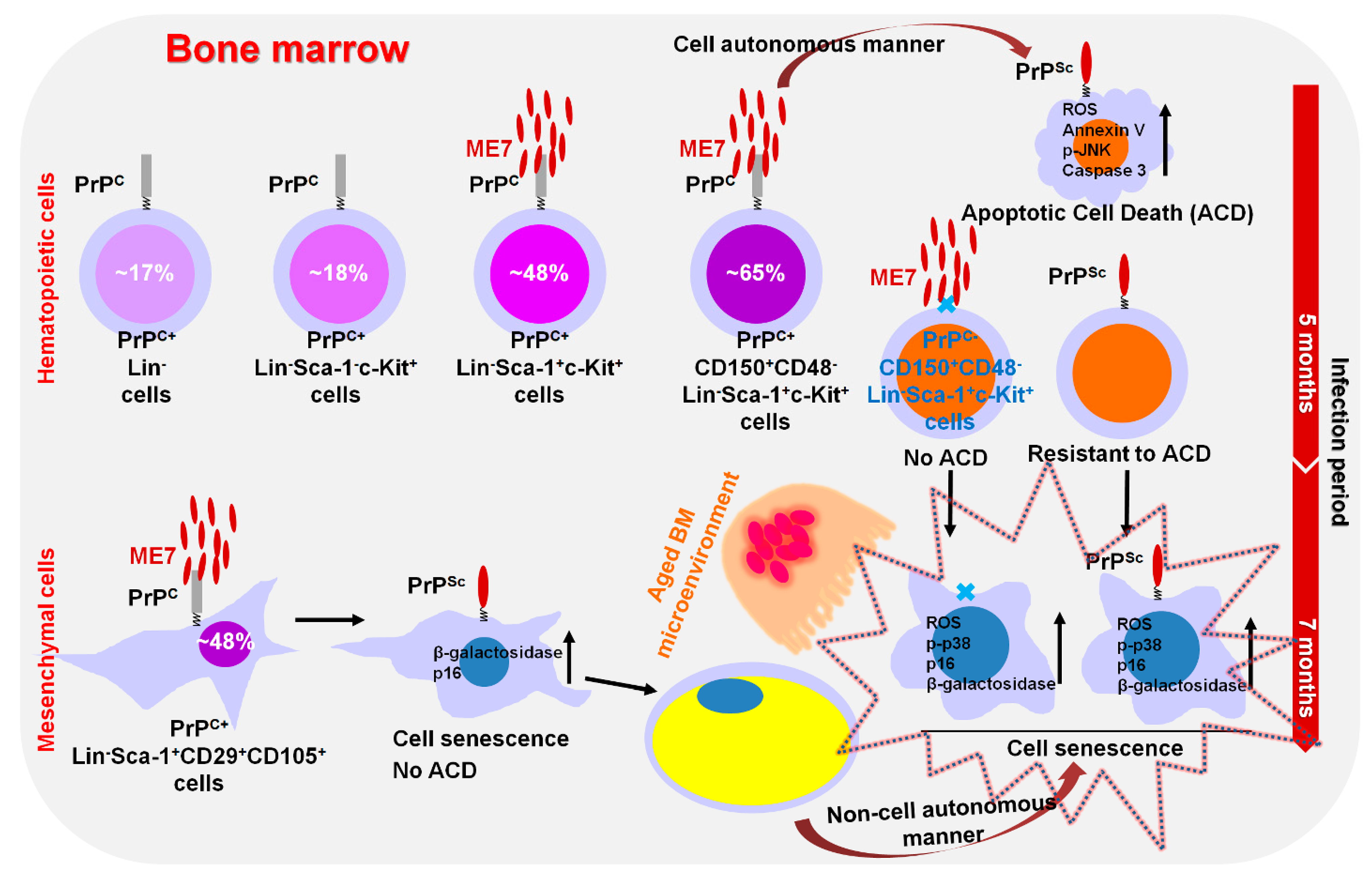
7. Modulation of Hematopoietic Stem/Progenitor Cell Fate by Prion Disease
8. MSCs as a Potential Therapy for Prion Diseases
9. NSCs as a Potential Therapy for Prion Diseases
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rivera-Milla, E.; Oidtmann, B.; Panagiotidis, C.H.; Baier, M.; Sklaviadis, T.; Hoffmann, R.; Zhou, Y.; Solis, G.P.; Stuermer, C.A.; Málaga-Trillo, E. Disparate evolution of prion protein domains and the distinct origin of Doppel- and prion-related loci revealed by fish-to-mammal comparisons. FASEB J. 2006, 20, 317–319. [Google Scholar] [CrossRef] [PubMed]
- Bendheim, P.E.; Brown, H.R.; Rudelli, R.D.; Scala, L.J.; Goller, N.L.; Wen, G.Y.; Kascsak, R.J.; Cashman, N.R.; Bolton, D.C. Nearly ubiquitous tissue distribution of the scrapie agent precursor protein. Neurology 1992, 42, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Steele, A.D.; Emsley, J.G.; Ozdinler, P.H.; Lindquist, S.; Macklis, J.D. Prion protein (PrPc) positively regulates neural precursor proliferation during developmental and adult mammalian neurogenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 3416–3421. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.H.; Hajj, G.N.; Muras, A.G.; Mancini, G.L.; Castro, R.M.; Ribeiro, K.C.; Brentani, R.R.; Linden, R.; Martins, V.R. Interaction of cellular prion and stress-inducible protein 1 promotes neuritogenesis and neuroprotection by distinct signaling pathways. J. Neurosci. 2005, 25, 11330–11339. [Google Scholar] [CrossRef] [PubMed]
- Linden, R.; Martins, V.R.; Prado, M.A.; Cammarota, M.; Izquierdo, I.; Brentani, R.R. Physiology of the prion protein. Physiol. Rev. 2008, 88, 673–728. [Google Scholar] [CrossRef]
- Aguzzi, A.; Baumann, F.; Bremer, J. The prion’s elusive reason for being. Annu. Rev. Neurosci. 2008, 31, 439–477. [Google Scholar] [CrossRef]
- Hajj, G.N.; Lopes, M.H.; Mercadante, A.F.; Veiga, S.S.; da Silveira, R.B.; Santos, T.G.; Ribeiro, K.C.; Juliano, M.A.; Jacchieri, S.G.; Zanata, S.M.; et al. Cellular prion protein interaction with vitronectin supports axonal growth and is compensated by integrins. J. Cell Sci. 2007, 120 Pt 11, 1915–1926. [Google Scholar] [CrossRef]
- Zhang, C.C.; Steele, A.D.; Lindquist, S.; Lodish, H.F. Prion protein is expressed on long-term repopulating hematopoietic stem cells and is important for their self-renewal. Proc. Natl. Acad. Sci. USA 2006, 103, 2184–2189. [Google Scholar] [CrossRef]
- Martellucci, S.; Manganelli, V.; Santacroce, C.; Santilli, F.; Piccoli, L.; Sorice, M.; Mattei, V. Role of Prion protein-EGFR multimolecular complex during neuronal differentiation of human dental pulp-derived stem cells. Prion 2018, 12, 117–126. [Google Scholar] [CrossRef]
- Scheckel, C.; Aguzzi, A. Prions, prionoids and protein misfolding disorders. Nat. Rev. Genet. 2018, 19, 405–418. [Google Scholar] [CrossRef]
- Watts, J.C.; Balachandran, A.; Westaway, D. The expanding universe of prion diseases. PLoS Pathog. 2006, 2, e26. [Google Scholar] [CrossRef] [PubMed]
- Jeong, B.H.; Kim, Y.S. Genetic studies in human prion diseases. J. Korean Med. Sci. 2014, 29, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Krance, S.H.; Luke, R.; Shenouda, M.; Israwi, A.R.; Colpitts, S.J.; Darwish, L.; Strauss, M.; Watts, J.C. Cellular models for discovering prion disease therapeutics: Progress and challenges. J. Neurochem. 2020, 153, 150–172. [Google Scholar] [CrossRef] [PubMed]
- Jeong, B.H.; Kim, N.H.; Jin, J.K.; Choi, J.K.; Lee, Y.J.; Kim, J.I.; Choi, E.K.; Carp, R.I.; Kim, Y.S. Reduction of prion infectivity and levels of scrapie prion protein by lithium aluminum hydride: Implications for RNA in prion diseases. J. Neuropathol. Exp. Neurol. 2009, 68, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Barreca, M.L.; Iraci, N.; Biggi, S.; Cecchetti, V.; Biasini, E. Pharmacological Agents Targeting the Cellular Prion Protein. Pathogens 2018, 7, 27. [Google Scholar] [CrossRef]
- Zakrzewski, W.; Dobrzyński, M.; Szymonowicz, M.; Rybak, Z. Stem cells: Past, present, and future. Stem Cell Res. Ther. 2019, 10, 68. [Google Scholar] [CrossRef]
- Zayed, M.; Iohara, K.; Watanabe, H.; Ishikawa, M.; Tominaga, M.; Nakashima, M. Characterization of stable hypoxia-preconditioned dental pulp stem cells compared with mobilized dental pulp stem cells for application for pulp regenerative therapy. Stem Cell Res. Ther. 2021, 12, 302. [Google Scholar] [CrossRef]
- Colby, D.W.; Prusiner, S.B. Prions. Cold Spring Harb. Perspect. Biol. 2011, 3, a006833. [Google Scholar] [CrossRef]
- Prusiner, S.B. Molecular biology of prion diseases. Science 1991, 252, 1515–1522. [Google Scholar] [CrossRef]
- Kim, Y.C.; Jeong, B.H. Creutzfeldt-Jakob Disease Incidence, South Korea, 2001–2019. Emerg Infect Dis 2022, 28, 1863–1866. [Google Scholar] [CrossRef]
- Will, R.G.; Ironside, J.W.; Zeidler, M.; Cousens, S.N.; Estibeiro, K.; Alperovitch, A.; Poser, S.; Pocchiari, M.; Hofman, A.; Smith, P.G. A new variant of Creutzfeldt-Jakob disease in the UK. Lancet 1996, 347, 921–925. [Google Scholar] [CrossRef]
- Zahn, R.; Liu, A.; Lührs, T.; Riek, R.; von Schroetter, C.; López García, F.; Billeter, M.; Calzolai, L.; Wider, G.; Wüthrich, K. NMR solution structure of the human prion protein. Proc. Natl. Acad. Sci. USA 2000, 97, 145–150. [Google Scholar] [CrossRef]
- Westergard, L.; Christensen, H.M.; Harris, D.A. The cellular prion protein (PrP(C)): Its physiological function and role in disease. Biochim. Biophys. Acta 2007, 1772, 629–644. [Google Scholar] [CrossRef]
- Martellucci, S.; Santacroce, C.; Santilli, F.; Manganelli, V.; Sorice, M.; Mattei, V. Prion Protein in Stem Cells: A Lipid Raft Component Involved in the Cellular Differentiation Process. Int. J. Mol. Sci. 2020, 21, 4168. [Google Scholar] [CrossRef] [PubMed]
- Kretzschmar, H.A.; Prusiner, S.B.; Stowring, L.E.; DeArmond, S.J. Scrapie prion proteins are synthesized in neurons. Am. J. Pathol. 1986, 122, 1–5. [Google Scholar]
- Moser, M.; Colello, R.J.; Pott, U.; Oesch, B. Developmental expression of the prion protein gene in glial cells. Neuron 1995, 14, 509–517. [Google Scholar] [CrossRef]
- Aguzzi, A.; Polymenidou, M. Mammalian prion biology: One century of evolving concepts. Cell 2004, 116, 313–327. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Chen, M.; Zhu, C. Neuroinflammation in Prion Disease. Int. J. Mol. Sci. 2021, 22, 2196. [Google Scholar] [CrossRef] [PubMed]
- Soto, C.; Satani, N. The intricate mechanisms of neurodegeneration in prion diseases. Trends Mol. Med. 2011, 17, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Funez, P.; Casas-Tinto, S.; Zhang, Y.; Gómez-Velazquez, M.; Morales-Garza, M.A.; Cepeda-Nieto, A.C.; Castilla, J.; Soto, C.; Rincon-Limas, D.E. In vivo generation of neurotoxic prion protein: Role for hsp70 in accumulation of misfolded isoforms. PLoS Genet. 2009, 5, e1000507. [Google Scholar] [CrossRef]
- Jeong, B.H.; Ju, W.K.; Huh, K.; Lee, E.A.; Choi, I.S.; Im, J.H.; Choi, E.K.; Kim, Y.S. Molecular analysis of prion protein gene (PRNP) in Korean patients with Creutzfeldt-Jakob disease. J. Korean Med. Sci. 1998, 13, 234–240. [Google Scholar] [CrossRef]
- Kim, Y.C.; Park, K.J.; Hwang, J.Y.; Park, H.C.; Kang, H.E.; Sohn, H.J.; Jeong, B.H. In-depth examination of PrP(Sc) in Holstein cattle carrying the E211K somatic mutation of the bovine prion protein gene (PRNP). Transbound. Emerg. Dis. 2022, 69, e356–e361. [Google Scholar] [CrossRef]
- Kim, Y.C.; Won, S.Y.; Jeong, B.H. Altered expression of glymphatic system-related proteins in prion diseases: Implications for the role of the glymphatic system in prion diseases. Cell Mol. Immunol. 2021, 18, 2281–2283. [Google Scholar] [CrossRef] [PubMed]
- Vanni, I.; Iacobone, F.; D’Agostino, C.; Giovannelli, M.; Pirisinu, L.; Altmeppen, H.C.; Castilla, J.; Torres, J.M.; Agrimi, U.; Nonno, R. An optimized Western blot assay provides a comprehensive assessment of the physiological endoproteolytic processing of the prion protein. J. Biol. Chem. 2023, 299, 102823. [Google Scholar] [CrossRef] [PubMed]
- Le, N.T.T.; Wu, B.; Harris, D.A. Prion neurotoxicity. Brain Pathol. 2019, 29, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Büeler, H.; Aguzzi, A.; Sailer, A.; Greiner, R.A.; Autenried, P.; Aguet, M.; Weissmann, C. Mice devoid of PrP are resistant to scrapie. Cell 1993, 73, 1339–1347. [Google Scholar] [CrossRef] [PubMed]
- Brandner, S.; Isenmann, S.; Raeber, A.; Fischer, M.; Sailer, A.; Kobayashi, Y.; Marino, S.; Weissmann, C.; Aguzzi, A. Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature 1996, 379, 339–343. [Google Scholar] [CrossRef]
- Mallucci, G.; Dickinson, A.; Linehan, J.; Klöhn, P.C.; Brandner, S.; Collinge, J. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science 2003, 302, 871–874. [Google Scholar] [CrossRef]
- Ferreira, N.C.; Marques, I.A.; Conceição, W.A.; Macedo, B.; Machado, C.S.; Mascarello, A.; Chiaradia-Delatorre, L.D.; Yunes, R.A.; Nunes, R.J.; Hughson, A.G.; et al. Anti-prion activity of a panel of aromatic chemical compounds: In vitro and in silico approaches. PLoS ONE 2014, 9, e84531. [Google Scholar] [CrossRef]
- Sweeney, P.; Park, H.; Baumann, M.; Dunlop, J.; Frydman, J.; Kopito, R.; McCampbell, A.; Leblanc, G.; Venkateswaran, A.; Nurmi, A.; et al. Protein misfolding in neurodegenerative diseases: Implications and strategies. Transl. Neurodegener. 2017, 6, 6. [Google Scholar] [CrossRef]
- Raymond, G.J.; Zhao, H.T.; Race, B.; Raymond, L.D.; Williams, K.; Swayze, E.E.; Graffam, S.; Le, J.; Caron, T.; Stathopoulos, J.; et al. Antisense oligonucleotides extend survival of prion-infected mice. JCI Insight 2019, 5, e131175. [Google Scholar] [CrossRef]
- Burke, C.M.; Walsh, D.J.; Steele, A.D.; Agrimi, U.; Di Bari, M.A.; Watts, J.C.; Supattapone, S. Full restoration of specific infectivity and strain properties from pure mammalian prion protein. PLoS Pathog. 2019, 15, e1007662. [Google Scholar] [CrossRef]
- Shim, K.H.; Sharma, N.; An, S.S.A. Prion therapeutics: Lessons from the past. Prion 2022, 16, 265–294. [Google Scholar] [CrossRef]
- Cashman, N.R.; Caughey, B. Prion diseases—close to effective therapy? Nat. Rev. Drug Discov. 2004, 3, 874–884. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, M.; Candelise, N.; Llorens, F.; Zerr, I. Amplification and Detection of Minuscule Amounts of Misfolded Prion Protein by Using the Real-Time Quaking-Induced Conversion. Methods Mol. Biol. 2018, 1779, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Foutz, A.; Appleby, B.S.; Hamlin, C.; Liu, X.; Yang, S.; Cohen, Y.; Chen, W.; Blevins, J.; Fausett, C.; Wang, H.; et al. Diagnostic and prognostic value of human prion detection in cerebrospinal fluid. Ann. Neurol. 2017, 81, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Hyeon, J.W.; Kim, S.Y.; Lee, S.M.; Lee, J.; An, S.S.A.; Lee, M.K.; Lee, Y.S. Anti-Prion Screening for Acridine, Dextran, and Tannic Acid using Real Time–Quaking Induced Conversion: A Comparison with PrPSc-Infected Cell Screening. PLoS ONE 2017, 12, e0170266. [Google Scholar] [CrossRef] [PubMed]
- Karagianni, K.; Pettas, S.; Kanata, E.; Lioulia, E.; Thune, K.; Schmitz, M.; Tsamesidis, I.; Lymperaki, E.; Xanthopoulos, K.; Sklaviadis, T.; et al. Carnosic Acid and Carnosol Display Antioxidant and Anti-Prion Properties in In Vitro and Cell-Free Models of Prion Diseases. Antioxidants 2022, 11, 726. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, M.; Candelise, N.; Kanata, E.; Llorens, F.; Thüne, K.; Villar-Piqué, A.; da Silva Correia, S.M.; Dafou, D.; Sklaviadis, T.; Appelhans, D.; et al. Validation of Poly(Propylene Imine) Glycodendrimers Towards Their Anti-prion Conversion Efficiency. Mol. Neurobiol. 2020, 57, 1863–1874. [Google Scholar] [CrossRef]
- Schmitz, M.; Cramm, M.; Llorens, F.; Candelise, N.; Müller-Cramm, D.; Varges, D.; Schulz-Schaeffer, W.J.; Zafar, S.; Zerr, I. Application of an in vitro-amplification assay as a novel pre-screening test for compounds inhibiting the aggregation of prion protein scrapie. Sci. Rep. 2016, 6, 28711. [Google Scholar] [CrossRef]
- Vascellari, S.; Orrù, C.D.; Caughey, B. Real-Time Quaking-Induced Conversion Assays for Prion Diseases, Synucleinopathies, and Tauopathies. Front. Aging Neurosci. 2022, 14, 853050. [Google Scholar] [CrossRef] [PubMed]
- Uliassi, E.; Nikolic, L.; Bolognesi, M.L.; Legname, G. Therapeutic strategies for identifying small molecules against prion diseases. Cell Tissue Res. 2023, 392, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Zaccagnini, L.; Rossetti, G.; Tran, T.H.; Salzano, G.; Gandini, A.; Colini Baldeschi, A.; Bolognesi, M.L.; Carloni, P.; Legname, G. In silico/in vitro screening and hit evaluation identified new phenothiazine anti-prion derivatives. Eur. J. Med. Chem. 2020, 196, 112295. [Google Scholar] [CrossRef] [PubMed]
- Staderini, M.; Vanni, S.; Baldeschi, A.C.; Giachin, G.; Zattoni, M.; Celauro, L.; Ferracin, C.; Bistaffa, E.; Moda, F.; Pérez, D.I.; et al. Bifunctional carbazole derivatives for simultaneous therapy and fluorescence imaging in prion disease murine cell models. Eur. J. Med. Chem. 2023, 245, 114923. [Google Scholar] [CrossRef]
- Moreno, J.A.; Halliday, M.; Molloy, C.; Radford, H.; Verity, N.; Axten, J.M.; Ortori, C.A.; Willis, A.E.; Fischer, P.M.; Barrett, D.A.; et al. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci. Transl. Med. 2013, 5, 206ra138. [Google Scholar] [CrossRef] [PubMed]
- Jheng, C.-P.; Lee, C.-I. Combination of structure-based virtual screening, molecular docking and molecular dynamics approaches for the discovery of anti-prion fibril flavonoids. Front. Mol. Biosci. 2023, 9, 1088733. [Google Scholar] [CrossRef]
- Hoang, D.M.; Pham, P.T.; Bach, T.Q.; Ngo, A.T.L.; Nguyen, Q.T.; Phan, T.T.K.; Nguyen, G.H.; Le, P.T.T.; Hoang, V.T.; Forsyth, N.R.; et al. Stem cell-based therapy for human diseases. Signal Transduct. Target. Ther. 2022, 7, 272. [Google Scholar] [CrossRef]
- Lo, B.; Parham, L. Ethical issues in stem cell research. Endocr. Rev. 2009, 30, 204–213. [Google Scholar] [CrossRef]
- Al Abbar, A.; Ngai, S.C.; Nograles, N.; Alhaji, S.Y.; Abdullah, S. Induced Pluripotent Stem Cells: Reprogramming Platforms and Applications in Cell Replacement Therapy. Biores Open Access 2020, 9, 121–136. [Google Scholar] [CrossRef]
- Qiao, Y.; Agboola, O.S.; Hu, X.; Wu, Y.; Lei, L. Tumorigenic and Immunogenic Properties of Induced Pluripotent Stem Cells: A Promising Cancer Vaccine. Stem Cell Rev. Rep. 2020, 16, 1049–1061. [Google Scholar] [CrossRef]
- Keating, A. Mesenchymal stromal cells. Curr. Opin. Hematol. 2006, 13, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Zayed, M.; Caniglia, C.; Misk, N.; Dhar, M.S. Donor-Matched Comparison of Chondrogenic Potential of Equine Bone Marrow- and Synovial Fluid-Derived Mesenchymal Stem Cells: Implications for Cartilage Tissue Regeneration. Front. Vet. Sci. 2017, 3, 121. [Google Scholar] [CrossRef] [PubMed]
- Friedenstein, A.J.; Chailakhyan, R.K.; Gerasimov, U.V. Bone marrow osteogenic stem cells: In vitro cultivation and transplantation in diffusion chambers. Cell Tissue Kinet. 1987, 20, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Li, X.; Zhang, Y.; Han, Y.; Chang, F.; Ding, J. Mesenchymal Stem Cells for Regenerative Medicine. Cells 2019, 8, 886. [Google Scholar] [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Chan, J.L.; Tang, K.C.; Patel, A.P.; Bonilla, L.M.; Pierobon, N.; Ponzio, N.M.; Rameshwar, P. Antigen-presenting property of mesenchymal stem cells occurs during a narrow window at low levels of interferon-gamma. Blood 2006, 107, 4817–4824. [Google Scholar] [CrossRef]
- MacQueen, L.; Sun, Y.; Simmons, C.A. Mesenchymal stem cell mechanobiology and emerging experimental platforms. J. R. Soc. Interface 2013, 10, 20130179. [Google Scholar] [CrossRef]
- Seo, Y.; Shin, T.H.; Kim, H.S. Current Strategies to Enhance Adipose Stem Cell Function: An Update. Int. J. Mol. Sci. 2019, 20, 3827. [Google Scholar] [CrossRef]
- Mabuchi, Y.; Okawara, C.; Méndez-Ferrer, S.; Akazawa, C. Cellular Heterogeneity of Mesenchymal Stem/Stromal Cells in the Bone Marrow. Front. Cell Dev. Biol. 2021, 9, 689366. [Google Scholar] [CrossRef]
- Zayed, M.N.; Schumacher, J.; Misk, N.; Dhar, M.S. Effects of pro-inflammatory cytokines on chondrogenesis of equine mesenchymal stromal cells derived from bone marrow or synovial fluid. Vet. J. 2016, 217, 26–32. [Google Scholar] [CrossRef]
- Hoogduijn, M.J.; Dor, F.J. Mesenchymal stem cells: Are we ready for clinical application in transplantation and tissue regeneration? Front. Immunol. 2013, 4, 144. [Google Scholar] [CrossRef] [PubMed]
- Dufrane, D. Impact of Age on Human Adipose Stem Cells for Bone Tissue Engineering. Cell Transplant. 2017, 26, 1496–1504. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Greenberger, J.S.; Epperly, M.W.; Goff, J.P.; Adler, C.; Leboff, M.S.; Glowacki, J. Age-related intrinsic changes in human bone-marrow-derived mesenchymal stem cells and their differentiation to osteoblasts. Aging Cell 2008, 7, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Maldonado-Soto, A.R.; Oakley, D.H.; Wichterle, H.; Stein, J.; Doetsch, F.K.; Henderson, C.E. Stem cells in the nervous system. Am. J. Phys. Med. Rehabil. 2014, 93 (Suppl. S3), S132–S144. [Google Scholar] [CrossRef]
- Hass, R.; Kasper, C.; Böhm, S.; Jacobs, R. Different populations and sources of human mesenchymal stem cells (MSC): A comparison of adult and neonatal tissue-derived MSC. Cell Commun. Signal. 2011, 9, 12. [Google Scholar] [CrossRef]
- Sagaradze, G.D.; Basalova, N.A.; Efimenko, A.Y.; Tkachuk, V.A. Mesenchymal Stromal Cells as Critical Contributors to Tissue Regeneration. Front. Cell Dev. Biol. 2020, 8, 576176. [Google Scholar] [CrossRef]
- Liang, X.; Ding, Y.; Zhang, Y.; Tse, H.F.; Lian, Q. Paracrine mechanisms of mesenchymal stem cell-based therapy: Current status and perspectives. Cell Transplant. 2014, 23, 1045–1059. [Google Scholar] [CrossRef]
- Keshtkar, S.; Azarpira, N.; Ghahremani, M.H. Mesenchymal stem cell-derived extracellular vesicles: Novel frontiers in regenerative medicine. Stem Cell Res. Ther. 2018, 9, 63. [Google Scholar] [CrossRef]
- Gnecchi, M.; Zhang, Z.; Ni, A.; Dzau, V.J. Paracrine mechanisms in adult stem cell signaling and therapy. Circ. Res. 2008, 103, 1204–1219. [Google Scholar] [CrossRef]
- Gao, F.; Chiu, S.M.; Motan, D.A.L.; Zhang, Z.; Chen, L.; Ji, H.L.; Tse, H.F.; Fu, Q.L.; Lian, Q. Mesenchymal stem cells and immunomodulation: Current status and future prospects. Cell Death Dis. 2016, 7, e2062. [Google Scholar] [CrossRef]
- Li, M.; Sun, X.; Kuang, X.; Liao, Y.; Li, H.; Luo, D. Mesenchymal stem cells suppress CD8+ T cell-mediated activation by suppressing natural killer group 2, member D protein receptor expression and secretion of prostaglandin E2, indoleamine 2, 3-dioxygenase and transforming growth factor-β. Clin. Exp. Immunol. 2014, 178, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Luz-Crawford, P.; Kurte, M.; Bravo-Alegría, J.; Contreras, R.; Nova-Lamperti, E.; Tejedor, G.; Noël, D.; Jorgensen, C.; Figueroa, F.; Djouad, F.; et al. Mesenchymal stem cells generate a CD4+CD25+Foxp3+ regulatory T cell population during the differentiation process of Th1 and Th17 cells. Stem Cell Res. Ther. 2013, 4, 65. [Google Scholar] [CrossRef] [PubMed]
- Volpe, G.; Bernstock, J.D.; Peruzzotti-Jametti, L.; Pluchino, S. Modulation of host immune responses following non-hematopoietic stem cell transplantation: Translational implications in progressive multiple sclerosis. J. Neuroimmunol. 2019, 331, 11–27. [Google Scholar] [CrossRef] [PubMed]
- Gnecchi, M.; Danieli, P.; Malpasso, G.; Ciuffreda, M.C. Paracrine Mechanisms of Mesenchymal Stem Cells in Tissue Repair. Methods Mol. Biol. 2016, 1416, 123–146. [Google Scholar] [CrossRef]
- Zayed, M.; Iohara, K. Immunomodulation and Regeneration Properties of Dental Pulp Stem Cells: A Potential Therapy to Treat Coronavirus Disease 2019. Cell Transpl. 2020, 29, 963689720952089. [Google Scholar] [CrossRef]
- Kou, M.; Huang, L.; Yang, J.; Chiang, Z.; Chen, S.; Liu, J.; Guo, L.; Zhang, X.; Zhou, X.; Xu, X.; et al. Mesenchymal stem cell-derived extracellular vesicles for immunomodulation and regeneration: A next generation therapeutic tool? Cell Death Dis. 2022, 13, 580. [Google Scholar] [CrossRef]
- Pathan, M.; Fonseka, P.; Chitti, S.V.; Kang, T.; Sanwlani, R.; Van Deun, J.; Hendrix, A.; Mathivanan, S. Vesiclepedia 2019: A compendium of RNA, proteins, lipids and metabolites in extracellular vesicles. Nucleic Acids Res 2019, 47, D516–D519. [Google Scholar] [CrossRef]
- Bazzoni, R.; Takam Kamga, P.; Tanasi, I.; Krampera, M. Extracellular Vesicle-Dependent Communication Between Mesenchymal Stromal Cells and Immune Effector Cells. Front. Cell Dev. Biol. 2020, 8, 596079. [Google Scholar] [CrossRef]
- Gowen, A.; Shahjin, F.; Chand, S.; Odegaard, K.E.; Yelamanchili, S.V. Mesenchymal Stem Cell-Derived Extracellular Vesicles: Challenges in Clinical Applications. Front. Cell Dev. Biol. 2020, 8, 149. [Google Scholar] [CrossRef]
- Zhou, X.; Jin, N.; Wang, F.; Chen, B. Mesenchymal stem cells: A promising way in therapies of graft-versus-host disease. Cancer Cell Int. 2020, 20, 114. [Google Scholar] [CrossRef]
- Yao, L.; Hu, X.; Dai, K.; Yuan, M.; Liu, P.; Zhang, Q.; Jiang, Y. Mesenchymal stromal cells: Promising treatment for liver cirrhosis. Stem Cell Res. Ther. 2022, 13, 308. [Google Scholar] [CrossRef] [PubMed]
- Torres-Torrillas, M.; Rubio, M.; Damia, E.; Cuervo, B.; Del Romero, A.; Peláez, P.; Chicharro, D.; Miguel, L.; Sopena, J.J. Adipose-Derived Mesenchymal Stem Cells: A Promising Tool in the Treatment of Musculoskeletal Diseases. Int. J. Mol. Sci. 2019, 20, 3105. [Google Scholar] [CrossRef] [PubMed]
- Brandner, S.; Jaunmuktane, Z. Prion disease: Experimental models and reality. Acta Neuropathol. 2017, 133, 197–222. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, Y.; Kawagoe, K.; Chen, C.J.; Teruya, K.; Sakasegawa, Y.; Doh-ura, K. Orally administered amyloidophilic compound is effective in prolonging the incubation periods of animals cerebrally infected with prion diseases in a prion strain-dependent manner. J. Virol. 2007, 81, 12889–12898. [Google Scholar] [CrossRef]
- Vorberg, I.; Chiesa, R. Experimental models to study prion disease pathogenesis and identify potential therapeutic compounds. Curr. Opin. Pharmacol. 2019, 44, 28–38. [Google Scholar] [CrossRef]
- Berry, D.B.; Lu, D.; Geva, M.; Watts, J.C.; Bhardwaj, S.; Oehler, A.; Renslo, A.R.; DeArmond, S.J.; Prusiner, S.B.; Giles, K. Drug resistance confounding prion therapeutics. Proc. Natl. Acad. Sci. USA 2013, 110, E4160–E4169. [Google Scholar] [CrossRef]
- Giles, K.; Berry, D.B.; Condello, C.; Dugger, B.N.; Li, Z.; Oehler, A.; Bhardwaj, S.; Elepano, M.; Guan, S.; Silber, B.M.; et al. Optimization of Aryl Amides that Extend Survival in Prion-Infected Mice. J. Pharmacol. Exp. Ther. 2016, 358, 537–547. [Google Scholar] [CrossRef]
- Lu, D.; Giles, K.; Li, Z.; Rao, S.; Dolghih, E.; Gever, J.R.; Geva, M.; Elepano, M.L.; Oehler, A.; Bryant, C.; et al. Biaryl amides and hydrazones as therapeutics for prion disease in transgenic mice. J. Pharmacol. Exp. Ther. 2013, 347, 325–338. [Google Scholar] [CrossRef]
- Lawson, V.A.; Vella, L.J.; Stewart, J.D.; Sharples, R.A.; Klemm, H.; Machalek, D.M.; Masters, C.L.; Cappai, R.; Collins, S.J.; Hill, A.F. Mouse-adapted sporadic human Creutzfeldt-Jakob disease prions propagate in cell culture. Int. J. Biochem. Cell Biol. 2008, 40, 2793–2801. [Google Scholar] [CrossRef]
- Giles, K.; Berry, D.B.; Condello, C.; Hawley, R.C.; Gallardo-Godoy, A.; Bryant, C.; Oehler, A.; Elepano, M.; Bhardwaj, S.; Patel, S.; et al. Different 2-Aminothiazole Therapeutics Produce Distinct Patterns of Scrapie Prion Neuropathology in Mouse Brains. J. Pharmacol. Exp. Ther. 2015, 355, 2–12. [Google Scholar] [CrossRef]
- Ghaemmaghami, S.; Phuan, P.W.; Perkins, B.; Ullman, J.; May, B.C.; Cohen, F.E.; Prusiner, S.B. Cell division modulates prion accumulation in cultured cells. Proc. Natl. Acad. Sci. USA 2007, 104, 17971–17976. [Google Scholar] [CrossRef] [PubMed]
- Watts, J.C.; Prusiner, S.B. Mouse models for studying the formation and propagation of prions. J. Biol. Chem. 2014, 289, 19841–19849. [Google Scholar] [CrossRef] [PubMed]
- Follet, J.; Lemaire-Vieille, C.; Blanquet-Grossard, F.; Podevin-Dimster, V.; Lehmann, S.; Chauvin, J.P.; Decavel, J.P.; Varea, R.; Grassi, J.; Fontès, M.; et al. PrP expression and replication by Schwann cells: Implications in prion spreading. J. Virol. 2002, 76, 2434–2439. [Google Scholar] [CrossRef] [PubMed]
- Oboznaya, M.B.; Gilch, S.; Titova, M.A.; Koroev, D.O.; Volkova, T.D.; Volpina, O.M.; Schätzl, H.M. Antibodies to a nonconjugated prion protein peptide 95-123 interfere with PrPSc propagation in prion-infected cells. Cell Mol. Neurobiol. 2007, 27, 271–284. [Google Scholar] [CrossRef]
- Uryu, M.; Karino, A.; Kamihara, Y.; Horiuchi, M. Characterization of prion susceptibility in Neuro2a mouse neuroblastoma cell subclones. Microbiol. Immunol. 2007, 51, 661–669. [Google Scholar] [CrossRef]
- Herbst, A.; Banser, P.; Velasquez, C.D.; Mays, C.E.; Sim, V.L.; Westaway, D.; Aiken, J.M.; McKenzie, D. Infectious prions accumulate to high levels in non proliferative C2C12 myotubes. PLoS Pathog. 2013, 9, e1003755. [Google Scholar] [CrossRef]
- Mahal, S.P.; Baker, C.A.; Demczyk, C.A.; Smith, E.W.; Julius, C.; Weissmann, C. Prion strain discrimination in cell culture: The cell panel assay. Proc. Natl. Acad. Sci. USA 2007, 104, 20908–20913. [Google Scholar] [CrossRef]
- Iwamaru, Y.; Takenouchi, T.; Ogihara, K.; Hoshino, M.; Takata, M.; Imamura, M.; Tagawa, Y.; Hayashi-Kato, H.; Ushiki-Kaku, Y.; Shimizu, Y.; et al. Microglial cell line established from prion protein-overexpressing mice is susceptible to various murine prion strains. J. Virol. 2007, 81, 1524–1527. [Google Scholar] [CrossRef]
- Nishida, N.; Harris, D.A.; Vilette, D.; Laude, H.; Frobert, Y.; Grassi, J.; Casanova, D.; Milhavet, O.; Lehmann, S. Successful transmission of three mouse-adapted scrapie strains to murine neuroblastoma cell lines overexpressing wild-type mouse prion protein. J. Virol. 2000, 74, 320–325. [Google Scholar] [CrossRef]
- Marshall, K.E.; Hughson, A.; Vascellari, S.; Priola, S.A.; Sakudo, A.; Onodera, T.; Baron, G.S. PrP Knockout Cells Expressing Transmembrane PrP Resist Prion Infection. J. Virol. 2017, 91, e01686-16. [Google Scholar] [CrossRef]
- Milhavet, O.; McMahon, H.E.; Rachidi, W.; Nishida, N.; Katamine, S.; Mangé, A.; Arlotto, M.; Casanova, D.; Riondel, J.; Favier, A.; et al. Prion infection impairs the cellular response to oxidative stress. Proc. Natl. Acad. Sci. USA 2000, 97, 13937–13942. [Google Scholar] [CrossRef] [PubMed]
- Martins-Taylor, K.; Xu, R.H. Concise review: Genomic stability of human induced pluripotent stem cells. Stem Cells 2012, 30, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, S.T.; Cairney, C.J.; Chantry, A.D.; Madan, S.; Fernandes, J.A.; Howe, S.J.; Moore, H.D.; Thompson, M.J.; Chen, B.; Thrasher, A.; et al. A small molecule modulator of prion protein increases human mesenchymal stem cell lifespan, ex vivo expansion, and engraftment to bone marrow in NOD/SCID mice. Stem Cells 2012, 30, 1134–1143. [Google Scholar] [CrossRef] [PubMed]
- Takakura, Y.; Yamaguchi, N.; Nakagaki, T.; Satoh, K.; Kira, J.; Nishida, N. Bone marrow stroma cells are susceptible to prion infection. Biochem. Biophys. Res. Commun. 2008, 377, 957–961. [Google Scholar] [CrossRef]
- Lyahyai, J.; Mediano, D.R.; Ranera, B.; Sanz, A.; Remacha, A.R.; Bolea, R.; Zaragoza, P.; Rodellar, C.; Martín-Burriel, I. Isolation and characterization of ovine mesenchymal stem cells derived from peripheral blood. BMC Vet. Res. 2012, 8, 169. [Google Scholar] [CrossRef]
- Hernaiz, A.; Cobeta, P.; Marín, B.; Vázquez, F.J.; Badiola, J.J.; Zaragoza, P.; Bolea, R.; Martín-Burriel, I. Susceptibility of Ovine Bone Marrow-Derived Mesenchymal Stem Cell Spheroids to Scrapie Prion Infection. Animals 2023, 13, 1043. [Google Scholar] [CrossRef]
- García-Mendívil, L.; Mediano, D.R.; Hernaiz, A.; Sanz-Rubio, D.; Vázquez, F.J.; Marín, B.; López-Pérez, Ó.; Otero, A.; Badiola, J.J.; Zaragoza, P.; et al. Effect of Scrapie Prion Infection in Ovine Bone Marrow-Derived Mesenchymal Stem Cells and Ovine Mesenchymal Stem Cell-Derived Neurons. Animals 2021, 11, 1137. [Google Scholar] [CrossRef]
- Krejciova, Z.; Alibhai, J.; Zhao, C.; Krencik, R.; Rzechorzek, N.M.; Ullian, E.M.; Manson, J.; Ironside, J.W.; Head, M.W.; Chandran, S. Human stem cell-derived astrocytes replicate human prions in a PRNP genotype-dependent manner. J. Exp. Med. 2017, 214, 3481–3495. [Google Scholar] [CrossRef]
- Sim, H.-J.; Kim, Y.-C.; Bhattarai, G.; Won, S.-Y.; Lee, J.-C.; Jeong, B.-H.; Kook, S.-H. Prion infection modulates hematopoietic stem/progenitor cell fate through cell-autonomous and non-autonomous mechanisms. Leukemia 2023, 37, 877–887. [Google Scholar] [CrossRef]
- Mahar, M.; Cavalli, V. Intrinsic mechanisms of neuronal axon regeneration. Nat. Rev. Neurosci. 2018, 19, 323–337. [Google Scholar] [CrossRef]
- Li, Z.; Dong, X.; Tian, M.; Liu, C.; Wang, K.; Li, L.; Liu, Z.; Liu, J. Stem cell-based therapies for ischemic stroke: A systematic review and meta-analysis of clinical trials. Stem Cell Res. Ther. 2020, 11, 252. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Y.; Yang, L.P.; Zhao, L. Stem cell therapy for Alzheimer’s disease. World J. Stem Cells 2020, 12, 787–802. [Google Scholar] [CrossRef] [PubMed]
- Heris, R.M.; Shirvaliloo, M.; Abbaspour-Aghdam, S.; Hazrati, A.; Shariati, A.; Youshanlouei, H.R.; Niaragh, F.J.; Valizadeh, H.; Ahmadi, M. The potential use of mesenchymal stem cells and their exosomes in Parkinson’s disease treatment. Stem Cell Res. Ther. 2022, 13, 371. [Google Scholar] [CrossRef] [PubMed]
- Shang, Z.; Wang, M.; Zhang, B.; Wang, X.; Wanyan, P. Clinical translation of stem cell therapy for spinal cord injury still premature: Results from a single-arm meta-analysis based on 62 clinical trials. BMC Med. 2022, 20, 284. [Google Scholar] [CrossRef]
- Available online: https://clinicaltrials.gov/ (accessed on 16 August 2023).
- Song, C.H.; Honmou, O.; Ohsawa, N.; Nakamura, K.; Hamada, H.; Furuoka, H.; Hasebe, R.; Horiuchi, M. Effect of transplantation of bone marrow-derived mesenchymal stem cells on mice infected with prions. J. Virol. 2009, 83, 5918–5927. [Google Scholar] [CrossRef] [PubMed]
- Shan, Z.; Hirai, Y.; Nakayama, M.; Hayashi, R.; Yamasaki, T.; Hasebe, R.; Song, C.H.; Horiuchi, M. Therapeutic effect of autologous compact bone-derived mesenchymal stem cell transplantation on prion disease. J. Gen. Virol. 2017, 98, 2615–2627. [Google Scholar] [CrossRef] [PubMed]
- Relaño-Ginés, A.; Lehmann, S.; Bencsik, A.; Herva, M.E.; Torres, J.M.; Crozet, C.A. Stem Cell Therapy Extends Incubation and Survival Time in Prion-Infected Mice in a Time Window–Dependant Manner. J. Infect. Dis. 2011, 204, 1038–1045. [Google Scholar] [CrossRef]
- Hay, A.J.D.; Latham, A.S.; Mumford, G.; Hines, A.D.; Risen, S.; Gordon, E.; Siebenaler, C.; Gilberto, V.S.; Zabel, M.D.; Moreno, J.A. Intranasally delivered mesenchymal stromal cells decrease glial inflammation early in prion disease. Front. Neurosci. 2023, 17, 1158408. [Google Scholar] [CrossRef]
- Song, C.H.; Honmou, O.; Furuoka, H.; Horiuchi, M. Identification of chemoattractive factors involved in the migration of bone marrow-derived mesenchymal stem cells to brain lesions caused by prions. J. Virol. 2011, 85, 11069–11078. [Google Scholar] [CrossRef]
- Hay, A.J.D.; Murphy, T.J.; Popichak, K.A.; Zabel, M.D.; Moreno, J.A. Adipose-derived mesenchymal stromal cells decrease prion-induced glial inflammation in vitro. Sci. Rep. 2022, 12, 22567. [Google Scholar] [CrossRef]
- Galiakberova, A.A.; Dashinimaev, E.B. Neural Stem Cells and Methods for Their Generation From Induced Pluripotent Stem Cells in vitro. Front. Cell Dev. Biol. 2020, 8, 815. [Google Scholar] [CrossRef] [PubMed]
- De Gioia, R.; Biella, F.; Citterio, G.; Rizzo, F.; Abati, E.; Nizzardo, M.; Bresolin, N.; Comi, G.P.; Corti, S. Neural Stem Cell Transplantation for Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 3103. [Google Scholar] [CrossRef] [PubMed]
- Castle, A.R.; Gill, A.C. Physiological Functions of the Cellular Prion Protein. Front. Mol. Biosci. 2017, 4, 19. [Google Scholar] [CrossRef] [PubMed]
- Milhavet, O.; Casanova, D.; Chevallier, N.; McKay, R.D.; Lehmann, S. Neural stem cell model for prion propagation. Stem Cells 2006, 24, 2284–2291. [Google Scholar] [CrossRef] [PubMed]
- Relaño-Ginès, A.; Gabelle, A.; Hamela, C.; Belondrade, M.; Casanova, D.; Mourton-Gilles, C.; Lehmann, S.; Crozet, C. Prion replication occurs in endogenous adult neural stem cells and alters their neuronal fate: Involvement of endogenous neural stem cells in prion diseases. PLoS Pathog. 2013, 9, e1003485. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.L.; Brown, J.; Ritchie, D.L.; Sales, J.; Fraser, J.R. Fetal cell grafts provide long-term protection against scrapie induced neuronal loss. Neuroreport 2001, 12, 77–82. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Preclinical Study | Cell Source | Species | Outcome | Reference |
|---|---|---|---|---|
| Effect of transplantation of bone marrow-derived mesenchymal stem cells on mice infected with prions. | Immortalized human bone marrow-derived MSCs | Mice infected with Obihiro/Chandler scrapie strain | Prolonged survival time Produced trophic factors and differentiated into neuronal lineages | [126] |
| The therapeutic effect of autologous compact bone-derived mesenchymal stem cell transplantation on prion disease. | Autologous compact bone-derived MSCs | Mice infected with Obihiro/Chandler scrapie strain | Enhanced microglial activation MSCs migrate to brain lesions | [127] |
| Stem cell therapy extends incubation and survival time in prion-infected mice in a time window–dependent manner. | Fetal NSCs | RML strains of mouse-adapted prions | Increased incubation (20.1%) and survival times (13.6%) Reduction in the number of astrocytes | [128] |
| Intranasally delivered mesenchymal stromal cells decrease glial inflammation early in prion disease. | Adipose-derived MSCs | RML strains of mouse-adapted prions | Decreased vacuolization Promoting a quiescent state in hippocampal microglia Decrease in reactive astrocytes | [129] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zayed, M.; Kook, S.-H.; Jeong, B.-H. Potential Therapeutic Use of Stem Cells for Prion Diseases. Cells 2023, 12, 2413. https://doi.org/10.3390/cells12192413
Zayed M, Kook S-H, Jeong B-H. Potential Therapeutic Use of Stem Cells for Prion Diseases. Cells. 2023; 12(19):2413. https://doi.org/10.3390/cells12192413
Chicago/Turabian StyleZayed, Mohammed, Sung-Ho Kook, and Byung-Hoon Jeong. 2023. "Potential Therapeutic Use of Stem Cells for Prion Diseases" Cells 12, no. 19: 2413. https://doi.org/10.3390/cells12192413