
Metformin: A New Inhibitor of the Wnt Signaling Pathway in Cancer
 , ,
, ,
Abstract
:1. Introduction
2. The Wnt Signaling Pathway
2.1. The Canonical Wnt Signaling Pathway
2.2. The Non-Canonical Wnt Signaling Pathway
3. Molecular Players Involved in the Wnt/β-Catenin-Mediated Anticancer Activity of Metformin
3.1. DVL3
3.2. MMP26
3.3. HNF4α
3.4. Wnt3a
3.5. Intracellular Acidification and ER Stress
3.6. PPARGC1A
3.7. Klotho
3.8. miRNAs
4. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- He, L.; Wondisford, F.E. Metformin Action: Concentrations Matter. Cell Metab. 2015, 21, 159–162. [Google Scholar] [CrossRef] [PubMed]
- King, P.; Peacock, I.; Donnelly, R. The UK Prospective Diabetes Study (UKPDS): Clinical and therapeutic implications for type 2 diabetes. Br. J. Clin. Pharmacol. 1999, 48, 643. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Rangel, E.; Inzucchi, S.E. Metformin: Clinical use in type 2 diabetes. Diabetologia 2017, 60, 1586–1593. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.-L.; Yin, J.; Alimujiang, M.; Yu, X.-Y.; Ai, L.-G.; Bao, Y.-Q.; Liu, F.; Jia, W.-P. Inhibition of mitochondrial complex I improves glucose metabolism independently of AMPK activation. J. Cell. Mol. Med. 2018, 22, 1316–1328. [Google Scholar] [CrossRef] [PubMed]
- Ben Sahra, I.; Regazzetti, C.; Robert, G.; Laurent, K.; Le Marchand-Brustel, Y.; Auberger, P.; Tanti, J.-F.; Giorgetti-Peraldi, S.; Bost, F. Metformin, Independent of AMPK, Induces mTOR Inhibition and Cell-Cycle Arrest through REDD1. Cancer Res. 2011, 71, 4366–4372. [Google Scholar] [CrossRef]
- Musi, N.; Hirshman, M.F.; Nygren, J.; Svanfeldt, M.; Bavenholm, P.; Rooyackers, O.; Zhou, G.; Williamson, J.M.; Ljunqvist, O.; Efendic, S.; et al. Metformin Increases AMP-Activated Protein Kinase Activity in Skeletal Muscle of Subjects With Type 2 Diabetes. Diabetes 2002, 51, 2074–2081. [Google Scholar] [CrossRef]
- Vasamsetti, S.B.; Karnewar, S.; Kanugula, A.K.; Thatipalli, A.R.; Kumar, J.M.; Kotamraju, S. Metformin Inhibits Monocyte-to-Macrophage Differentiation via AMPK-Mediated Inhibition of STAT3 Activation: Potential Role in Atherosclerosis. Diabetes 2015, 64, 2028–2041. [Google Scholar] [CrossRef]
- Eisenreich, A.; Leppert, U. Update on the Protective Renal Effects of Metformin in Diabetic Nephropathy. Curr. Med. Chem. 2017, 24, 3397–3412. [Google Scholar] [CrossRef]
- Lord, J.M.; Flight, I.H.K.; Norman, R.J. Metformin in polycystic ovary syndrome: Systematic review and meta-analysis. BMJ 2003, 327, 951–953. [Google Scholar] [CrossRef]
- El Massry, M.; Alaeddine, L.M.; Ali, L.; Saad, C.; Eid, A.A. Metformin: A Growing Journey from Glycemic Control to the Treatment of Alzheimer’s Disease and Depression. Curr. Med. Chem. 2021, 28, 2328–2345. [Google Scholar] [CrossRef]
- Salvatore, T.; Galiero, R.; Caturano, A.; Vetrano, E.; Rinaldi, L.; Coviello, F.; Di Martino, A.; Albanese, G.; Marfella, R.; Sardu, C.; et al. Effects of Metformin in Heart Failure: From Pathophysiological Rationale to Clinical Evidence. Biomolecules 2021, 11, 1834. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.M.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. Br. Med. J. 2005, 330, 1304–1305. [Google Scholar] [CrossRef] [PubMed]
- Zi, F.; Zi, H.; Li, Y.; He, J.; Shi, Q.; Cai, Z. Metformin and cancer: An existing drug for cancer prevention and therapy. Oncol. Lett. 2018, 15, 683–690. [Google Scholar] [CrossRef]
- Kasznicki, J.; Sliwinska, A.; Drzewoski, J. Metformin in cancer prevention and therapy. Ann. Transl. Med. 2014, 2, 57. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Lee, S.; Chun, K.H.; Jeon, J.Y.; Han, S.J.; Kim, D.J.; Kim, Y.S.; Woo, J.T.; Nam, M.S.; Baik, S.H.; et al. Metformin reduces the risk of cancer in patients with type 2 diabetes: An analysis based on the Korean National Diabetes Program Cohort. Medicine 2018, 97, e0036. [Google Scholar] [CrossRef]
- Conza, D.; Mirra, P.; Calì, G.; Insabato, L.; Fiory, F.; Beguinot, F.; Ulianich, L. Metformin Dysregulates the Unfolded Protein Response and the WNT/β-Catenin Pathway in Endometrial Cancer Cells through an AMPK-Independent Mechanism. Cells 2021, 10, 1067. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, X. Advances of Wnt Signalling Pathway in Colorectal Cancer. Cells 2023, 12, 447. [Google Scholar] [CrossRef]
- Clevers, H.; Nusse, R. Wnt/Beta-Catenin Signaling and Disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef]
- Cheng, X.; Xu, X.; Chen, D.; Zhao, F.; Wang, W. Therapeutic Potential of Targeting the Wnt/Beta-Catenin Signaling Pathway in Colorectal Cancer. Biomed Pharmacoth. 2019, 110, 473–481. [Google Scholar] [CrossRef]
- Li, V.S.W.; Ng, S.S.; Boersema, P.J.; Low, T.Y.; Karthaus, W.R.; Gerlach, J.P.; Mohammed, S.; Heck, A.J.R.; Maurice, M.M.; Mahmoudi, T.; et al. Wnt Signaling through Inhibition of Beta-Catenin Degradation in an Intact Axin1 Complex. Cell 2012, 149, 1245–1256. [Google Scholar] [CrossRef]
- Mantilla, C.; Suárez Mellado, I.; Duque Jaramillo, A.; Navas, M.C. β-catenin signaling mechanisms and its role in carcinogenesis. CES Med. 2015, 29, 109–127. [Google Scholar]
- Kim, N.G.; Xu, C.; Gumbiner, B.M. Identification of Targets of the Wnt Pathway Destruction Complex in Addition to Beta-Catenin. Proc. Natl. Acad. Sci. USA 2009, 106, 5165–5170. [Google Scholar] [CrossRef]
- Latres, E.; Chiaur, D.S.; Pagano, M. The Human F Box Protein Beta-Trcp Associates with the Cul1/Skp1 Complex and Regulates the Stability of Beta-Catenin. Oncogene 1999, 18, 849–854. [Google Scholar] [CrossRef] [PubMed]
- Koch, S.; Acebron, S.P.; Herbst, J.; Hatiboglu, G.; Niehrs, C. Post-transcriptional Wnt Signaling Governs Epididymal Sperm Maturation. Cell 2015, 163, 1225–1236. [Google Scholar] [CrossRef] [PubMed]
- Behrens, J.; Von Kries, J.P.; Kühl, M.; Bruhn, L.; Wedlich, D.; Grosschedl, R.; Birchmeier, W. Functional interaction of β-catenin with the transcription factor LEF-1. Nature 1996, 382, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Lustig, B.; Jerchow, B.; Sachs, M.; Weiler, S.; Pietsch, T.; Karsten, U.; van de Wetering, M.; Clevers, H.; Schlag, P.M.; Birchmeier, W.; et al. Negative Feedback Loop of Wnt Signaling through Upregulation of Conductin/Axin2 in Colorectal and Liver Tumors. Mol. Cell. Biol. 2002, 22, 1184–1193. [Google Scholar] [CrossRef]
- Liu, C.; Li, Y.; Semenov, M.; Han, C.; Baeg, G.H.; Tan, Y.; Zhang, Z.; Lin, X.; He, X. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 2002, 108, 837–847. [Google Scholar] [CrossRef]
- He, T.-C.; Sparks, A.B.; Rago, C.; Hermeking, H.; Zawel, L.; da Costa, L.T.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Identification of c- MYC as a Target of the APC Pathway. Science 1998, 281, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Kawano, Y.; Kypta, R. Secreted antagonists of the Wnt signalling pathway. J. Cell Sci. 2003, 116, 2627–2634. [Google Scholar] [CrossRef]
- Li, X.; Zhang, C.; Yuan, Y.; Wang, Y.; Lu, S.; Zhou, Z.; Zhen, P.; Zhou, M. Downregulation of ARMC8 promotes tumorigenesis through activating Wnt/β-catenin pathway and EMT in cutaneous squamous cell carcinomas. J. Dermatol. Sci. 2021, 102, 184–192. [Google Scholar] [CrossRef]
- Heuberger, J.; Birchmeier, W. Interplay of Cadherin-Mediated Cell Adhesion and Canonical Wnt Signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a002915. [Google Scholar] [CrossRef] [PubMed]
- Margulis, A.; Zhang, W.; Alt-Holland, A.; Crawford, H.C.; Fusenig, N.E.; Garlick, J.A. E-cadherin Suppression Accelerates Squamous Cell Carcinoma Progression in Three-Dimensional, Human Tissue Constructs. Cancer Res 2005, 65, 1783–1791. [Google Scholar] [CrossRef]
- Conacci-Sorrell, M.; Simcha, I.; Ben-Yedidia, T.; Blechman, J.; Savagner, P.; Ben-Ze’ev, A. Autoregulation of E-cadherin expression by cadherin-cadherin interactions: The roles of beta-catenin signaling, Slug, and MAPK. J. Cell Biol. 2003, 163, 847–857. [Google Scholar] [CrossRef]
- Khan, M.A.; Chen, H.-C.; Zhang, D.; Fu, J. Twist: A molecular target in cancer therapeutics. Tumour Biol. 2013, 34, 2497–2506. [Google Scholar] [CrossRef]
- Hlubek, F.; Spaderna, S.; Jung, A.; Kirchner, T.; Brabletz, T. Beta-catenin activates a coordinated expression of the proinvasive factors laminin-5 gamma2 chain and MT1-MMP in colorectal carcinomas. Int. J. Cancer 2004, 108, 321–326. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt Signaling in Cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Sheldahl, L.C.; Slusarski, D.C.; Pandur, P.; Miller, J.R.; Kühl, M.; Moon, R.T. Dishevelled activates Ca2+ flux, PKC, and CamKII in vertebrate embryos. J. Cell Biol. 2003, 161, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Ishitani, T.; Kishida, S.; Hyodo-Miura, J.; Ueno, N.; Yasuda, J.; Waterman, M.; Shibuya, H.; Moon, R.T.; Ninomiya-Tsuji, J.; Matsumoto, K. The TAK1-NLK mitogen-activated protein kinase cascade functions in the Wnt-5a/Ca(2+) pathway to antagonize Wnt/beta-catenin signaling. Mol. Cell Biol. 2003, 23, 131–139. [Google Scholar] [CrossRef]
- Schlessinger, K.; Hall, A.; Tolwinski, N. Wnt signaling pathways meet Rho GTPases. Genes Dev. 2009, 23, 265–277. [Google Scholar] [CrossRef]
- Yamanaka, H.; Moriguchi, T.; Masuyama, N.; Kusakabe, M.; Hanafusa, H.; Takada, R.; Takada, S.; Nishida, E. JNK functions in the non-canonical Wnt pathway to regulate convergent extension movements in vertebrates. EMBO Rep. 2002, 3, 69–75. [Google Scholar] [CrossRef]
- Habas, R.; Kato, Y.; He, X. Wnt/Frizzled Activation of Rho Regulates Vertebrate Gastrulation and Requires a Novel Formin Homology Protein Daam1. Cell 2001, 107, 843–854. [Google Scholar] [CrossRef]
- Endo, Y.; Wolf, V.; Muraiso, K.; Kamijo, K.; Soon, L.; Üren, A.; Barshishat-Küpper, M.; Rubin, J.S. Wnt-3a-dependent Cell Motility Involves RhoA Activation and Is Specifically Regulated by Dishevelled-2. J. Biol. Chem. 2005, 280, 777–786. [Google Scholar] [CrossRef] [PubMed]
- Schlessinger, K.; McManus, E.J.; Hall, A. Cdc42 and noncanonical Wnt signal transduction pathways cooperate to promote cell polarity. J. Cell Biol. 2007, 178, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Nhieu, J.T.; Renard, C.A.; Wei, Y.; Cherqui, D.; Zafrani, E.S.; Buendia, M.A. Nuclear accumulation of mutated beta-catenin in hepatocellular carcinoma is associated with increased cell proliferation. Am. J. Pathol. 1999, 155, 703–710. [Google Scholar] [CrossRef]
- Maiti, S.; Alam, R.; Amos, C.I.; Huff, V. Frequent association of beta-catenin and WT1 mutations in Wilms tumors. Cancer Res. 2000, 60, 6288–6292. [Google Scholar]
- Palacios, J.; Gamallo, C. Mutations in the beta-catenin gene (CTNNB1) in endometrioid ovarian carcinomas. Cancer Res. 1998, 58, 1344–1347. [Google Scholar] [PubMed]
- Voeller, H.J.; Truica, C.I.; Gelmann, E.P. Beta-catenin mutations in human prostate cancer. Cancer Res. 1998, 58, 2520–2523. [Google Scholar]
- Zurawel, R.H.; Chiappa, S.A.; Allen, C.; Raffel, C. Sporadic medulloblastomas contain oncogenic beta-catenin mutations. Cancer Res. 1998, 58, 896–899. [Google Scholar]
- Zaman, G.J.R.; de Roos, J.A.D.M.; Libouban, M.A.A.; Prinsen, M.B.W.; de Man, J.; Buijsman, R.C.; Uitdehaag, J.C.M. TTK inhibitors as a targeted therapy for CTNNB1 (beta-catenin) mutant cancers. Mol. Cancer Ther. 2017, 16, 2609–2617. [Google Scholar] [CrossRef]
- Garcia-Rostan, G.; Tallini, G.; Herrero, A.; D’Aquila, T.G.; Carcangiu, M.L.; Rimm, D.L. Frequent mutation and nuclear localization of beta-catenin in anaplastic thyroid carcinoma. Cancer Res 1999, 59, 1811–1815. [Google Scholar]
- Tammela, T.; Sanchez-Rivera, F.J.; Cetinbas, N.M.; Wu, K.; Joshi, N.S.; Helenius, K.; Park, Y.; Azimi, R.; Kerper, N.R.; Wesselhoeft, R.A.; et al. A Wnt-producing niche drives proliferative potential and progression in lung adenocarcinoma. Nature 2017, 545, 355–359. [Google Scholar] [CrossRef]
- Kwan, H.T.; Chan, D.W.; Cai, P.C.H.; Mak, C.S.L.; Yung, M.M.H.; Leung, T.H.Y.; Wong, O.G.W.; Cheung, A.N.Y.; Ngan, H.Y.S. AMPK Activators Suppress Cervical Cancer Cell Growth through Inhibition of DVL3 Mediated Wnt/β-Catenin Signaling Activity. PLoS ONE 2013, 8, e53597. [Google Scholar] [CrossRef]
- Zou, Y.-F.; Xie, C.-W.; Yang, S.-X.; Xiong, J.-P. AMPK activators suppress breast cancer cell growth by inhibiting DVL3-facilitated Wnt/β-catenin signaling pathway activity. Mol. Med. Rep. 2017, 1, 899–907. [Google Scholar] [CrossRef]
- Banerjee, P.; Dutta, S.; Rajarshi, P. Dysregulation of Wnt-Signaling and a Candidate Set of miRNAs Underlie the Effect of Metformin on Neural Crest Cell Development. Stem Cells 2016, 34, 334–345. [Google Scholar] [CrossRef]
- Salatino, A.; Mirabelli, M.; Chiefari, E.; Greco, M.; Di Vito, A.; Bonapace, G.; Brunetti, F.S.; Crocerossa, F.; Epstein, A.L.; Foti, D.P.; et al. The anticancer effects of Metformin in the male germ tumor SEM-1 cell line are mediated by HMGA1. Front. Endocrinol. 2022, 13, 1051988. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhang, H.; Sun, H. Metformin inhibits the proliferation and invasion of ovarian cancer cells by suppressing tripartite motif-containing 37-induced tumor necrosis factor receptor-associated factor 2 ubiquitination. Cancer Sci. 2022, 113, 3776–3786. [Google Scholar] [CrossRef]
- Hwang, Y.P.; Jeong, H.G. Metformin blocks migration and invasion of tumour cells by inhibition of matrix metalloproteinase-9 activation through a calcium and protein kinase Calpha-dependent pathway: Phorbol-12-myristate-13-acetate-induced/extracellular signal-regulated kinase/activator protein-1. Br. J. Pharmacol. 2010, 160, 1195–1211. [Google Scholar]
- Trinh, S.X.; Nguyen, H.T.B.; Saimuang, K.; Prachayasittikul, V.; Chan-On, W. Metformin Inhibits Migration and Invasion of Cholangiocarcinoma Cells. Asian Pac. J. Cancer Prev. 2017, 18, 473–477. [Google Scholar] [CrossRef]
- Cerezo, M.; Tichet, M.; Abbe, P.; Ohanna, M.; Lehraiki, A.; Rouaud, F.; Allegra, M.; Giacchero, D.; Bahadoran, P.; Bertolotto, C.; et al. Metformin blocks melanoma invasion and metastasis development in AMPK/p53-dependent manner. Mol. Cancer Ther. 2013, 12, 1605–1615. [Google Scholar] [CrossRef]
- Tan, B.K.; Adya, R.; Chen, J.; Lehnert, H.; Cassia, L.J.S.; Randeva, H.S. Metformin Treatment Exerts Antiinvasive and Antimetastatic Effects in Human Endometrial Carcinoma Cells. J. Clin. Endocrinol. Metab. 2011, 96, 808–816. [Google Scholar] [CrossRef]
- Yenmis, G.; Sarac, E.Y.; Besli, N.; Soydas, T.; Tastan, C.; Kancagi, D.D.; Yilanci, M.; Senol, K.; Karagulle, O.O.; Ekmekci, C.G.; et al. Anti-cancer effect of metformin on the metastasis and invasion of primary breast cancer cells through mediating NF-kB activity. Acta Histochem. 2021, 123, 151709. [Google Scholar] [CrossRef]
- Liang, F.; Wang, Y.-G.; Wang, C. Metformin Inhibited Growth, Invasion and Metastasis of Esophageal Squamous Cell Carcinoma in Vitro and in Vivo. Cell. Physiol. Biochem. 2018, 51, 1276–1286. [Google Scholar] [CrossRef]
- Xu, X.; Ma, J.; Li, C.; Zhao, W.; Xu, Y. Regulation of chondrosarcoma invasion by MMP26. Tumor Biol. 2014, 36, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.R.; Nam, S.; Kook, M.C.; Kim, K.T.; Liu, X.; Yao, H.; Jung, H.R.; Jr, L.R.; Seo, H.H.; Park, H.S.; et al. HNF4α is a therapeutic target that links AMPK to WNT signalling in early-stage gastric cancer. Gut 2016, 65, 19–32. [Google Scholar] [CrossRef]
- Hong, Y.H.; Varanasi, U.S.; Yang, W.; Leff, T. AMP-activated Protein Kinase Regulates HNF4α Transcriptional Activity by Inhibiting Dimer Formation and Decreasing Protein Stability. J. Biol. Chem. 2003, 278, 27495–27501. [Google Scholar] [CrossRef]
- Ribeiro, A.; Archer, A.; Le Beyec, J.; Cattin, A.-L.; Saint-Just, S.; Pinçon-Raymond, M.; Chambaz, J.; Lacasa, M.; Cardot, P. Hepatic Nuclear Factor-4, a Key Transcription Factor at the Crossroads Between Architecture and Function of Epithelia. Recent Pat. Endocr. Metab. Immune Drug Discov. 2007, 1, 166–175. [Google Scholar] [CrossRef]
- Kim, D.Y.; Park, E.Y.; Chang, E.; Kang, H.G.; Koo, Y.; Lee, E.J.; Ko, J.Y.; Kong, H.K.; Chun, K.H.; Park, J.H. A novel mir-34a target, protein kinase d1, stimulates cancer stemness and drug resistance through gsk3/beta-catenin signaling in breast cancer. Oncotarget 2016, 7, 14791–14802. [Google Scholar] [CrossRef] [PubMed]
- Wiese, M.; Walther, N.; Diederichs, C.; Schill, F.; Monecke, S.; Salinas, G.; Sturm, D.; Pfister, S.M.; Dressel, R.; Johnsen, S.A.; et al. The beta-catenin/cbp-antagonist icg-001 inhibits pediatric glioma tumorigenicity in a wnt-independent manner. Oncotarget 2017, 8, 27300–27313. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, Y. Metformin attenuates cells stemness and epithelial-mesenchymal transition in colorectal cancer cells by inhibiting the Wnt3a/β-catenin pathway. Mol. Med. Rep. 2019, 19, 1203–1209. [Google Scholar] [CrossRef]
- Tian, Y.; Tang, B.; Wang, C.; Sun, D.; Zhang, R.; Luo, N.; Han, Z.; Liang, R.; Gao, Z.; Wang, L. Metformin mediates resensitivity to 5-fluorouracil in hepatocellular carcinoma via the suppression of YAP. Oncotarget 2016, 7, 46230–46241. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Lanza-Jacoby, S. Metformin decreases growth of pancreatic cancer cells by decreasing reactive oxygen species: Role of NOX4. Biochem. Biophys. Res. Commun. 2015, 465, 41–46. [Google Scholar] [CrossRef]
- Liu, Y.; He, C.; Huang, X. Metformin partially reverses the carboplatin-resistance in NSCLC by inhibiting glucose metabolism. Oncotarget 2017, 8, 75206–75216. [Google Scholar] [CrossRef]
- Justus, C.R.; Dong, L.; Yang, L.V. Acidic tumor microenvironment and pH sensing G protein-coupled receptors. Front. Physiol. 2013, 4, 354. [Google Scholar] [CrossRef]
- Janzer, A.; German, N.J.; Gonzalez-Herrera, K.N.; Asara, J.M.; Haigis, M.C.; Struhl, K. Metformin and phenformin deplete tricarboxylic acid cycle and glycolytic intermediates during cell transformation and NTPs in cancer stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, 10574–10579. [Google Scholar] [CrossRef]
- Melnik, S.; Dvornikov, M.; Müller-Decker, K.; Depner, S.; Stannek, P.; Meister, M.; Warth, A.; Thomas, M.; Muley, T.; Risch, A.; et al. Cancer cell specific inhibition of Wnt/β-catenin signaling by forced intracellular acidification. Cell Discov. 2018, 4, 37. [Google Scholar] [PubMed]
- Schröder, M.; Kaufman, R. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef]
- Treglia, A.S.; Turco, S.; Ulianich, L.; Ausiello, P.; Lofrumento, D.D.; Nicolardi, G.; Miele, C.; Garbi, C.; Beguinot, F.; Di Jeso, B. Cell fate following ER stress: Just a matter of “quo ante” recovery or death? Histol. Histopathol. 2012, 27, 1–12. [Google Scholar] [PubMed]
- Quentin, T.; Steinmetz, M.; Poppe, A.; Thoms, S. Metformin differentially activates ER stress signaling pathways without inducing apoptosis. Dis. Model. Mech. 2012, 5, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Horndasch, M.; Lienkamp, S.; Springer, E.; Schmitt, A.; Pavenstädt, H.; Walz, G.; Gloy, J. The C/EBP homologous protein CHOP (GADD153) is an inhibitor of Wnt/TCF signals. Oncogene 2006, 25, 3397–3407. [Google Scholar]
- Vervoort, S.J.; Lourenco, A.R.; van Boxtel, R.; Coffer, P.J. SOX4 mediates TGF-beta-induced expression of mesenchymal markers during mammary cell epithelial to mesenchymal transition. PLoS ONE. 2013, 8, e53238. [Google Scholar]
- Bifulco, G.; Miele, C.; Di Jeso, B.; Beguinot, F.; Nappi, C.; Di Carlo, C.; Capuozzo, S.; Terrazzano, G.; Insabato, L.; Ulianich, L. Endoplasmic reticulum stress is activated in endometrial adenocarcinoma. Gynecol. Oncol. 2012, 125, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Calì, G.; Insabato, L.; Conza, D.; Bifulco, G.; Parrillo, L.; Mirra, P.; Fiory, F.; Miele, C.; Raciti, G.A.; Di Jeso, B.; et al. GRP78 Mediates Cell Growth and Invasiveness in Endometrial Cancer. J. Cell. Physiol. 2014, 229, 1417–1426. [Google Scholar] [CrossRef]
- Ulianich, L.; Insabato, L. Endoplasmic Reticulum Stress in Endometrial Cancer. Front. Med. 2014, 1, 55. [Google Scholar] [CrossRef]
- Ma, T.; Meng, L.; Wang, X.; Tian, Z.; Wang, J.; Liu, X.; Zhang, W.; Zhang, Y. TNFSF13B and PPARGC1A expression is associated with tumor-infiltrating immune cell abundance and prognosis in clear cell renal cell carcinoma. Am. J. Transl. Res. 2021, 13, 11048–11064. [Google Scholar]
- Huang, X.; Pan, L.; Zuo, Z.; Li, M.; Zeng, L.; Li, R.; Ye, Y.; Zhang, J.; Wu, G.; Bai, R.; et al. LINC00842 inactivates transcription co-regulator PGC-1α to promote pancreatic cancer malignancy through metabolic remodelling. Nat. Commun. 2021, 12, 3830. [Google Scholar] [CrossRef] [PubMed]
- Andrzejewski, S.; Klimcakova, E.; Johnson, R.M.; Tabariès, S.; Annis, M.G.; McGuirk, S.; Northey, J.J.; Chénard, V.; Sriram, U.; Papadopoli, D.J.; et al. PGC-1α Promotes Breast Cancer Metastasis and Confers Bioenergetic Flexibility against Metabolic Drugs. Cell Metab. 2017, 26, 778–787.e5. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, Y.; Guo, Y.; Tang, H.; Li, M.; Liu, L. A novel machine learning derived RNA-binding protein gene–based score system predicts prognosis of hepatocellular carcinoma patients. PeerJ 2021, 9, e12572. [Google Scholar] [CrossRef]
- Zuo, Q.; He, J.; Zhang, S.; Wang, H.; Jin, G.; Jin, H.; Cheng, Z.; Tao, X.; Yu, C.; Li, B.; et al. PPARgamma Coactivator-1alpha suppresses metastasis of hepatocellular carcinoma by inhibiting Warburg effect by PPARgamma-dependent WNT/beta-Catenin/Pyruvate Dehydrogenase Kinase Isozyme 1 Axis. Hepatology 2021, 73, 644–660. [Google Scholar]
- Zhang, Q.; Xiong, L.; Wei, T.; Liu, Q.; Yan, L.; Chen, J.; Dai, L.; Shi, L.; Zhang, W.; Yang, J.; et al. Hypoxia-responsive PPARGC1A/BAMBI/ACSL5 axis promotes progression and resistance to lenvatinib in hepatocellular carcinoma. Oncogene 2023, 42, 1509–1523. [Google Scholar] [CrossRef]
- Prud’homme, G.J.; Kurt, M.; Wang, Q. Pathobiology of the Klotho Antiaging Protein and Therapeutic Considerations. Front. Aging 2022, 3, 931331. [Google Scholar] [CrossRef]
- Urakawa, I.; Yamazaki, Y.; Shimada, T.; Iijima, K.; Hasegawa, H.; Okawa, K.; Fujita, T.; Fukumoto, S.; Yamashita, T. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 2006, 444, 770–774. [Google Scholar] [CrossRef]
- Dalton, G.D.; Xie, J.; An, S.-W.; Huang, C.-L. New Insights into the Mechanism of Action of Soluble Klotho. Front. Endocrinol. 2017, 8, 323. [Google Scholar] [CrossRef]
- Xuan, N.T.; Hai, N.V. Changes in expression of klotho affect physiological processes, diseases, and cancer. Iran. J. Basic Med. Sci. 2018, 21, 3–8. [Google Scholar]
- Abolghasemi, M.; Yousefi, T.; Maniati, M.; Qujeq, D. The interplay of Klotho with signaling pathway and microRNAs in cancers. J. Cell. Biochem. 2019, 120, 14306–14317. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, X.; Wang, X.; Jie, P.; Lu, H.; Zhang, S.; Lin, X.; Lam, E.K.; Cui, Y.; Yu, J.; et al. Klotho is silenced through promoter hypermethylation in gastric cancer. Am. J. Cancer Res. 2010, 1, 111–119. [Google Scholar]
- Liu, H.; Fergusson, M.M.; Castilho, R.M.; Liu, J.; Cao, L.; Chen, J.; Malide, D.; Rovira, I.I.; Schimel, D.; Kuo, C.J.; et al. Augmented Wnt Signaling in a Mammalian Model of Accelerated Aging. Science 2007, 317, 803–806. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, Z. Current understanding of klotho. Ageing Res. Rev. 2009, 8, 43–51. [Google Scholar] [CrossRef]
- Alimoradi, N.; Firouzabadi, N.; Fatehi, R. How metformin affects various malignancies by means of microRNAs: A brief review. Cancer Cell Int. 2021, 21, 207. [Google Scholar]
- Nangia-Makker, P.; Yu, Y.; Vasudevan, A.; Farhana, L.; Rajendra, S.G.; Levi, E.; Majumdar, A.P.N. Metformin: A Potential Therapeutic Agent for Recurrent Colon Cancer. PLoS ONE 2014, 9, e84369. [Google Scholar] [PubMed]
- Yu, Y.; Kanwar, S.S.; Patel, B.B.; Oh, P.S.; Nautiyal, J.; Fazlul, H.S.; Adhip, P.N.M. miRNA-21 induces stemness by downregulating transforming growth factor beta receptor 2 (TGFbetaR2) in colon cancer cells. Carcinogenesis 2012, 33, 68–76. [Google Scholar]


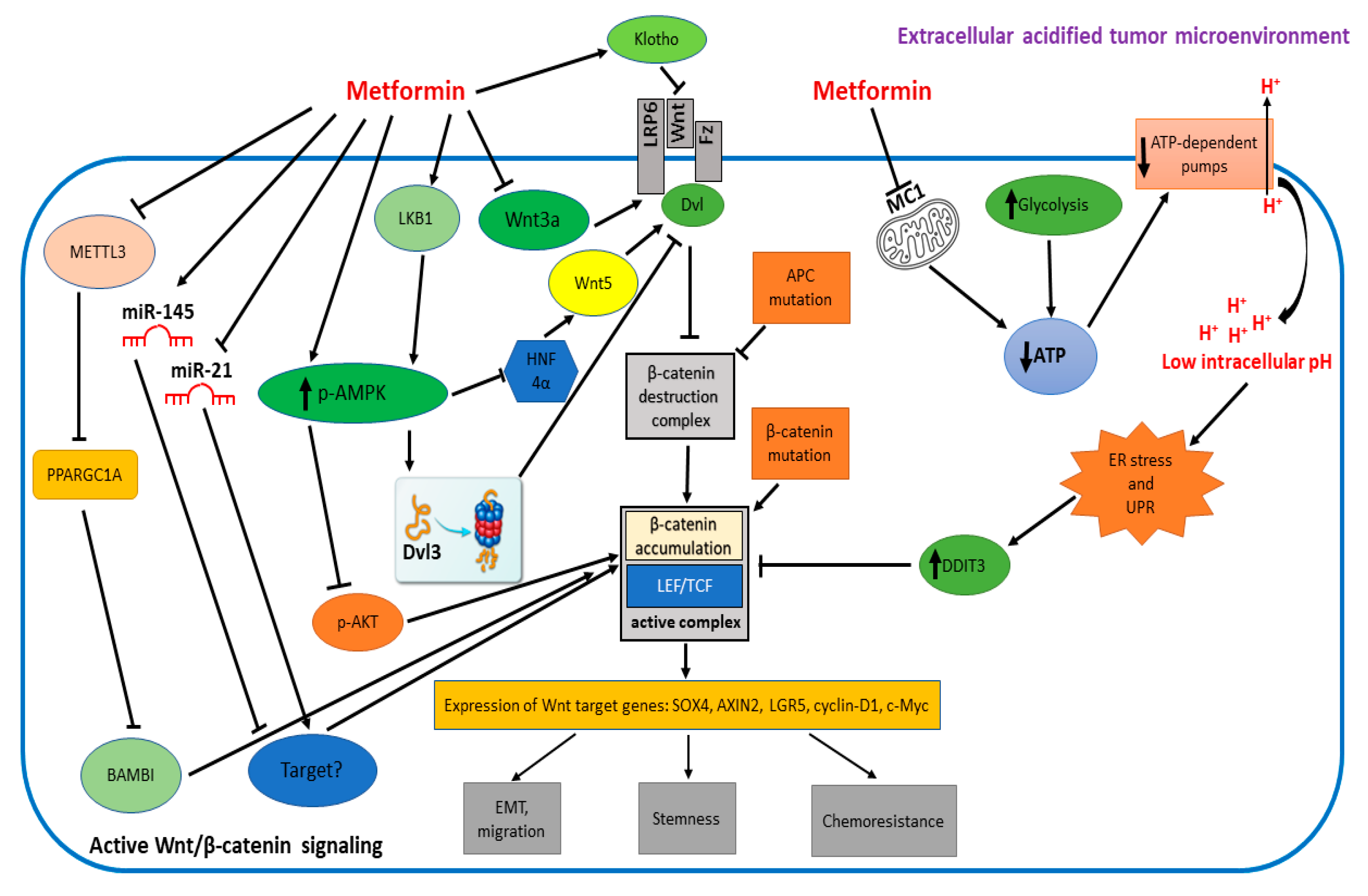{kind=link}
| Mediator | Cancer Type | Mechanism | Reference |
|---|---|---|---|
| Dv13 | Cervical, breast | Increased Dv13 proteasomal degradation | [53,54] |
| MMP26 | Chondrosarcoma | Increased β-catenin phosphorylation | [57] |
| HNF4α | Gastric | Wnt5 downregulation | [65] |
| Wnt3a | Colorectal | Wnt3a and β-catenin downregulation | [70] |
| DDIT3 | Lung, breast, colon, prostate, melanoma, glioblastoma | Inhibitory binding to LEF/TCF complex | [76,79,80] |
| PPARGC1A | Hepatocellular | BAMBI repression | [89,90] |
| Klotho | Gastric | Competitive binding to Wnt ligands | [97,98] |
| miR-21 | Colon | Reduced β-catenin activity | [33,92] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conza, D.; Mirra, P.; Fiory, F.; Insabato, L.; Nicolò, A.; Beguinot, F.; Ulianich, L. Metformin: A New Inhibitor of the Wnt Signaling Pathway in Cancer. Cells 2023, 12, 2182. https://doi.org/10.3390/cells12172182
Conza D, Mirra P, Fiory F, Insabato L, Nicolò A, Beguinot F, Ulianich L. Metformin: A New Inhibitor of the Wnt Signaling Pathway in Cancer. Cells. 2023; 12(17):2182. https://doi.org/10.3390/cells12172182
Chicago/Turabian StyleConza, Domenico, Paola Mirra, Francesca Fiory, Luigi Insabato, Antonella Nicolò, Francesco Beguinot, and Luca Ulianich. 2023. "Metformin: A New Inhibitor of the Wnt Signaling Pathway in Cancer" Cells 12, no. 17: 2182. https://doi.org/10.3390/cells12172182