

Regulation of Microglial Signaling by Lyn and SHIP-1 in the Steady-State Adult Mouse Brain
 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Behavior Testing
2.3. Transcardial Perfusion and Tissue Fixation
2.4. Immunofluorescence
2.5. Microglia Morphology Analysis
2.6. Cresyl Violet Staining
2.7. Flow Cytometry Analysis
2.8. Intracellular Cytokine Flow Cytometry
2.9. Quantitative Real-Time Polymerase Chain Reaction (qPCR)
2.10. Statistical Analysis
3. Results
3.1. Subsection
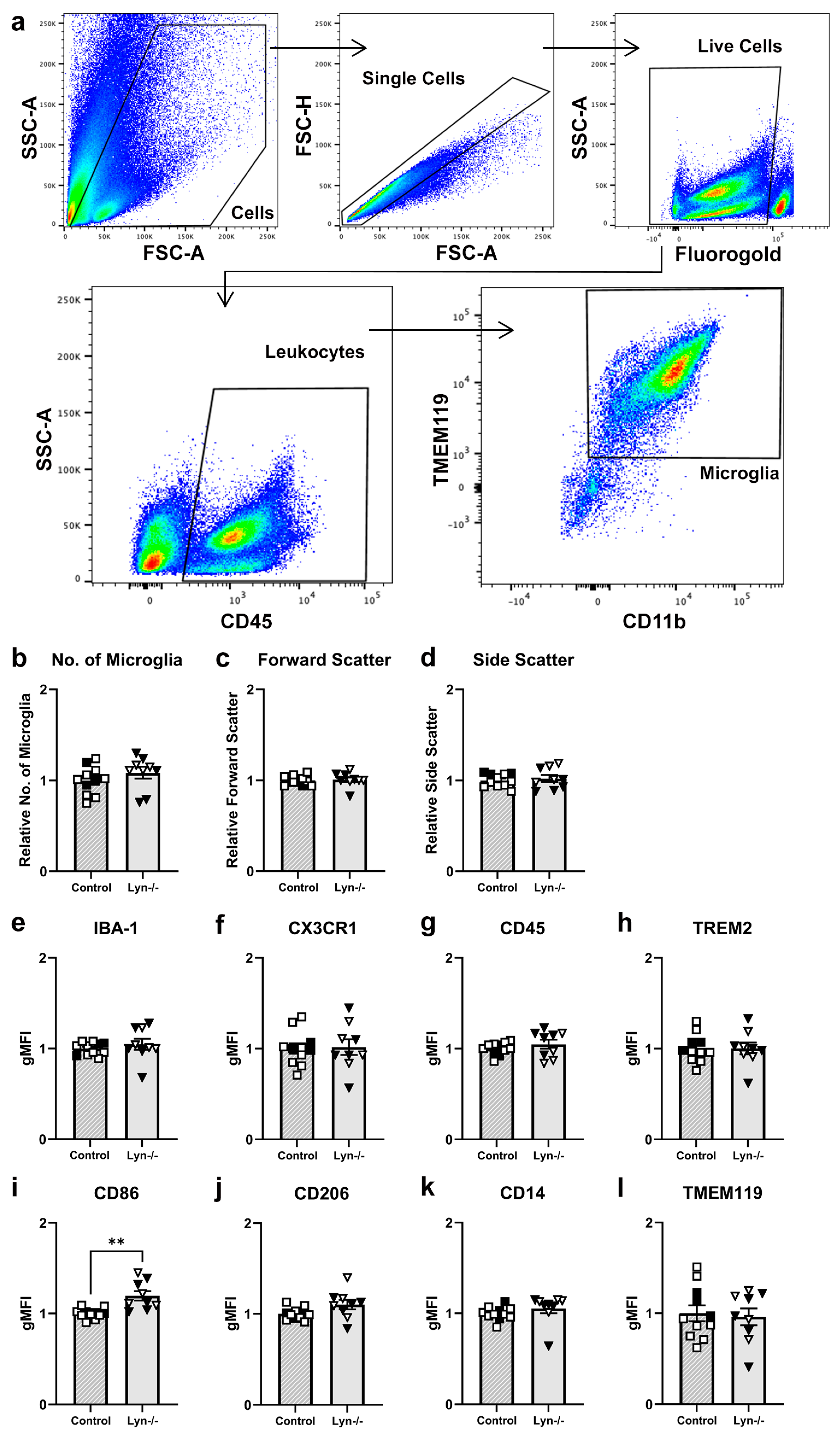
3.1.1. Young Adult Lyn−/− Mice Have Normal Morphological Characteristics and Behavioral Phenotype
3.1.2. Microglia Activation Was Unchanged in Lyn-Deficient Mice
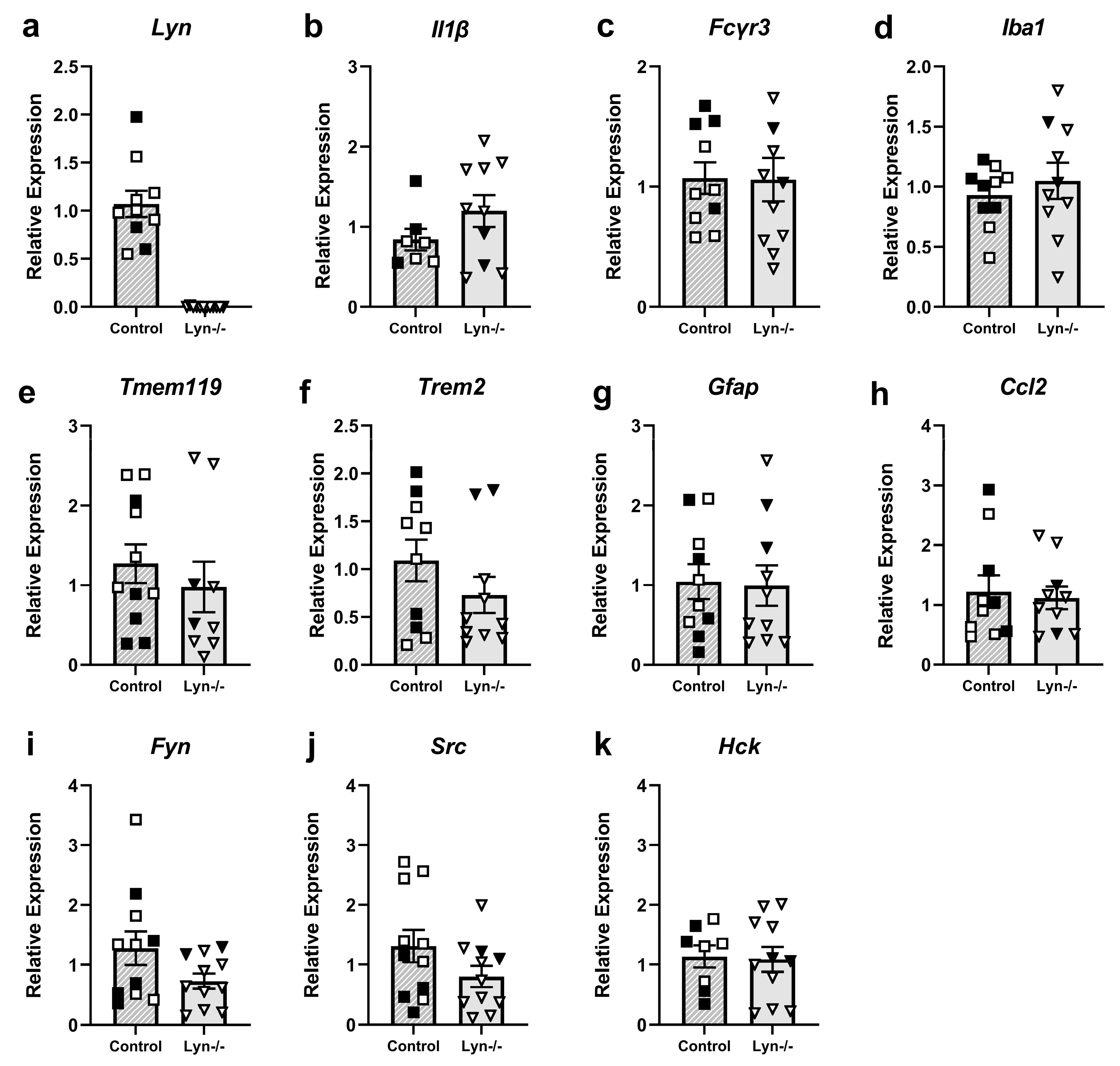
3.1.3. Lyn Deficiency Does Not Alter the Steady-State Microglial Phenotype
3.1.4. Lyn Deficiency Does Not Alter Expression of Neuroimmune Response Genes
3.1.5. Brain Volume of SHIP-1−/− Mice Is Unchanged but SHIP1−/− Mice Display Behavioral Differences
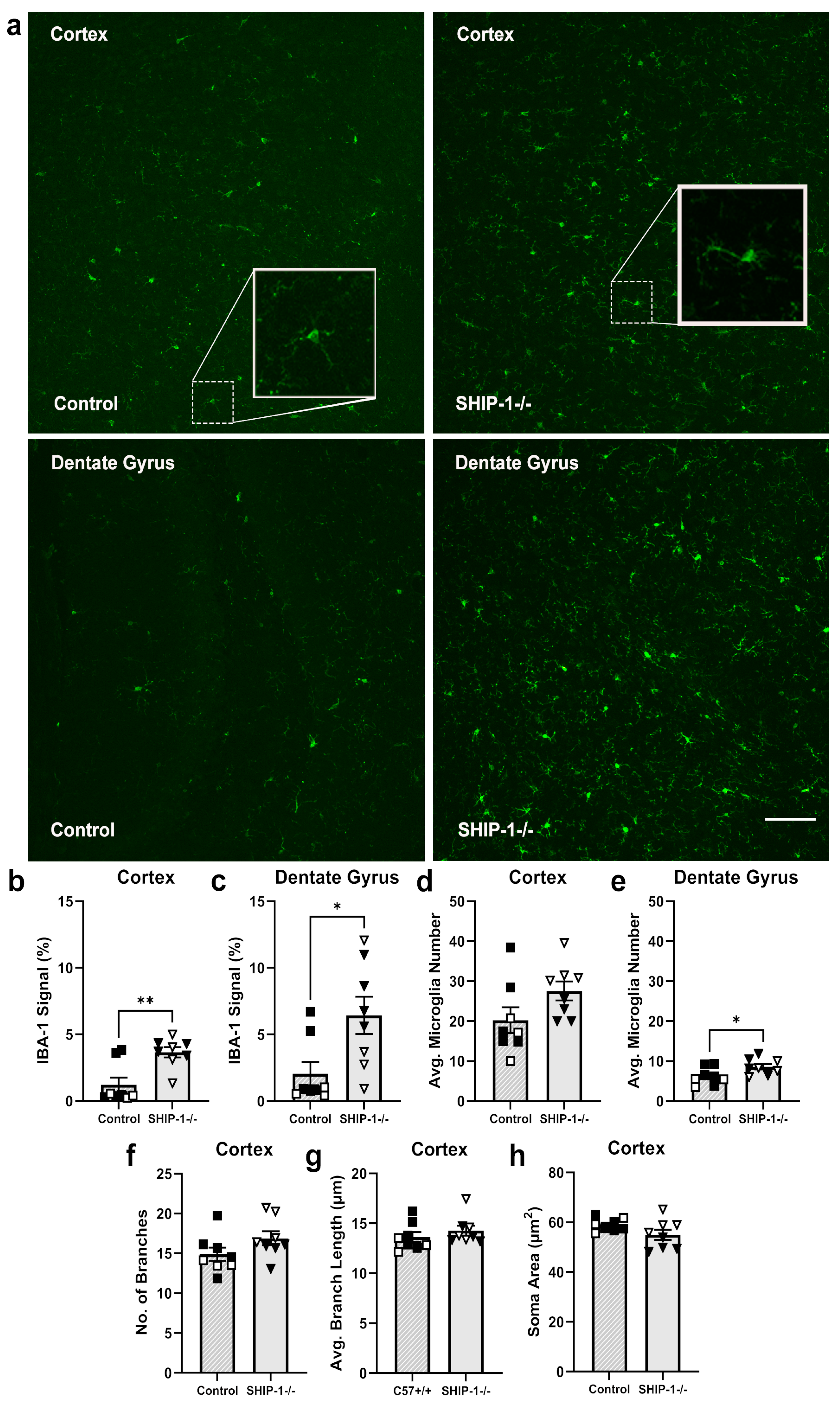
3.1.6. SHIP-1 Deficiency Altered Microglial Phenotype in the Cortex and Hippocampus
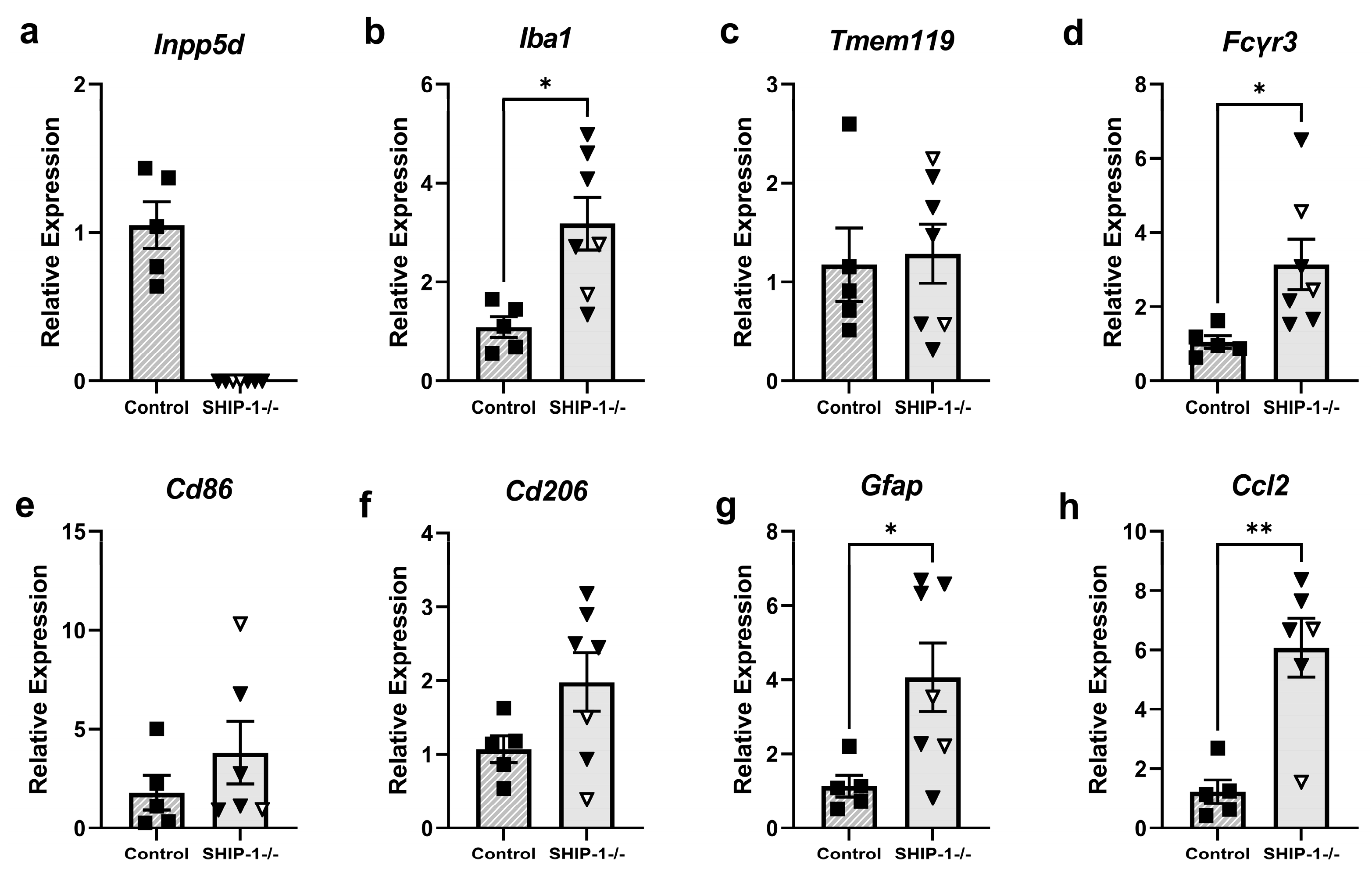
3.1.7. SHIP-1-Deficient Microglia Exhibit an Altered Cell Surface Phenotype
3.1.8. Increased Inflammatory Gene Expression in Cortical Brain Tissue of SHIP-1-Deficient Mice
3.1.9. Microglia from Lyn−/− and SHIP-1−/− Mice Are Hyper-Responsive to LPS Stimulation
4. Discussion
4.1. Lyn Deficiency Did Not Impact Neuroimmune Responses
4.2. SHIP-1 Deficiency Exacerbated Neuroinflammation and Microglial Activation
4.3. Limitations and Future Directions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wyss-Coray, T.; Mucke, L. Inflammation in Neurodegenerative Disease—A Double-Edged Sword. Neuron 2002, 35, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, R.; Samokhina, E.; Rossetti, I.; Morley, J.W.; Buskila, Y. Neuromodulation of Glial Function During Neurodegeneration. Front. Cell. Neurosci. 2020, 14, 278. [Google Scholar] [CrossRef] [PubMed]
- Chu, E.; Mychasiuk, R.; Hibbs, M.L.; Semple, B.D. Dysregulated phosphoinositide 3-kinase signaling in microglia: Shaping chronic neuroinflammation. J. Neuroinflamm. 2021, 18, 276. [Google Scholar] [CrossRef]
- Yamanaka, K.; Chun, S.J.; Boillee, S.; Fujimori-Tonou, N.; Yamashita, H.; Gutmann, D.H.; Takahashi, R.; Misawa, H.; Cleveland, D.W. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat. Neurosci. 2008, 11, 251–253. [Google Scholar] [CrossRef] [PubMed]
- Weichhart, T.; Säemann, M.D. The PI3K/Akt/mTOR pathway in innate immune cells: Emerging therapeutic applications. Ann. Rheum. Dis. 2008, 67, iii70–iii74. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef]
- Sotirellis, N.; Johnson, T.M.; Hibbs, M.L.; Stanley, I.J.; Stanley, E.; Dunn, A.R.; Cheng, H.-C. Autophosphorylation Induces Autoactivation and a Decrease in the Src Homology 2 Domain Accessibility of the Lyn Protein Kinase (∗). J. Biol. Chem. 1995, 270, 29773–29780. [Google Scholar] [CrossRef]
- Nunes de Miranda, S.M.; Wilhelm, T.; Huber, M.; Zorn, C.N. Differential Lyn-dependence of the SHIP1-deficient mast cell phenotype. Cell. Commun. Signal. 2016, 14, 12. [Google Scholar] [CrossRef]
- Harder, K.W.; Parsons, L.M.; Armes, J.; Evans, N.; Kountouri, N.; Clark, R.; Quilici, C.; Grail, D.; Hodgson, G.S.; Dunn, A.R.; et al. Gain- and loss-of-function Lyn mutant mice define a critical inhibitory role for Lyn in the myeloid lineage. Immunity 2001, 15, 603–615. [Google Scholar] [CrossRef]
- Hibbs, M.L.; Tarlinton, D.M.; Armes, J.; Grail, D.; Hodgson, G.; Maglitto, R.; Stacker, S.A.; Dunn, A.R. Multiple defects in the immune system of Lyn-deficient mice, culminating in autoimmune disease. Cell 1995, 83, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, M.J.; Duan, M.; Armes, J.E.; Anderson, G.P.; Tarlinton, D.M.; Hibbs, M.L. Genetic segregation of inflammatory lung disease and autoimmune disease severity in SHIP-1−/− mice. J. Immunol. 2011, 186, 7164–7175. [Google Scholar] [CrossRef] [PubMed]
- Helgason, C.D.; Damen, J.E.; Rosten, P.; Grewal, R.; Sorensen, P.; Chappel, S.M.; Borowski, A.; Jirik, F.; Krystal, G.; Humphries, R.K. Targeted disruption of SHIP leads to hemopoietic perturbations, lung pathology, and a shortened life span. Genes Dev. 1998, 12, 1610–1620. [Google Scholar] [CrossRef]
- Liu, Q.; Sasaki, T.; Kozieradzki, I.; Wakeham, A.; Itie, A.; Dumont, D.J.; Penninger, J.M. SHIP is a negative regulator of growth factor receptor-mediated PKB/Akt activation and myeloid cell survival. Genes Dev. 1999, 13, 786–791. [Google Scholar] [CrossRef]
- Hibbs, M.L.; Harder, K.W. The duplicitous nature of the Lyn tyrosine kinase in growth factor signaling. Growth Factors 2006, 24, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Hibbs, M.L.; Raftery, A.L.; Tsantikos, E. Regulation of hematopoietic cell signaling by SHIP-1 inositol phosphatase: Growth factors and beyond. Growth Factors 2018, 36, 213–231. [Google Scholar] [CrossRef]
- Takeshita, S.; Namba, N.; Zhao, J.J.; Jiang, Y.; Genant, H.K.; Silva, M.J.; Brodt, M.D.; Helgason, C.D.; Kalesnikoff, J.; Rauh, M.J.; et al. SHIP-deficient mice are severely osteoporotic due to increased numbers of hyper-resorptive osteoclasts. Nat. Med. 2002, 8, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Kerr, W.G.; Park, M.-Y.; Maubert, M.; Engelman, R.W. SHIP deficiency causes Crohn’s disease-like ileitis. Gut 2011, 60, 177–188. [Google Scholar] [CrossRef]
- McLarren, K.W.; Cole, A.E.; Weisser, S.B.; Voglmaier, N.S.; Conlin, V.S.; Jacobson, K.; Popescu, O.; Boucher, J.-L.; Sly, L.M. SHIP-Deficient Mice Develop Spontaneous Intestinal Inflammation and Arginase-Dependent Fibrosis. Am. J. Pathol. 2011, 179, 180–188. [Google Scholar] [CrossRef]
- Hayashi, T.; Umemori, H.; Mishina, M.; Yamamoto, T. The AMPA receptor interacts with and signals through the protein tyrosine kinase Lyn. Nature 1999, 397, 72–76. [Google Scholar] [CrossRef]
- Umemori, H.; Ogura, H.; Tozawa, N.; Mikoshiba, K.; Nishizumi, H.; Yamamoto, T. Impairment of N-methyl-D-aspartate receptor-controlled motor activity in LYN-deficient mice. Neuroscience 2003, 118, 709–713. [Google Scholar] [CrossRef]
- Umemori, H.; Wanaka, A.; Kato, H.; Takeuchi, M.; Tohyama, M.; Yamamoto, T. Specific expressions of Fyn and Lyn, lymphocyte antigen receptor-associated tyrosine kinases, in the central nervous system. Brain Res. Mol. Brain Res. 1992, 16, 303–310. [Google Scholar] [CrossRef]
- Metere, A.; Mallozzi, C.; Minetti, M.; Domenici, M.R.; Pèzzola, A.; Popoli, P.; Di Stasi, A.M. Quinolinic acid modulates the activity of src family kinases in rat striatum: In vivo and in vitro studies. J. Neurochem. 2006, 97, 1327–1336. [Google Scholar] [CrossRef] [PubMed]
- Gibb, S.L.; Jeanblanc, J.; Barak, S.; Yowell, Q.V.; Yaka, R.; Ron, D. Lyn kinase regulates mesolimbic dopamine release: Implication for alcohol reward. J. Neurosci. 2011, 31, 2180–2187. [Google Scholar] [CrossRef] [PubMed]
- Portugal, C.C.; Almeida, T.O.; Socodato, R.; Relvas, J.B. Src family kinases (SFKs): Critical regulators of microglial homeostatic functions and neurodegeneration in Parkinson’s and Alzheimer’s diseases. FEBS J. 2022, 289, 7760–7775. [Google Scholar] [CrossRef]
- Colognato, H.; Ramachandrappa, S.; Olsen, I.M.; Ffrench-Constant, C. Integrins direct Src family kinases to regulate distinct phases of oligodendrocyte development. J. Cell Biol. 2004, 167, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Watzlawik, J.O.; Warrington, A.E.; Rodriguez, M. PDGF is Required for Remyelination-Promoting IgM Stimulation of Oligodendrocyte Progenitor Cell Proliferation. PLoS ONE 2013, 8, e55149. [Google Scholar] [CrossRef] [PubMed]
- Pedicone, C.; Fernandes, S.; Dungan, O.M.; Dormann, S.M.; Viernes, D.R.; Adhikari, A.A.; Choi, L.B.; De Jong, E.P.; Chisholm, J.D.; Kerr, W.G. Pan-SHIP1/2 inhibitors promote microglia effector functions essential for CNS homeostasis. J. Cell Sci. 2020, 133, jcs238030. [Google Scholar] [CrossRef]
- Pedicone, C.; Fernandes, S.; Matera, A.; Meyer, S.T.; Loh, S.; Ha, J.H.; Bernard, D.; Chisholm, J.D.; Paolicelli, R.C.; Kerr, W.G. Discovery of a novel SHIP1 agonist that promotes degradation of lipid-laden phagocytic cargo by microglia. iScience 2022, 25, 104170. [Google Scholar] [CrossRef]
- Tang, H.; Mao, J.; Ye, X.; Zhang, F.; Kerr, W.G.; Zheng, T.; Zhu, Z. SHIP-1, a target of miR-155, regulates endothelial cell responses in lung fibrosis. FASEB J. 2020, 34, 2011–2023. [Google Scholar] [CrossRef]
- Weerawarna, P.M.; Richardson, T.I. Lyn Kinase Structure, Regulation, and Involvement in Neurodegenerative Diseases: A Mini Review. Kinases Phosphatases 2023, 1, 23–38. [Google Scholar] [CrossRef]
- Gwon, Y.; Kim, S.H.; Kim, H.T.; Kam, T.I.; Park, J.; Lim, B.; Cha, H.; Chang, H.J.; Hong, Y.R.; Jung, Y.K. Amelioration of amyloid β-FcγRIIb neurotoxicity and tau pathologies by targeting LYN. FASEB J. 2019, 33, 4300–4313. [Google Scholar] [CrossRef] [PubMed]
- Sondag, C.M.; Dhawan, G.; Combs, C.K. Beta amyloid oligomers and fibrils stimulate differential activation of primary microglia. J. Neuroinflamm. 2009, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, G.; Floden, A.M.; Combs, C.K. Amyloid-β oligomers stimulate microglia through a tyrosine kinase dependent mechanism. Neurobiol. Aging 2012, 33, 2247–2261. [Google Scholar] [CrossRef]
- Manocha, G.D.; Puig, K.L.; Austin, S.A.; Seyb, K.; Glicksman, M.A.; Combs, C.K. Characterization of Novel Src Family Kinase Inhibitors to Attenuate Microgliosis. PLoS ONE 2015, 10, e0132604. [Google Scholar] [CrossRef]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef]
- Jing, H.; Zhu, J.-X.; Wang, H.-F.; Zhang, W.; Zheng, Z.-J.; Kong, L.-L.; Tan, C.-C.; Wang, Z.-X.; Tan, L.; Tan, L. INPP5D rs35349669 polymorphism with late-onset Alzheimer’s disease: A replication study and meta-analysis. Oncotarget 2016, 7, 69225–69230. [Google Scholar] [CrossRef]
- Yoshino, Y.; Yamazaki, K.; Ozaki, Y.; Sao, T.; Yoshida, T.; Mori, T.; Mori, Y.; Ochi, S.; Iga, J.I.; Ueno, S.I. INPP5D mRNA Expression and Cognitive Decline in Japanese Alzheimer’s Disease Subjects. J. Alzheimer’s Dis. JAD 2017, 58, 687–694. [Google Scholar] [CrossRef]
- Efthymiou, A.G.; Goate, A.M. Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol. Neurodegener. 2017, 12, 43. [Google Scholar] [CrossRef]
- Zajac, D.J.; Simpson, J.; Zhang, E.; Parikh, I.; Estus, S. Expression of INPP5D Isoforms in Human Brain: Impact of Alzheimer’s Disease Neuropathology and Genetics. Genes 2023, 14, 763. [Google Scholar] [CrossRef]
- Tsantikos, E.; Oracki, S.A.; Quilici, C.; Anderson, G.P.; Tarlinton, D.M.; Hibbs, M.L. Autoimmune disease in Lyn-deficient mice is dependent on an inflammatory environment established by IL-6. J. Immunol. 2010, 184, 1348–1360. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Casillas-Espinosa, P.M.; Dill, L.K.; Rewell, S.S.J.; Hudson, M.R.; O’Brien, T.J.; Shultz, S.R.; Semple, B.D. Pediatric traumatic brain injury and a subsequent transient immune challenge independently influenced chronic outcomes in male mice. Brain Behav. Immun. 2022, 100, 29–47. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.L.; Dill, L.K.; Wood, R.J.; Wang, S.; Robertson, K.; Murray, S.S.; Zamani, A.; Semple, B.D. Acute treatment with TrkB agonist LM22A-4 confers neuroprotection and preserves myelin integrity in a mouse model of pediatric traumatic brain injury. Exp. Neurol. 2021, 339, 113652. [Google Scholar] [CrossRef] [PubMed]
- Dill, L.K.; Teymornejad, S.; Sharma, R.; Bozkurt, S.; Christensen, J.; Chu, E.; Rewell, S.S.; Shad, A.; Mychasiuk, R.; Semple, B.D. Modulating chronic outcomes after pediatric traumatic brain injury: Distinct effects of social and environmental enrichment. Exp. Neurol. 2023, 364, 114407. [Google Scholar] [CrossRef]
- Semple, B.D.; O’Brien, T.J.; Gimlin, K.; Wright, D.K.; Kim, S.E.; Casillas-Espinosa, P.M.; Webster, K.M.; Petrou, S.; Noble-Haeusslein, L.J. Interleukin-1 Receptor in Seizure Susceptibility after Traumatic Injury to the Pediatric Brain. J. Neurosci. 2017, 37, 7864–7877. [Google Scholar] [CrossRef]
- Sharma, R.; Chu, E.; Dill, L.K.; Shad, A.; Zamani, A.; O’Brien, T.J.; Casillas-Espinosa, P.M.; Shultz, S.R.; Semple, B.D. Ccr2 Gene Ablation Does Not Influence Seizure Susceptibility, Tissue Damage, or Cellular Inflammation after Murine Pediatric Traumatic Brain Injury. J. Neurotrauma 2023, 40, 365–382. [Google Scholar] [CrossRef]
- Zamani, A.; Powell, K.L.; May, A.; Semple, B.D. Validation of reference genes for gene expression analysis following experimental traumatic brain injury in a pediatric mouse model. Brain Res. Bull. 2020, 156, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Tsantikos, E.; Maxwell, M.J.; Putoczki, T.; Ernst, M.; Rose-John, S.; Tarlinton, D.M.; Hibbs, M.L. Interleukin-6 trans-signaling exacerbates inflammation and renal pathology in lupus-prone mice. Arthritis Rheum. 2013, 65, 2691–2702. [Google Scholar] [CrossRef]
- Baran, C.P.; Tridandapani, S.; Helgason, C.D.; Humphries, R.K.; Krystal, G.; Marsh, C.B. The inositol 5’-phosphatase SHIP-1 and the Src kinase Lyn negatively regulate macrophage colony-stimulating factor-induced Akt activity. J. Biol. Chem. 2003, 278, 38628–38636. [Google Scholar] [CrossRef]
- Harder, K.W.; Quilici, C.; Naik, E.; Inglese, M.; Kountouri, N.; Turner, A.; Zlatic, K.; Tarlinton, D.M.; Hibbs, M.L. Perturbed myelo/erythropoiesis in Lyn-deficient mice is similar to that in mice lacking the inhibitory phosphatases SHP-1 and SHIP-1. Blood 2004, 104, 3901–3910. [Google Scholar] [CrossRef]
- Tsantikos, E.; Maxwell, M.J.; Kountouri, N.; Harder, K.W.; Tarlinton, D.M.; Hibbs, M.L. Genetic interdependence of Lyn and negative regulators of B cell receptor signaling in autoimmune disease development. J. Immunol. 2012, 189, 1726–1736. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Fang, L.; Pu, Q.; Lin, P.; Hoggarth, A.; Huang, H.; Li, X.; Li, G.; Wu, M. Lyn prevents aberrant inflammatory responses to Pseudomonas infection in mammalian systems by repressing a SHIP-1-associated signaling cluster. Signal Transduct. Target. Ther. 2016, 1, 16032. [Google Scholar] [CrossRef] [PubMed]
- Sly, L.M.; Rauh, M.J.; Kalesnikoff, J.; Song, C.H.; Krystal, G. LPS-induced upregulation of SHIP is essential for endotoxin tolerance. Immunity 2004, 21, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Keck, S.; Freudenberg, M.; Huber, M. Activation of murine macrophages via TLR2 and TLR4 is negatively regulated by a Lyn/PI3K module and promoted by SHIP1. J. Immunol. 2010, 184, 5809–5818. [Google Scholar] [CrossRef]
- Wu, X.; Zhu, D.; Jiang, X.; Okagaki, P.; Mearow, K.; Zhu, G.; McCall, S.; Banaudha, K.; Lipsky, R.H.; Marini, A.M. AMPA protects cultured neurons against glutamate excitotoxicity through a phosphatidylinositol 3-kinase-dependent activation in extracellular signal-regulated kinase to upregulate BDNF gene expression. J. Neurochem. 2004, 90, 807–818. [Google Scholar] [CrossRef]
- Bailey, K.R.; Crawley, J.N. Anxiety-Related Behaviors in Mice. In Methods of Behavior Analysis in Neuroscience, 2nd ed.; Buccafusco, J.J., Ed.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2009. [Google Scholar]
- Salomons, A.R.; Arndt, S.S.; Ohl, F. Impact of anxiety profiles on cognitive performance in BALB/c and 129P2 mice. Cogn. Affect. Behav. Neurosci. 2012, 12, 794–803. [Google Scholar] [CrossRef]
- Sierksma, A.; Lu, A.; Mancuso, R.; Fattorelli, N.; Thrupp, N.; Salta, E.; Zoco, J.; Blum, D.; Buée, L.; De Strooper, B.; et al. Novel Alzheimer risk genes determine the microglia response to amyloid-β but not to TAU pathology. EMBO Mol. Med. 2020, 12, e10606. [Google Scholar] [CrossRef]
- Zhou, T.; Huang, Z.; Sun, X.; Zhu, X.; Zhou, L.; Li, M.; Cheng, B.; Liu, X.; He, C. Microglia Polarization with M1/M2 Phenotype Changes in rd1 Mouse Model of Retinal Degeneration. Front. Neuroanat. 2017, 11, 77. [Google Scholar] [CrossRef]
- Li, R.; Liu, W.; Yin, J.; Chen, Y.; Guo, S.; Fan, H.; Li, X.; Zhang, X.; He, X.; Duan, C. TSG-6 attenuates inflammation-induced brain injury via modulation of microglial polarization in SAH rats through the SOCS3/STAT3 pathway. J. Neuroinflamm. 2018, 15, 231. [Google Scholar] [CrossRef]
- Hyakkoku, K.; Hamanaka, J.; Tsuruma, K.; Shimazawa, M.; Tanaka, H.; Uematsu, S.; Akira, S.; Inagaki, N.; Nagai, H.; Hara, H. Toll-like receptor 4 (TLR4), but not TLR3 or TLR9, knock-out mice have neuroprotective effects against focal cerebral ischemia. Neuroscience 2010, 171, 258–267. [Google Scholar] [CrossRef]
- Scapini, P.; Pereira, S.; Zhang, H.; Lowell, C.A. Multiple roles of Lyn kinase in myeloid cell signaling and function. Immunol. Rev. 2009, 228, 23–40. [Google Scholar] [CrossRef] [PubMed]
- Brodie, E.J.; Infantino, S.; Low, M.S.Y.; Tarlinton, D.M. Lyn, Lupus, and (B) Lymphocytes, a Lesson on the Critical Balance of Kinase Signaling in Immunity. Front. Immunol. 2018, 9, 401. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Xiao, Y.; Lin, G.; Guo, H.; Deng, H.-X.; Tu, S.; Langdon, W.Y.; Yang, H.; Tao, L.; Li, Y.; et al. Tyrosine phosphorylation of NLRP3 by the Src family kinase Lyn suppresses the activity of the NLRP3 inflammasome. Sci. Signal. 2021, 14, eabe3410. [Google Scholar] [CrossRef] [PubMed]
- Panicker, N.; Saminathan, H.; Jin, H.; Neal, M.; Harischandra, D.S.; Gordon, R.; Kanthasamy, K.; Lawana, V.; Sarkar, S.; Luo, J.; et al. Fyn Kinase Regulates Microglial Neuroinflammatory Responses in Cell Culture and Animal Models of Parkinson’s Disease. J. Neurosci. 2015, 35, 10058–10077. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, L.; Zang, C.; Wang, Y.; Shang, J.; Zhang, Z.; Liu, H.; Bao, X.; Wang, X.; Zhang, D. Src Inhibition Attenuates Neuroinflammation and Protects Dopaminergic Neurons in Parkinson’s Disease Models. Front. Neurosci. 2020, 14, 45. [Google Scholar] [CrossRef] [PubMed]
- Ohsawa, K.; Imai, Y.; Sasaki, Y.; Kohsaka, S. Microglia/macrophage-specific protein Iba1 binds to fimbrin and enhances its actin-bundling activity. J. Neurochem. 2004, 88, 844–856. [Google Scholar] [CrossRef] [PubMed]
- Salemi, J.; Obregon, D.F.; Cobb, A.; Reed, S.; Sadic, E.; Jin, J.; Fernandez, F.; Tan, J.; Giunta, B. Flipping the switches: CD40 and CD45 modulation of microglial activation states in HIV associated dementia (HAD). Mol. Neurodegener. 2011, 6, 3. [Google Scholar] [CrossRef]
- Kigerl, K.A.; Gensel, J.C.; Ankeny, D.P.; Alexander, J.K.; Donnelly, D.J.; Popovich, P.G. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J. Neurosci. 2009, 29, 13435–13444. [Google Scholar] [CrossRef]
- Fadini, G.P.; Cappellari, R.; Mazzucato, M.; Agostini, C.; Vigili de Kreutzenberg, S.; Avogaro, A. Monocyte-macrophage polarization balance in pre-diabetic individuals. Acta Diabetol. 2013, 50, 977–982. [Google Scholar] [CrossRef]
- Green, T.R.F.; Murphy, S.M.; Moreno-Montano, M.P.; Audinat, E.; Rowe, R.K. Reactive morphology of dividing microglia following kainic acid administration. Front. Neurosci. 2022, 16, 972138. [Google Scholar] [CrossRef]
- Ritzel, R.M.; Patel, A.R.; Grenier, J.M.; Crapser, J.; Verma, R.; Jellison, E.R.; McCullough, L.D. Functional differences between microglia and monocytes after ischemic stroke. J. Neuroinflamm. 2015, 12, 106. [Google Scholar] [CrossRef] [PubMed]
- Samuels, J.D.; Moore, K.A.; Ennerfelt, H.E.; Johnson, A.M.; Walsh, A.E.; Price, R.J.; Lukens, J.R. The Alzheimer’s disease risk factor INPP5D restricts neuroprotective microglial responses in amyloid beta-mediated pathology. Alzheimer’s Dement. 2023. [Google Scholar] [CrossRef] [PubMed]
- Castranio, E.L.; Hasel, P.; Haure-Mirande, J.V.; Ramirez Jimenez, A.V.; Hamilton, B.W.; Kim, R.D.; Glabe, C.G.; Wang, M.; Zhang, B.; Gandy, S.; et al. Microglial INPP5D limits plaque formation and glial reactivity in the PSAPP mouse model of Alzheimer’s disease. Alzheimer’s Dement. 2023, 19, 2239–2252. [Google Scholar] [CrossRef] [PubMed]
- Galandrini, R.; Tassi, I.; Mattia, G.; Lenti, L.; Piccoli, M.; Frati, L.; Santoni, A. SH2-containing inositol phosphatase (SHIP-1) transiently translocates to raft domains and modulates CD16-mediated cytotoxicity in human NK cells. Blood 2002, 100, 4581–4589. [Google Scholar] [CrossRef]
- Townsend, K.P.; Town, T.; Mori, T.; Lue, L.F.; Shytle, D.; Sanberg, P.R.; Morgan, D.; Fernandez, F.; Flavell, R.A.; Tan, J. CD40 signaling regulates innate and adaptive activation of microglia in response to amyloid beta-peptide. Eur. J. Immunol. 2005, 35, 901–910. [Google Scholar] [CrossRef]
- Townsend, K.P.; Shytle, D.R.; Bai, Y.; San, N.; Zeng, J.; Freeman, M.; Mori, T.; Fernandez, F.; Morgan, D.; Sanberg, P.; et al. Lovastatin modulation of microglial activation via suppression of functional CD40 expression. J. Neurosci. Res. 2004, 78, 167–176. [Google Scholar] [CrossRef]
- Calingasan, N.Y.; Erdely, H.A.; Altar, C.A. Identification of CD40 ligand in Alzheimer’s disease and in animal models of Alzheimer’s disease and brain injury. Neurobiol. Aging 2002, 23, 31–39. [Google Scholar] [CrossRef]
- Togo, T.; Akiyama, H.; Kondo, H.; Ikeda, K.; Kato, M.; Iseki, E.; Kosaka, K. Expression of CD40 in the brain of Alzheimer’s disease and other neurological diseases. Brain Res. 2000, 885, 117–121. [Google Scholar] [CrossRef]
- Priller, J.; Flügel, A.; Wehner, T.; Boentert, M.; Haas, C.A.; Prinz, M.; Fernández-Klett, F.; Prass, K.; Bechmann, I.; de Boer, B.A.; et al. Targeting gene-modified hematopoietic cells to the central nervous system: Use of green fluorescent protein uncovers microglial engraftment. Nat. Med. 2001, 7, 1356–1361. [Google Scholar] [CrossRef]
- Sevenich, L. Brain-Resident Microglia and Blood-Borne Macrophages Orchestrate Central Nervous System Inflammation in Neurodegenerative Disorders and Brain Cancer. Front. Immunol. 2018, 9, 697. [Google Scholar] [CrossRef]
- An, H.; Xu, H.; Zhang, M.; Zhou, J.; Feng, T.; Qian, C.; Qi, R.; Cao, X. Src homology 2 domain-containing inositol-5-phosphatase 1 (SHIP1) negatively regulates TLR4-mediated LPS response primarily through a phosphatase activity- and PI-3K-independent mechanism. Blood 2005, 105, 4685–4692. [Google Scholar] [CrossRef] [PubMed]
- Laird, M.H.W.; Rhee, S.H.; Perkins, D.J.; Medvedev, A.E.; Piao, W.; Fenton, M.J.; Vogel, S.N. TLR4/MyD88/PI3K interactions regulate TLR4 signaling. J. Leukoc. Biol. 2009, 85, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Ceasrine, A.M.; Bilbo, S.D. Primetime for microglia: When stress and infection collide. Neuron 2021, 109, 2503–2505. [Google Scholar] [CrossRef] [PubMed]
- Chou, V.; Fancher, S.B.; Pearse, R.V., II; Lee, H.; Lam, M.; Seyfried, N.T.; Bennett, D.A.; Jager, P.L.D.; Menon, V.; Young-Pearse, T.L. INPP5D/SHIP1 regulates inflammasome activation in human microglia. bioRxiv 2023. [Google Scholar] [CrossRef]
- Kwon, H.S.; Koh, S.-H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef]
- Tsantikos, E.; Lau, M.; Castelino, C.M.; Maxwell, M.J.; Passey, S.L.; Hansen, M.J.; McGregor, N.E.; Sims, N.A.; Steinfort, D.P.; Irving, L.B.; et al. Granulocyte-CSF links destructive inflammation and comorbidities in obstructive lung disease. J. Clin. Investig. 2018, 128, 2406–2418. [Google Scholar] [CrossRef]
- Haddon, D.; Antignano, F.; Hughes, M.; Blanchet, M.-R.; Zbytnuik, L.; Krystal, G.; McNagny, K. SHIP1 Is a Repressor of Mast Cell Hyperplasia, Cytokine Production, and Allergic Inflammation In Vivo. J. Immunol. 2009, 183, 228–236. [Google Scholar] [CrossRef]
- Kanaya, A.; Yang, M.; Emala, C.; Mikami, M. Chronic allergic lung inflammation negatively influences neurobehavioral outcomes in mice. J. Neuroinflamm. 2022, 19, 210. [Google Scholar] [CrossRef]
- Craig, C.F.; Filippone, R.T.; Stavely, R.; Bornstein, J.C.; Apostolopoulos, V.; Nurgali, K. Neuroinflammation as an etiological trigger for depression comorbid with inflammatory bowel disease. J. Neuroinflamm. 2022, 19, 4. [Google Scholar] [CrossRef]
- Koellhoffer, E.C.; McCullough, L.D.; Ritzel, R.M. Old Maids: Aging and Its Impact on Microglia Function. Int. J. Mol. Sci. 2017, 18, 769. [Google Scholar] [CrossRef]
- Angelova, D.M.; Brown, D.R. Microglia and the aging brain: Are senescent microglia the key to neurodegeneration? J. Neurochem. 2019, 151, 676–688. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chu, E.; Mychasiuk, R.; Tsantikos, E.; Raftery, A.L.; L’Estrange-Stranieri, E.; Dill, L.K.; Semple, B.D.; Hibbs, M.L. Regulation of Microglial Signaling by Lyn and SHIP-1 in the Steady-State Adult Mouse Brain. Cells 2023, 12, 2378. https://doi.org/10.3390/cells12192378
Chu E, Mychasiuk R, Tsantikos E, Raftery AL, L’Estrange-Stranieri E, Dill LK, Semple BD, Hibbs ML. Regulation of Microglial Signaling by Lyn and SHIP-1 in the Steady-State Adult Mouse Brain. Cells. 2023; 12(19):2378. https://doi.org/10.3390/cells12192378
Chicago/Turabian StyleChu, Erskine, Richelle Mychasiuk, Evelyn Tsantikos, April L. Raftery, Elan L’Estrange-Stranieri, Larissa K. Dill, Bridgette D. Semple, and Margaret L. Hibbs. 2023. "Regulation of Microglial Signaling by Lyn and SHIP-1 in the Steady-State Adult Mouse Brain" Cells 12, no. 19: 2378. https://doi.org/10.3390/cells12192378