
Microvascular Thrombosis and Liver Fibrosis Progression: Mechanisms and Clinical Applications
 , , , and
, , , and
Abstract
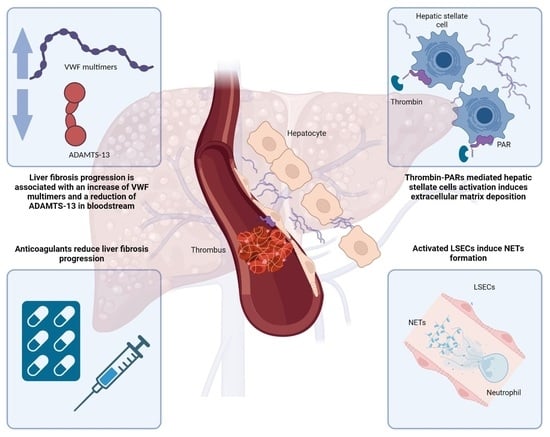
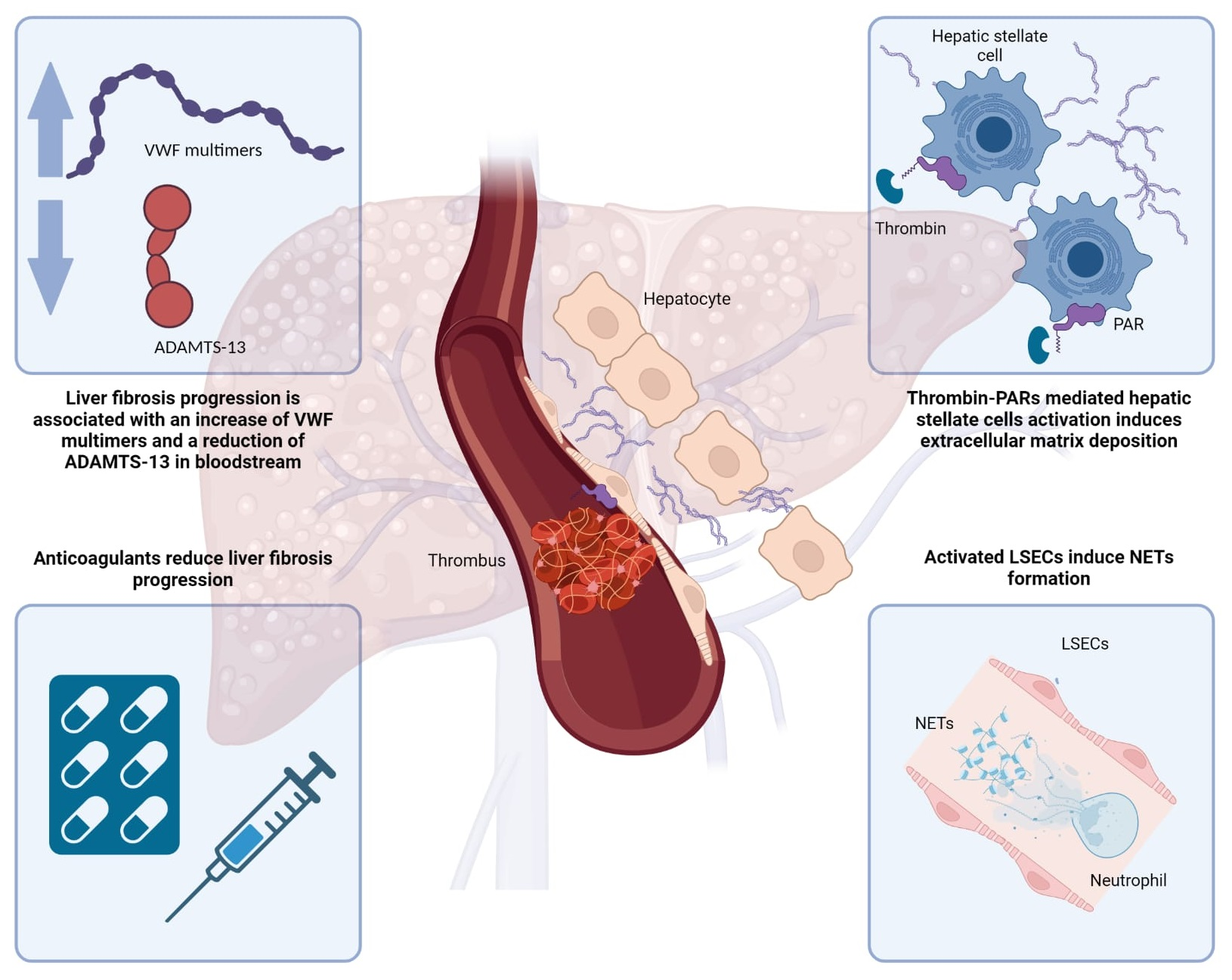
:
1. Introduction
2. From Microvascular Thrombosis to Hepatic Fibrosis
3. Hepatic Stellate Cells and Protease Activated Receptor
4. The Role of Liver Sinusoidal Endothelial Cells and Neutrophil Extracellular Traps
5. Platelets: Not Just Hemostatic Functions
6. ADAMTS 13—Von Willebrand Factor, a Bridge between Coagulation and Fibrosis
7. Hemostatic Balance in Liver Disease: Translational Implications
7.1. Correlation with Liver-Related Outcomes
7.2. The Gut–Liver Axis Is Associated with Alteration of Hemostatic Factors
8. Therapeutic Perspectives
8.1. Low Molecular Weight Heparin
8.2. Direct Oral Anticoagulants
8.3. Antiplatelet Agents
8.4. Statins

{kind=link}
{kind=link}
| Study | Model | Drug | Results |
|---|---|---|---|
| Assy et al., 2007 [137] | TAA-induced liver damage in rats | Enoxaparin | ↓ liver fibrosis severity (METAVIR score), ↓ total serum bilirubin, ↑ liver regeneration |
| Abe et al., 2006 [138] | CCl4-induced liver damage in rats | Dalteparin | ↓ liver fibrosis progression, ↑ hepatocyte growth factor (HGF), inhibition of HSCs, ↑ liver regeneration |
| Abdel-Salam et al., 2005 [139] | Bile duct ligation in mice | Enoxaparin | ↓ liver necrosis |
| ↓ fibrosis | |||
| Cerini et al., 2016 [23] | CCl4- and TAA-induced cirrhosis in rats | Enoxaparin | ↓ hepatic venule resistance, ↓ portal pressure, ↓ hepatic fibrin deposition, ↓ HSCs activation, ↓ liver fibrosis |
| Villa et al., 2012 [141] | Patients with cirrhosis (Child–Pugh B-C) | Enoxaparin (4000 UI qd) for 48 weeks | No patients in the treatment group developed PVT, ↓ decompensation, ↑ liver function, ↑ survival, ↓ inflammatory markers |
| Lee et al., 2018 [145] | TAA-induced liver damage in rats | Dabigatran | ↓ collagen and fibrin deposition in the liver |
| Mahmoud et al., 2019 [146] | CCl4-induced liver damage in rats | Rivaroxaban | ↓ inflammatory and fibrosis markers, ↓ liver fibrosis |
| Assy et al., 2007 [137] | TAA-induced liver damage in rats | Aspirin | ↓ liver fibrosis severity (METAVIR score), ↓ total serum bilirubin, ↑ liver regeneration |
| Sitia et al., 2012 [85] | Mice infected with HBV | Antiplatelet drugs (aspirin or clopidogrel) | ↓ intrahepatic inflammatory cells, ↓ liver fibrosis severity, ↓ HCC risk |
| Fujita et al., 2008 [149] | Murine model of NAFLD | Aspirin, ticlopidine, or cilostazol | ↓ inflammatory cells, ↓ procollagen proteins, ↓ hepatic steatosis, ↓ inflammation, ↓ fibrosis |
| Poujol-Robert et al., 2016 [150] | Retrospective study in patients with recurrent HCV infection after liver transplantation | Low-dose Aspirin | ↓ fibrosis progression |
9. Discussion
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Rockey, D.C.; Bell, P.D.; Hill, J.A. Fibrosis—A Common Pathway to Organ Injury and Failure. N. Engl. J. Med. 2015, 372, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- Jourde-Chiche, N.; Fakhouri, F.; Dou, L.; Bellien, J.; Burtey, S.; Frimat, M.; Jarrot, P.-A.; Kaplanski, G.; Le Quintrec, M.; Pernin, V.; et al. Endothelium Structure and Function in Kidney Health and Disease. Nat. Rev. Nephrol. 2019, 15, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Chambers, R.C.; Scotton, C.J. Coagulation Cascade Proteinases in Lung Injury and Fibrosis. Proc. Am. Thorac. Soc. 2012, 9, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Macphee, P.J.; Dindzans, V.J.; Fung, L.-S.; Levy, G.A. Acute and Chronic Changes in the Microcirculation of the Liver in Inbred Strains of Mice Following Infection with Mouse Hepatitis Virus Type 3. Hepatology 1985, 5, 649–660. [Google Scholar] [CrossRef]
- MacPhee, P.J.; Schmidt, E.E.; Keown, P.A.; Groom, A.C. Microcirculatory Changes in Livers of Mice Infected with Murine Hepatitis Virus. Evidence from Microcorrosion Casts and Measurements of Red Cell Velocity. Microvasc. Res. 1988, 36, 140–149. [Google Scholar] [CrossRef]
- Levy, G.A.; Macphee, P.J.; Fung, L.S.; Fisher, M.M.; Rappaport, A.M. The Effect of Mouse Hepatitis Virus Infection on the Microcirculation of the Liver. Hepatology 2007, 3, 964–973. [Google Scholar] [CrossRef]
- Wanless, I.R.; Liu, J.J.; Butany, J. Role of Thrombosis in the Pathogenesis of Congestive Hepatic Fibrosis (Cardiac Cirrhosis). Hepatology 1995, 21, 1232–1237. [Google Scholar] [CrossRef]
- Wanless, I.R. The Role of Vascular Injury and Congestion in the Pathogenesis of Cirrhosis: The Congestive Escalator and the Parenchymal Extinction Sequence. Curr. Hepatol. Rep. 2020, 19, 40–53. [Google Scholar] [CrossRef] [Green Version]
- Bissell, D. Inflammation and Hepatic Fibrosis. Semin. Liver Dis. 2010, 30, 211–214. [Google Scholar] [CrossRef]
- Simonetto, D.A.; Yang, H.; Yin, M.; de Assuncao, T.M.; Kwon, J.H.; Hilscher, M.; Pan, S.; Yang, L.; Bi, Y.; Beyder, A.; et al. Chronic Passive Venous Congestion Drives Hepatic Fibrogenesis via Sinusoidal Thrombosis and Mechanical Forces. Hepatology 2015, 61, 648–659. [Google Scholar] [CrossRef] [Green Version]
- Martinod, K.; Deppermann, C. Immunothrombosis and Thromboinflammation in Host Defense and Disease. Platelets 2021, 32, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Wang, H.; Billiar, T.R.; Kroemer, G.; Kang, R. Emerging Mechanisms of Immunocoagulation in Sepsis and Septic Shock. Trends Immunol. 2021, 42, 508–522. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Tyagi, T.; Antoniak, S. Platelet in Thrombo-Inflammation: Unraveling New Therapeutic Targets. Front. Immunol. 2022, 13, 1039843. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.; Park, H.E.; Song, M.S.; Kim, H.; Baek, J.-H. The Therapeutic Potential of Anticoagulation in Organ Fibrosis. Front. Med. 2022, 9, 866746. [Google Scholar] [CrossRef]
- Nalugo, M.; Schulte, L.J.; Masood, M.F.; Zayed, M.A. Microvascular Angiopathic Consequences of COVID-19. Front. Cardiovasc. Med. 2021, 8, 636843. [Google Scholar] [CrossRef]
- Leo, M.; Galante, A.; Pagnamenta, A.; Ruinelli, L.; Ponziani, F.R.; Gasbarrini, A.; De Gottardi, A. Hepatocellular Liver Injury in Hospitalized Patients Affected by COVID-19: Presence of Different Risk Factors at Different Time Points. Dig. Liver Dis. 2022, 54, 565–571. [Google Scholar] [CrossRef]
- Ponziani, F.R.; Del Zompo, F.; Nesci, A.; Santopaolo, F.; Ianiro, G.; Pompili, M.; Gasbarrini, A. “Gemelli against COVID-19” group. Liver Involvement Is Not Associated with Mortality: Results from a Large Cohort of SARS-CoV-2 Positive Patients. Aliment. Pharmacol. Ther. 2020, 52, 1060–1068. [Google Scholar] [CrossRef]
- Sonzogni, A.; Previtali, G.; Seghezzi, M.; Grazia Alessio, M.; Gianatti, A.; Licini, L.; Morotti, D.; Zerbi, P.; Carsana, L.; Rossi, R.; et al. Liver Histopathology in Severe COVID 19 Respiratory Failure Is Suggestive of Vascular Alterations. Liver Int. 2020, 40, 2110–2116. [Google Scholar] [CrossRef]
- Kondo, R.; Kawaguchi, N.; McConnell, M.J.; Sonzogni, A.; Licini, L.; Valle, C.; Bonaffini, P.A.; Sironi, S.; Alessio, M.G.; Previtali, G.; et al. Pathological Characteristics of Liver Sinusoidal Thrombosis in COVID-19 Patients: A Series of 43 Cases. Hepatol. Res. 2021, 51, 1000–1006. [Google Scholar] [CrossRef]
- Del Zompo, F.; De Siena, M.; Ianiro, G.; Gasbarrini, A.; Pompili, M.; Ponziani, F.R. Prevalence of Liver Injury and Correlation with Clinical Outcomes in Patients with COVID-19: Systematic Review with Meta-Analysis. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 13072–13088. [Google Scholar] [CrossRef]
- Moya, L.; Farashi, S.; Suravajhala, P.; Janaththani, P.; Batra, J. Severe COVID-19 May Impact Hepatic Fibrosis /Hepatic Stellate Cells Activation as Indicated by a Pathway and Population Genetic Study. Genes 2022, 14, 22. [Google Scholar] [CrossRef]
- Targher, G.; Mantovani, A.; Byrne, C.D.; Wang, X.-B.; Yan, H.-D.; Sun, Q.-F.; Pan, K.-H.; Zheng, K.I.; Chen, Y.-P.; Eslam, M.; et al. Risk of Severe Illness from COVID-19 in Patients with Metabolic Dysfunction-Associated Fatty Liver Disease and Increased Fibrosis Scores. Gut 2020, 69, 1545–1547. [Google Scholar] [CrossRef]
- Cerini, F.; Vilaseca, M.; Lafoz, E.; García-Irigoyen, O.; García-Calderó, H.; Tripathi, D.M.; Avila, M.; Reverter, J.C.; Bosch, J.; Gracia-Sancho, J.; et al. Enoxaparin Reduces Hepatic Vascular Resistance and Portal Pressure in Cirrhotic Rats. J. Hepatol. 2016, 64, 834–842. [Google Scholar] [CrossRef] [PubMed]
- Vilaseca, M.; García-Calderó, H.; Lafoz, E.; García-Irigoyen, O.; Avila, M.A.; Reverter, J.C.; Bosch, J.; Hernández-Gea, V.; Gracia-Sancho, J.; García-Pagán, J.C. The Anticoagulant Rivaroxaban Lowers Portal Hypertension in Cirrhotic Rats Mainly by Deactivating Hepatic Stellate Cells. Hepatology 2017, 65, 2031–2044. [Google Scholar] [CrossRef] [Green Version]
- Liedtke, C.; Nevzorova, Y.A.; Luedde, T.; Zimmermann, H.; Kroy, D.; Strnad, P.; Berres, M.-L.; Bernhagen, J.; Tacke, F.; Nattermann, J.; et al. Liver Fibrosis—From Mechanisms of Injury to Modulation of Disease. Front. Med. 2022, 8, 814496. [Google Scholar] [CrossRef] [PubMed]
- Lee, U.E.; Friedman, S.L. Mechanisms of Hepatic Fibrogenesis. Best Pract. Res. Clin. Gastroenterol. 2011, 25, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Corpechot, C.; Barbu, V.; Wendum, D.; Kinnman, N.; Rey, C.; Poupon, R.; Housset, C.; Rosmorduc, O. Hypoxia-Induced VEGF and Collagen I Expressions Are Associated with Angiogenesis and Fibrogenesis in Experimental Cirrhosis: Hypoxia-Induced VEGF and Collagen I Expressions Are Associated with Angiogenesis and Fibrogenesis in Experimental Cirrhosis. Hepatology 2002, 35, 1010–1021. [Google Scholar] [CrossRef]
- Riewald, M. Orchestration of Coagulation Protease Signaling by Tissue Factor. Trends Cardiovasc. Med. 2002, 12, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Peach, C.J.; Edgington-Mitchell, L.E.; Bunnett, N.W.; Schmidt, B.L. Protease-Activated Receptors in Health and Disease. Physiol. Rev. 2023, 103, 717–785. [Google Scholar] [CrossRef]
- Kataoka, H.; Hamilton, J.R.; McKemy, D.D.; Camerer, E.; Zheng, Y.-W.; Cheng, A.; Griffin, C.; Coughlin, S.R. Protease-Activated Receptors 1 and 4 Mediate Thrombin Signaling in Endothelial Cells. Blood 2003, 102, 3224–3231. [Google Scholar] [CrossRef]
- Martorell, L.; Martínez-González, J.; Rodríguez, C.; Gentile, M.; Calvayrac, O.; Badimon, L. Thrombin and Protease-Activated Receptors (PARs) in Atherothrombosis. Thromb. Haemost. 2008, 99, 305–315. [Google Scholar] [CrossRef]
- Lin, C.; von der Thüsen, J.; Daalhuisen, J.; ten Brink, M.; Crestani, B.; van der Poll, T.; Borensztajn, K.; Spek, C.A. Protease-Activated Receptor (PAR)-2 Is Required for PAR-1 Signalling in Pulmonary Fibrosis. J. Cell Mol. Med. 2015, 19, 1346–1356. [Google Scholar] [CrossRef] [PubMed]
- Wygrecka, M.; Kwapiszewska, G.; Jablonska, E.; von Gerlach, S.; Henneke, I.; Zakrzewicz, D.; Guenther, A.; Preissner, K.T.; Markart, P. Role of Protease-Activated Receptor-2 in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2011, 183, 1703–1714. [Google Scholar] [CrossRef] [PubMed]
- Yokono, Y.; Hanada, K.; Narita, M.; Tatara, Y.; Kawamura, Y.; Miura, N.; Kitayama, K.; Nakata, M.; Nozaka, M.; Kato, T.; et al. Blockade of PAR-1 Signaling Attenuates Cardiac Hypertrophy and Fibrosis in Renin-Overexpressing Hypertensive Mice. J. Am. Heart Assoc. 2020, 9, e015616. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, P.; Dobie, R.; Wilson-Kanamori, J.R.; Dora, E.F.; Henderson, B.E.P.; Luu, N.T.; Portman, J.R.; Matchett, K.P.; Brice, M.; Marwick, J.A.; et al. Resolving the Fibrotic Niche of Human Liver Cirrhosis at Single-Cell Level. Nature 2019, 575, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Gaça, M.D.A.; Zhou, X.; Benyon, R.C. Regulation of Hepatic Stellate Cell Proliferation and Collagen Synthesis by Proteinase-Activated Receptors. J. Hepatol. 2002, 36, 362–369. [Google Scholar] [CrossRef]
- Chambers, R.C.; Dabbagh, K.; McANULTY, R.J.; Gray, A.J.; Blanc-Brude, O.P.; Laurent, G.J. Thrombin Stimulates Fibroblast Procollagen Production via Proteolytic Activation of Protease-Activated Receptor 1. Biochem. J. 1998, 333, 121–127. [Google Scholar] [CrossRef] [Green Version]
- Rullier, A.; Gillibert-Duplantier, J.; Costet, P.; Cubel, G.; Haurie, V.; Petibois, C.; Taras, D.; Dugot-Senant, N.; Deleris, G.; Bioulac-Sage, P.; et al. Protease-Activated Receptor 1 Knockout Reduces Experimentally Induced Liver Fibrosis. Am. J. Physiol.-Gastrointest. Liver Physiol. 2008, 294, G226–G235. [Google Scholar] [CrossRef] [PubMed]
- Dhar, A.; Sadiq, F.; Anstee, Q.M.; Levene, A.P.; Goldin, R.D.; Thursz, M.R. Thrombin and Factor Xa Link the Coagulation System with Liver Fibrosis. BMC Gastroenterol. 2018, 18, 60. [Google Scholar] [CrossRef]
- Fiorucci, S.; Antonelli, E.; Distrutti, E.; Severino, B.; Fiorentina, R.; Baldoni, M.; Caliendo, G.; Santagada, V.; Morelli, A.; Cirino, G. PAR1 Antagonism Protects against Experimental Liver Fibrosis. Role of Proteinase Receptors in Stellate Cell Activation. Hepatology 2004, 39, 365–375. [Google Scholar] [CrossRef]
- Poole, L.G.; Pant, A.; Cline-Fedewa, H.M.; Williams, K.J.; Copple, B.L.; Palumbo, J.S.; Luyendyk, J.P. Liver Fibrosis Is Driven by Protease-activated Receptor-1 Expressed by Hepatic Stellate Cells in Experimental Chronic Liver Injury. Res. Pract. Thromb. Haemost. 2020, 4, 906–917. [Google Scholar] [CrossRef] [PubMed]
- Kassel, K.M.; Sullivan, B.P.; Cui, W.; Copple, B.L.; Luyendyk, J.P. Therapeutic Administration of the Direct Thrombin Inhibitor Argatroban Reduces Hepatic Inflammation in Mice with Established Fatty Liver Disease. Am. J. Pathol. 2012, 181, 1287–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kassel, K.M.; Owens, A.P.; Rockwell, C.E.; Sullivan, B.P.; Wang, R.; Tawfik, O.; Li, G.; Guo, G.L.; Mackman, N.; Luyendyk, J.P. Protease-Activated Receptor 1 and Hematopoietic Cell Tissue Factor Are Required for Hepatic Steatosis in Mice Fed a Western Diet. Am. J. Pathol. 2011, 179, 2278–2289. [Google Scholar] [CrossRef]
- Nault, R.; Fader, K.A.; Kopec, A.K.; Harkema, J.R.; Zacharewski, T.R.; Luyendyk, J.P. From the Cover: Coagulation-Driven Hepatic Fibrosis Requires Protease Activated Receptor-1 (PAR-1) in a Mouse Model of TCDD-Elicited Steatohepatitis. Toxicol. Sci. 2016, 154, 381–391. [Google Scholar] [CrossRef] [Green Version]
- Martinelli, A.; Knapp, S.; Anstee, Q.; Worku, M.; Tommasi, A.; Zucoloto, S.; Goldin, R.; Thursz, M. Effect of a Thrombin Receptor (Protease-Activated Receptor 1, PAR-1) Gene Polymorphism in Chronic Hepatitis C Liver Fibrosis. J. Gastroenterol. Hepatol. 2008, 23, 1403–1409. [Google Scholar] [CrossRef]
- Rullier, A.; Senant, N.; Kisiel, W.; Bioulac-Sage, P.; Balabaud, C.; Le Bail, B.; Rosenbaum, J. Expression of Protease-Activated Receptors and Tissue Factor in Human Liver. Virchows Arch. 2006, 448, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, B.P.; Weinreb, P.H.; Violette, S.M.; Luyendyk, J.P. The Coagulation System Contributes to AVβ6 Integrin Expression and Liver Fibrosis Induced by Cholestasis. Am. J. Pathol. 2010, 177, 2837–2849. [Google Scholar] [CrossRef]
- Copple, B.L.; Moulin, F.; Hanumegowda, U.M.; Ganey, P.E.; Roth, R.A. Thrombin and Protease-Activated Receptor-1 Agonists Promote Lipopolysaccharide-Induced Hepatocellular Injury in Perfused Livers. J. Pharmacol. Exp. Ther. 2003, 305, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Du, W.; Wang, L. The Crosstalk Between Liver Sinusoidal Endothelial Cells and Hepatic Microenvironment in NASH Related Liver Fibrosis. Front. Immunol. 2022, 13, 936196. [Google Scholar] [CrossRef]
- Poisson, J.; Lemoinne, S.; Boulanger, C.; Durand, F.; Moreau, R.; Valla, D.; Rautou, P.-E. Liver Sinusoidal Endothelial Cells: Physiology and Role in Liver Diseases. J. Hepatol. 2017, 66, 212–227. [Google Scholar] [CrossRef] [Green Version]
- Peralta, C.; Jiménez-Castro, M.B.; Gracia-Sancho, J. Hepatic Ischemia and Reperfusion Injury: Effects on the Liver Sinusoidal Milieu. J. Hepatol. 2013, 59, 1094–1106. [Google Scholar] [CrossRef] [Green Version]
- Maslak, E.; Gregorius, A.; Chlopicki, S. Liver Sinusoidal Endothelial Cells (LSECs) Function and NAFLD; NO-Based Therapy Targeted to the Liver. Pharmacol. Rep. 2015, 67, 689–694. [Google Scholar] [CrossRef]
- Limmer, A.; Ohl, J.; Kurts, C.; Ljunggren, H.-G.; Reiss, Y.; Groettrup, M.; Momburg, F.; Arnold, B.; Knolle, P.A. Efficient Presentation of Exogenous Antigen by Liver Endothelial Cells to CD8+ T Cells Results in Antigen-Specific T-Cell Tolerance. Nat. Med. 2000, 6, 1348–1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shetty, S.; Weston, C.J.; Oo, Y.H.; Westerlund, N.; Stamataki, Z.; Youster, J.; Hubscher, S.G.; Salmi, M.; Jalkanen, S.; Lalor, P.F.; et al. Common Lymphatic Endothelial and Vascular Endothelial Receptor-1 Mediates the Transmigration of Regulatory T Cells across Human Hepatic Sinusoidal Endothelium. J. Immunol. 2011, 186, 4147–4155. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.-L. Fibrinogen-like Protein 2 Fibroleukin Expression and Its Correlation with Disease Progression in Murine Hepatitis Virus Type 3-Induced Fulminant Hepatitis and in Patients with Severe Viral Hepatitis B. World J. Gastroenterol. 2005, 11, 6936. [Google Scholar] [CrossRef]
- Zhu, C.; Sun, Y.; Luo, X.; Yan, W.; Xi, D.; Ning, Q. Novel Mfgl2 Antisense Plasmid Inhibits Murine Fgl2 Expression and Ameliorates Murine Hepatitis Virus Type 3-Induced Fulminant Hepatitis in BALB/CJ Mice. Hum. Gene Ther. 2006, 17, 589–600. [Google Scholar] [CrossRef]
- Marsden, P.A.; Ning, Q.; Fung, L.S.; Luo, X.; Chen, Y.; Mendicino, M.; Ghanekar, A.; Scott, J.A.; Miller, T.; Chan, C.W.Y.; et al. The Fgl2/Fibroleukin Prothrombinase Contributes to Immunologically Mediated Thrombosis in Experimental and Human Viral Hepatitis. J. Clin. Investig. 2003, 112, 58–66. [Google Scholar] [CrossRef]
- Hilscher, M.B.; Sehrawat, T.; Arab, J.P.; Zeng, Z.; Gao, J.; Liu, M.; Kostallari, E.; Gao, Y.; Simonetto, D.A.; Yaqoob, U.; et al. Mechanical Stretch Increases Expression of CXCL1 in Liver Sinusoidal Endothelial Cells to Recruit Neutrophils, Generate Sinusoidal Microthombi, and Promote Portal Hypertension. Gastroenterology 2019, 157, 193–209.e9. [Google Scholar] [CrossRef] [PubMed]
- van der Windt, D.J.; Sud, V.; Zhang, H.; Varley, P.R.; Goswami, J.; Yazdani, H.O.; Tohme, S.; Loughran, P.; O’Doherty, R.M.; Minervini, M.I.; et al. Neutrophil Extracellular Traps Promote Inflammation and Development of Hepatocellular Carcinoma in Nonalcoholic Steatohepatitis. Hepatology 2018, 68, 1347–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leppkes, M.; Knopf, J.; Naschberger, E.; Lindemann, A.; Singh, J.; Herrmann, I.; Stürzl, M.; Staats, L.; Mahajan, A.; Schauer, C.; et al. Vascular Occlusion by Neutrophil Extracellular Traps in COVID-19. EBioMedicine 2020, 58, 102925. [Google Scholar] [CrossRef]
- Middleton, E.A.; He, X.-Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil Extracellular Traps Contribute to Immunothrombosis in COVID-19 Acute Respiratory Distress Syndrome. Blood 2020, 136, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, D.; Kuriyama, N.; Ito, T.; Fujii, T.; Kato, H.; Mizuno, S.; Sakurai, H.; Isaji, S. Antiapoptotic Effect by PAR-1 Antagonist Protects Mouse Liver Against Ischemia-Reperfusion Injury. J. Surg. Res. 2020, 246, 568–583. [Google Scholar] [CrossRef]
- SenBanerjee, S.; Lin, Z.; Atkins, G.B.; Greif, D.M.; Rao, R.M.; Kumar, A.; Feinberg, M.W.; Chen, Z.; Simon, D.I.; Luscinskas, F.W.; et al. KLF2 Is a Novel Transcriptional Regulator of Endothelial Proinflammatory Activation. J. Exp. Med. 2004, 199, 1305–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nayak, L.; Lin, Z.; Jain, M.K. “Go With the Flow”: How Krüppel-Like Factor 2 Regulates the Vasoprotective Effects of Shear Stress. Antioxid. Redox Signal. 2011, 15, 1449–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Z.; Hamik, A.; Jain, R.; Kumar, A.; Jain, M.K. Kruppel-Like Factor 2 Inhibits Protease Activated Receptor-1 Expression and Thrombin-Mediated Endothelial Activation. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marrone, G.; Russo, L.; Rosado, E.; Hide, D.; García-Cardeña, G.; García-Pagán, J.C.; Bosch, J.; Gracia-Sancho, J. The Transcription Factor KLF2 Mediates Hepatic Endothelial Protection and Paracrine Endothelial–Stellate Cell Deactivation Induced by Statins. J. Hepatol. 2013, 58, 98–103. [Google Scholar] [CrossRef]
- Marrone, G.; Maeso-Díaz, R.; García-Cardena, G.; Abraldes, J.G.; García-Pagán, J.C.; Bosch, J.; Gracia-Sancho, J. KLF2 Exerts Antifibrotic and Vasoprotective Effects in Cirrhotic Rat Livers: Behind the Molecular Mechanisms of Statins. Gut 2015, 64, 1434–1443. [Google Scholar] [CrossRef]
- Marrone, G.; Shah, V.H.; Gracia-Sancho, J. Sinusoidal Communication in Liver Fibrosis and Regeneration. J. Hepatol. 2016, 65, 608–617. [Google Scholar] [CrossRef] [Green Version]
- Atkins, G.B.; Jain, M.K. Role of Krüppel-Like Transcription Factors in Endothelial Biology. Circ. Res. 2007, 100, 1686–1695. [Google Scholar] [CrossRef]
- Kamel, N.M.; El-Tanbouly, D.M.; Abdallah, D.M.; Sayed, H.M. PAR1, a Therapeutic Target for Remote Lung Injury Associated with Hind Limb Ischemia/Reperfusion: ERK5/KLF2-Dependent Lung Capillary Barrier Preservation. Chem. Biol. Interact. 2022, 354, 109809. [Google Scholar] [CrossRef]
- Chen, T.; Shi, Z.; Zhao, Y.; Meng, X.; Zhao, S.; Zheng, L.; Han, X.; Hu, Z.; Yao, Q.; Lin, H.; et al. LncRNA Airn Maintains LSEC Differentiation to Alleviate Liver Fibrosis via the KLF2-ENOS-SGC Pathway. BMC Med. 2022, 20, 335. [Google Scholar] [CrossRef]
- Sang, Y.; Roest, M.; de Laat, B.; de Groot, P.G.; Huskens, D. Interplay between Platelets and Coagulation. Blood Rev. 2021, 46, 100733. [Google Scholar] [CrossRef]
- Zagai, U.; Fredriksson, K.; Rennard, S.I.; Lundahl, J.; Sköld, C.M. Platelets Stimulate Fibroblast-Mediated Contraction of Collagen Gels. Respir. Res. 2003, 4, 13. [Google Scholar] [CrossRef] [Green Version]
- Kanikarla Marie, P.; Fowlkes, N.W.; Afshar-Kharghan, V.; Martch, S.L.; Sorokin, A.; Shen, J.P.; Morris, V.K.; Dasari, A.; You, N.; Sood, A.K.; et al. The Provocative Roles of Platelets in Liver Disease and Cancer. Front. Oncol. 2021, 11, 643815. [Google Scholar] [CrossRef]
- Morris, S.M.; Chauhan, A. The Role of Platelet Mediated Thromboinflammation in Acute Liver Injury. Front. Immunol. 2022, 13, 1037645. [Google Scholar] [CrossRef] [PubMed]
- Morishima, Y.; Shibutani, T.; Noguchi, K.; Ito, Y.; Honda, Y. Edoxaban, a Direct Oral Factor Xa Inhibitor, Ameliorates Coagulation, Microvascular Thrombus Formation, and Acute Liver Injury in a Lipopolysaccharide-Induced Coagulopathy Model in Rats. J. Thromb. Thrombolysis 2021, 52, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, H.; Yatomi, Y.; Yanase, M.; Satoh, H.; Maekawa, H.; Ogata, I.; Ozaki, Y.; Takuwa, Y.; Mochida, S.; Fujiwara, K. Biological Activities of Novel Lipid Mediator Sphingosine 1-Phosphate in Rat Hepatic Stellate Cells. Am. J. Physiol.-Gastrointest. Liver Physiol. 2000, 279, G304–G310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Jackson, M.L.; Goudswaard, L.J.; Moore, S.F.; Hutchinson, J.L.; Hers, I. Sphingosine-1-Phosphate Modulates PAR1-Mediated Human Platelet Activation in a Concentration-Dependent Biphasic Manner. Sci. Rep. 2021, 11, 15308. [Google Scholar] [CrossRef]
- Zaldivar, M.M.; Pauels, K.; von Hundelshausen, P.; Berres, M.-L.; Schmitz, P.; Bornemann, J.; Kowalska, M.A.; Gassler, N.; Streetz, K.L.; Weiskirchen, R.; et al. CXC Chemokine Ligand 4 (Cxcl4) Is a Platelet-Derived Mediator of Experimental Liver Fibrosis. Hepatology 2010, 51, 1345–1353. [Google Scholar] [CrossRef]
- Yoshida, S.; Ikenaga, N.; Liu, S.B.; Peng, Z.-W.; Chung, J.; Sverdlov, D.Y.; Miyamoto, M.; Kim, Y.O.; Ogawa, S.; Arch, R.H.; et al. Extrahepatic Platelet-Derived Growth Factor-β, Delivered by Platelets, Promotes Activation of Hepatic Stellate Cells and Biliary Fibrosis in Mice. Gastroenterology 2014, 147, 1378–1392. [Google Scholar] [CrossRef]
- Pinzani, M.; Gesualdo, L.; Sabbah, G.M.; Abboud, H.E. Effects of Platelet-Derived Growth Factor and Other Polypeptide Mitogens on DNA Synthesis and Growth of Cultured Rat Liver Fat-Storing Cells. J. Clin. Investig. 1989, 84, 1786–1793. [Google Scholar] [CrossRef] [Green Version]
- Karolczak, K.; Watala, C. Blood Platelets as an Important but Underrated Circulating Source of TGFβ. Int. J. Mol. Sci. 2021, 22, 4492. [Google Scholar] [CrossRef] [PubMed]
- Malehmir, M.; Pfister, D.; Gallage, S.; Szydlowska, M.; Inverso, D.; Kotsiliti, E.; Leone, V.; Peiseler, M.; Surewaard, B.G.J.; Rath, D.; et al. Platelet GPIbα Is a Mediator and Potential Interventional Target for NASH and Subsequent Liver Cancer. Nat. Med. 2019, 25, 641–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, P.; Martin Cramer, E. Platelet α-Granules. Blood Rev. 1993, 7, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Sitia, G.; Aiolfi, R.; Di Lucia, P.; Mainetti, M.; Fiocchi, A.; Mingozzi, F.; Esposito, A.; Ruggeri, Z.M.; Chisari, F.V.; Iannacone, M.; et al. Antiplatelet Therapy Prevents Hepatocellular Carcinoma and Improves Survival in a Mouse Model of Chronic Hepatitis B. Proc. Natl. Acad. Sci. USA 2012, 109, E2165–E2172. [Google Scholar] [CrossRef] [Green Version]
- Thongtan, T.; Deb, A.; Vutthikraivit, W.; Laoveeravat, P.; Mingbunjerdsuk, T.; Islam, S.; Islam, E. Antiplatelet Therapy Associated with Lower Prevalence of Advanced Liver Fibrosis in Non-Alcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Indian J. Gastroenterol. 2022, 41, 119–126. [Google Scholar] [CrossRef]
- Czajka, P.; Przybyłkowski, A.; Nowak, A.; Postula, M.; Wolska, M.; Mirowska-Guzel, D.; Czlonkowska, A.; Eyileten, C. Antiplatelet Drugs and Liver Fibrosis. Platelets 2022, 33, 219–228. [Google Scholar] [CrossRef]
- Murata, S.; Hashimoto, I.; Nakano, Y.; Myronovych, A.; Watanabe, M.; Ohkohchi, N. Single Administration of Thrombopoietin Prevents Progression of Liver Fibrosis and Promotes Liver Regeneration After Partial Hepatectomy in Cirrhotic Rats. Ann. Surg. 2008, 248, 821–828. [Google Scholar] [CrossRef]
- Ikeda, N.; Murata, S.; Maruyama, T.; Tamura, T.; Nozaki, R.; Kawasaki, T.; Fukunaga, K.; Oda, T.; Sasaki, R.; Homma, M.; et al. Platelet-Derived Adenosine 5′-Triphosphate Suppresses Activation of Human Hepatic Stellate Cell: In Vitro Study: Platelets Control Hepatic Stellate Cells. Hepatol. Res. 2012, 42, 91–102. [Google Scholar] [CrossRef] [Green Version]
- Kodama, T.; Takehara, T.; Hikita, H.; Shimizu, S.; Li, W.; Miyagi, T.; Hosui, A.; Tatsumi, T.; Ishida, H.; Tadokoro, S.; et al. Thrombocytopenia Exacerbates Cholestasis-Induced Liver Fibrosis in Mice. Gastroenterology 2010, 138, 2487–2498.e7. [Google Scholar] [CrossRef]
- Jiang, H.; Li, Y.; Sheng, Q.; Dou, X. Relationship between Hepatitis B Virus Infection and Platelet Production and Dysfunction. Platelets 2022, 33, 212–218. [Google Scholar] [CrossRef]
- Joshi, N.; Kopec, A.K.; O’Brien, K.M.; Towery, K.L.; Cline-Fedewa, H.; Williams, K.J.; Copple, B.L.; Flick, M.J.; Luyendyk, J.P. Coagulation-driven Platelet Activation Reduces Cholestatic Liver Injury and Fibrosis in Mice. J. Thromb. Haemost. 2015, 13, 57–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahn, M.L.; Zheng, Y.-W.; Huang, W.; Bigornia, V.; Zeng, D.; Moff, S.; Farese, R.V.; Tam, C.; Coughlin, S.R. A Dual Thrombin Receptor System for Platelet Activation. Nature 1998, 394, 690–694. [Google Scholar] [CrossRef]
- Tripodi, A.; Primignani, M.; Mannucci, P.M.; Caldwell, S.H. Changing concepts of cirrhotic coagulopathy. Am. J. Gastroenterol. 2017, 112, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Tripodi, A.; Primignani, M.; Chantarangkul, V.; Dell’Era, A.; Clerici, M.; de Franchis, R.; Colombo, M.; Mannucci, P.M. An imbalance of pro- vs anti-coagulation factors inplasma from patients with cirrhosis. Gastroenterology 2009, 137, 2105–2111. [Google Scholar] [CrossRef] [PubMed]
- Tripodi, A.; Primignani, M.; Lemma, L.; Chantarangkul, V.; Dell’Era, A.; Iannuzzi, F.; Aghemo, A.; Mannucci, P.M. Detection of the imbalance of procoagulant versus anticoagulant factors in cirrhosis by a simple laboratory method. Hepatology 2010, 52, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Tripodi, A.; Chantarangkul, V.; Primignani, M.; Clerici, M.; Dell’Era, A.; Aghemo, A.; Mannucci, P.M. Thrombin generation in plasma from patients with cirrhosis supplemented with normal plasma: Considerations on the efficacy of treatment with fresh-frozen plasma. Intern. Emerg. Med. 2012, 7, 139–144. [Google Scholar] [CrossRef]
- Tripodi, A.; Salerno, F.; Chantarangkul, V.; Clerici, M.; Cazzaniga, M.; Primignani, M.; Mannuccio Mannucci, P. Evidence of normal thrombin generation in cirrhosis despite abnormal conventional coagulation tests. Hepatology 2005, 41, 553–558. [Google Scholar] [CrossRef]
- Sinegre, T.; Duron, C.; Lecompte, T.; Pereira, B.; Massoulier, S.; Lamblin, G.; Abergel, A.; Lebreton, A. Increased factor VIII plays a significant role in plasma hypercoagulability phenotype of patients with cirrhosis. J. Thromb Haemost. 2018, 16, 1132–1140. [Google Scholar] [CrossRef] [Green Version]
- Zou, Z.; Yan, X.; Li, C.; Li, X.; Ma, X.; Zhang, C.; Ju, S.; Tian, J.; Qi, X. von Willebrand factor as a biomarker of clinically significant portal hypertension and severe portal hypertension: A systematic review and meta-analysis. BMJ Open 2019, 9, e025656. [Google Scholar] [CrossRef] [Green Version]
- Shahani, T.; Covens, K.; Lavend’homme, R.; Jazouli, N.; Sokal, E.; Peerlinck, K.; Jacquemin, M. Human liver sinusoidal endothelial cells but not hepatocytes contain factor VIII. J. Thromb Haemost. 2014, 12, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Tornai, I.; Hársfalvi, J.; Boda, Z.; Udvardy, M.; Pfliegler, G.; Rak, K. Endothelium releases more von Willebrand factor and tissue-type plasminogen activator upon venous occlusion in patients with liver cirrhosis than in normals. Haemostasis 1993, 23, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, A.; Ball, C.; Nolasco, L.; Moake, J.F.; Dong, J.F. Effects of inflammatory cytokines on the release and cleavage of the endothelial cell-derived ultralarge von Willebrand factor multimers under flow. Blood 2004, 104, 100–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Mura, V.; Reverter, J.C.; Flores-Arroyo, A.; Raffa, R.; Reverter, E.; Seijo, S.; Albrades, J.G.; Bosch, J.; Garcia-Pagan, J.C. Von Willebrand factor levels predict clinical outcome in patients with cirrhosis and portal hypertension. Gut 2011, 60, 1133–1138. [Google Scholar] [CrossRef] [PubMed]
- Ferlitsch, M.; Reiberger, T.; Hoke, M.; Salzl, P.; Schwengerer, B.; Ulbrich, G.; Payer, B.A.; Trauner, M.; Peck-Radosavljevic, M.; Ferlitsch, A. von Willebrand factor as new noninvasive predictor of portal hypertension, decompensation and mortality in patients with liver cirrhosis. Hepatology 2012, 56, 1439–1447. [Google Scholar] [CrossRef]
- Maieron, A.; Salzl, P.; Peck-Radosavljevic, M.; Trauner, M.; Hametner, S.; Schöfl, R.; Ferenci, P. Ferlitsch Von Willebrand Factor as a new marker for non-invasive assessment of liver fibrosis and cirrhosis in patients with chronic hepatitis C. Aliment. Pharmacol. Ther. 2014, 39, 331–338. [Google Scholar] [CrossRef]
- Lenting, P.J.; Christophe, O.D.; von Denis, C.V. Willebrand factor biosynthesis, secretion, and clearance: Connecting the far ends. Blood 2015, 125, 2019–2028. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.; Dinh, T.T.; Rajaraman, A.; Lee, M.; Scholz, A.; Czupalla, C.J.; Kiefel, H.; Zhu, L.; Xia, L.; Morser, J.; et al. Patterns of expression of factor VIII and von Willebrand factor by endothelial cell subsets in vivo. Blood 2016, 128, 104–109. [Google Scholar] [CrossRef]
- Zheng, X.; Chung, D.; Takayama, T.K.; Majerus, E.M.; Sadler, J.E.; Fujikawa, K. Structure of von Willebrand factor-cleaving protease (ADAMTS13), a metalloprotease involved in thrombotic thrombocytopenic purpura. J. Biol. Chem. 2001, 276, 41059–41063. [Google Scholar] [CrossRef] [Green Version]
- Soejima, K.; Mimura, N.; Hirashima, M.; Maeda, H.; Hamamoto, T.; Nakagaki, T.; Nozaki, C. A novel human metalloprotease synthesized in the liver and secreted into the blood: Possibly, the von Willebrand factor-cleaving protease? J. Biochem. 2001, 130, 475–480. [Google Scholar] [CrossRef]
- Uemura, M.; Tatsumi, K.; Matsumoto, M.; Fujimoto, M.; Matsuyama, T.; Ishikawa, M.; Iwamoto, T.A.; Mori, T.; Wanaka, A.; Fukui, H.; et al. Localization of ADAMTS13 to the stellate cells of human liver. Blood 2005, 106, 922–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, M.; Murata, M.; Matsubara, Y.; Uchida, T.; Ishihara, H.; Shibano, T.; Ashida, S.; Soejima, K.; Okada, Y.; Ikeda, Y. Detection of von Willebrand factor-cleaving protease (ADAMTS-13) in human platelets. Biochem. Biophys. Res. Commun. 2004, 313, 212–216. [Google Scholar] [CrossRef]
- Zhou, W.; Inada, M.; Lee, T.P.; Benten, D.; Lyubsky, S.; Bouhassira, E.E.; Gupta, S.; Tsai, H.M. ADAMTS13 is expressed in hepatic stellate cells. Lab. Investig. 2005, 85, 780–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uemura, M.; Fujimura, Y.; Matsumoto, M.; Ishizashi, H.; Kato, S.; Matsuyama, T.; Isonishi, A.; Ishikawa, M.; Yagita, M.; Morioka, C.; et al. Comprehensive analysis of ADAMTS13 in patients with liver cirrhosis. Thromb. Haemost. 2008, 99, 1019–1029. [Google Scholar] [PubMed]
- Levy, G.G.; Nichols, W.C.; Lian, E.C.; Foroud, T.; McClintick, J.N.; McGee, B.M.; Yang, A.Y.; Siemieniak, D.R.; Stark, K.R.; Gruppo, R.; et al. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature. 2001, 413, 488–494. [Google Scholar] [CrossRef] [Green Version]
- Kokame, K.; Matsumoto, M.; Soejima, K.; Yagi, H.; Ishizashi, H.; Funato, M.; Tamai, H.; Konno, M.; Kamide, K.; Kawano, Y.; et al. Mutations and common polymorphisms in ADAMTS13 gene responsible for von Willebrand factor-cleaving protease activity. Proc. Natl. Acad. Sci. USA 2002, 99, 11902–11907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feys, H.B.; Canciani, M.T.; Peyvandi, F.; Deckmyn, H.; Vanhoorelbeke, K.; Mannucci, P.M. ADAMTS13 activity to antigen ratio in physiological and pathological conditions associated with an increased risk of thrombosis. Br. J. Haematol. 2007, 138, 534–540. [Google Scholar] [CrossRef]
- Uemura, M.; Matsuyama, T.; Ishikawa, M.; Fujimoto, M.; Kojima, H.; Sakurai, S.; Ishii, S.; Toyohara, M.; Yamazaki, M.; Yoshiji, H.; et al. Decreased activity of plasma ADAMTS13 may contribute to the development of liver disturbance and multiorgan failure in patients with alcoholic hepatitis. Alcohol. Clin. Exp. Res. 2005, 29 (Suppl. 12), 264S–271S. [Google Scholar] [CrossRef]
- Matsuyama, T.; Uemura, M.; Ishikawa, M.; Matsumoto, M.; Ishizashi, H.; Kato, S.; Morioka, C.; Fujimoto, M.; Kojima, H.; Yoshiji, H.; et al. Increased von Willebrand factor over decreased ADAMTS13 activity may contribute to the development of liver disturbance and multiorgan failure in patients with alcoholic hepatitis. Alcohol. Clin. Exp. Res. 2007, 31 (Suppl. 1), S27–S35. [Google Scholar] [CrossRef]
- Wanless, I.R.; Wong, F.; Blendis, L.M.; Greig, P.; Heathcote, E.J.; Levy, G. Hepatic and portal vein thrombosis in cirrhosis: Possible role in development of parenchymal extinction and portal hypertension. Hepatology 1995, 21, 1238–1247. [Google Scholar]
- Moake, J.L. Thrombotic microangiopathies. N. Engl. J. Med. 2002, 347, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, Y.; Matsumoto, M.; Yagi, H.; Yoshioka, A.; Matsui, T.; Titani, K. Von Willebrand factor-cleaving protease and Upshaw-Schulman syndrome. Int. J. Hematol. 2002, 75, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Furlan, M. von Willebrand factor-cleaving protease in thrombotic thrombocytopenic purpura and hemolytic-uremic syndrome. Adv. Nephrol. Necker. Hosp. 2000, 30, 71–81. [Google Scholar]
- Kalambokis, G.N.; Oikonomou, A.; Christou, L.; Kolaitis, N.I.; Tsianos, E.V.; Christodoulou, D.; Baltayiannis, G. von Willebrand factor and procoagulant imbalance predict outcome in patients with cirrhosis and thrombocytopenia. J. Hepatol. 2016, 65, 921–928. [Google Scholar] [CrossRef]
- Scheiner, B.; Balcar, L.; Nussbaumer, R.J.; Weinzierl, J.; Paternostro, R.; Simbrunner, B.; Hartl, L.; Jachs, M.; Bauer, D.; Stättermayer, A.F.; et al. Factor VIII/protein C ratio independently predicts liver-related events but does not indicate a hypercoagulable state in ACLD. J. Hepatol. 2022, 76, 1090–1099. [Google Scholar] [CrossRef]
- Ponziani, F.R.; Santopaolo, F.; Gasbarrini, A.; De Cristofaro, R.; Pompili, M. From coagulation imbalance to prediction of advanced chronic liver disease decompensation: The wind of change? J. Hepatol. 2023, 76, E25–E27. [Google Scholar] [CrossRef]
- Takaya, H.; Namisaki, T.; Asada, S.; Iwai, S.; Kubo, T.; Suzuki, J.; Enomoto, M.; Tsuji, Y.; Fujinaga, Y.; Nishimura, N.; et al. ADAMTS13, VWF, and Endotoxin Are Interrelated and Associated with the Severity of Liver Cirrhosis via Hypercoagulability. J. Clin. Med. 2022, 11, 1835. [Google Scholar] [CrossRef] [PubMed]
- Mandorfer, M.; Schwabl, P.; Paternostro, R.; Pomej, K.; Bauer, D.; Thaler, J.; Ay, C.; Quehenberger, P.; Fritzer-Szekeres, M.; Peck-Radosavljevic, M.; et al. Vienna Hepatic Hemodynamic Lab. Von Willebrand factor indicates bacterial translocation, inflammation, and procoagulant imbalance and predicts complications independently of portal hypertension severity. Aliment. Pharmacol. Ther. 2018, 47, 980–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reuken, P.A.; Kussmann, A.; Kiehntopf, M.; Budde, U.; Stallmach, A.; Claus, R.A.; Bruns, T. Imbalance of von Willebrand factor and its cleaving protease ADAMTS13 during systemic inflammation superimposed on advanced cirrhosis. Liver Int. 2015, 35, 37–45. [Google Scholar] [CrossRef]
- Takaya, H.; Yoshiji, H.; Kawaratani, H.; Sakai, K.; Matsumoto, M.; Fujimura, Y.; Fukui, H. Decreased activity of plasma ADAMTS13 are related to enhanced cytokinemia and endotoxemia in patients with acute liver failure. Biomed. Rep. 2017, 7, 277–285. [Google Scholar] [CrossRef] [Green Version]
- Takaya, H.; Namisaki, T.; Enomoto, M.; Kubo, T.; Tsuji, Y.; Fujinaga, Y.; Nishimura, N.; Kaji, K.; Kawaratani, H.; Moriya, K.; et al. The Ratio of von Willebrand Factor Antigen to ADAMTS13 Activity: Usefulness as a Prognostic Biomarker in Acute-on-Chronic Liver Failure. Biology 2023, 12, 164. [Google Scholar] [CrossRef]
- Driever, E.G.; Stravitz, R.T.; Zhang, J.; Adelmeijer, J.; Durkalski, V.; Lee, W.M.; Lisman, T. VWF/ADAMTS13 Imbalance, But Not Global Coagulation or Fibrinolysis, Is Associated With Outcome and Bleeding in Acute Liver Failure. Hepatology 2021, 73, 1882–1891. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Kamath, P.S.; Reddy, K.R. The Evolving Challenge of Infections in Cirrhosis. N. Engl. J. Med. 2021, 384, 2317–2330. [Google Scholar] [CrossRef] [PubMed]
- Carnevale, R.; Raparelli, V.; Nocella, C.; Bartimoccia, S.; Novo, M.; Severino, A.; De Falco, E.; Cammisotto, V.; Pasquale, C.; Crescioli, C.; et al. Gut-derived endotoxin stimulates factor VIII secretion from endothelial cells. Implic. Hypercoagulability Cirrhosis. J Hepatol. 2017, 67, 950–956. [Google Scholar] [CrossRef]
- Ponziani, F.R.; Sacco, M.; Tardugno, M.; Santopaolo, F.; Marsico, A.; Manna, S.; Lancellotti, S.; Gasbarrini, A.; De Cristofaro, R.; Pompili, M. Low ADAMTS-13/VWF ratio and altered gut-liver axis predict complications of advanced chronic liver disease: A pilot study. Gastroenterol. Rep. 2022, 10, goac065. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, M.; Uemura, M.; Matsuyama, T.; Matsumoto, M.; Ishizashi, H.; Kato, S.; Morioka, C.; Fujimoto, M.; Kojima, H.; Yoshiji, H.; et al. Potential role of enhanced cytokinemia and plasma inhibitor on the decreased activity of plasma ADAMTS13 in patients with alcoholic hepatitis: Relationship to endotoxemia. Alcohol. Clin. Exp. Res. 2010, 34 (Suppl. 1), S25–S33. [Google Scholar] [CrossRef] [PubMed]
- Assy, N.; Hussein, O.; Khalil, A.; Luder, A.; Szvalb, S.; Paizi, M.; Spira, G. The beneficial effect of aspirin and enoxaparin on fibrosis progression and regenerative activity in a rat model of cirrhosis. Dig. Dis. Sci. 2007, 52, 1187–1193. [Google Scholar] [CrossRef]
- Abe, W.; Ikejima, K.; Lang, T.; Okumura, K.; Enomoto, N.; Kitamura, T.; Takei, Y.; Sato, N. Low molecular weight heparin prevents hepatic fibrogenesis caused by carbon tetrachloride in the rat. J. Hepatol. 2007, 46, 286–294. [Google Scholar] [CrossRef]
- Abdel-Salam, O.M.; Baiuomy, A.R.; Ameen, A.; Hassan, N.S. A study of unfractionated and low molecular weight heparins in a model of cholestatic liver injury in the rat. Pharmacol. Res. 2005, 51, 59–67. [Google Scholar] [CrossRef]
- Zhang, R.; Huang, X.; Jiang, Y.; Wang, J.; Chen, S. Effects of Anticoagulants on Experimental Models of Established Chronic Liver Diseases: A Systematic Review and Meta-Analysis. Can. J. Gastroenterol. Hepatol. 2020, 2020, 8887574. [Google Scholar] [CrossRef]
- Villa, E.; Cammà, C.; Marietta, M.; Luongo, M.; Critelli, R.; Colopi, S.; Tata, C.; Zecchini, R.; Gitto, S.; Petta, S.; et al. Enoxaparin prevents portal vein thrombosis and liver decompensation in patients with advanced cirrhosis. Gastroenterology 2012, 143, 1253–1260.e4. [Google Scholar] [CrossRef] [Green Version]
- Bechmann, L.P.; Sichau, M.; Wichert, M.; Gerken, G.; Kröger, K.; Hilgard, P. Low-molecular-weight heparin in patients with advanced cirrhosis. Liver Int. 2011, 31, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Senzolo, M.; Rodriguez-Castro, K.I.; Rossetto, V.; Radu, C.; Gavasso, S.; Carraro, P.; Zerbinati, P.; Sartori, M.T.; Simioni, P. Increased anticoagulant response to low-molecular-weight heparin in plasma from patients with advanced cirrhosis. J. Thromb. Haemost. 2012, 10, 1823–1829. [Google Scholar] [CrossRef]
- Fortea, J.I.; Zipprich, A.; Fernandez-Mena, C.; Puerto, M.; Bosoi, C.R.; Almagro, J.; Hollenbach, M.; Bañares, J.; Rodríguez-Sánchez, B.; Cercenado, E.; et al. Enoxaparin does not ameliorate liver fibrosis or portal hypertension in rats with advanced cirrhosis. Liver Int. 2018, 38, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.C.; Hsu, W.F.; Hsieh, Y.C.; Chan, C.C.; Yang, Y.Y.; Huang, Y.H.; Hou, M.C.; Lin, H.C. Dabigatran Reduces Liver Fibrosis in Thioacetamide-Injured Rats. Dig. Dis. Sci. 2019, 64, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, N.I.; Messiha, B.A.S.; Abo-Saif, A.A.; Abdel-Bakky, M.S. Inhibition of activated factor X; a new pathway in ameliorating carbon tetrachloride-induced liver fibrosis in rats. J. Biochem. Mol. Toxicol. 2019, 33, e22287. [Google Scholar] [CrossRef]
- Mahmoud, N.I.; Messiha, B.A.S.; Salehc, I.G.; Abo-Saif, A.A.; Abdel-Bakky, M.S. Interruption of platelets and thrombin function as a new approach against liver fibrosis induced experimentally in rats. Life Sci. 2019, 231, 116522. [Google Scholar] [CrossRef] [PubMed]
- Li, C.J.; Yang, Z.H.; Shi, X.L.; Liu, D.L. Effects of aspirin and enoxaparin in a rat model of liver fibrosis. World J. Gastroenterol. 2017, 23, 6412–6419. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Nozaki, Y.; Wada, K.; Yoneda, M.; Endo, H.; Takahashi, H.; Iwasaki, T.; Inamori, M.; Abe, Y.; Kobayashi, N.; et al. Effectiveness of antiplatelet drugs against experimental non-alcoholic fatty liver disease. Gut 2008, 57, 1583–1591. [Google Scholar] [CrossRef]
- Poujol-Robert, A.; Boëlle, P.Y.; Conti, F.; Durand, F.; Duvoux, C.; Wendum, D.; Paradis, V.; Mackiewicz, V.; Chazouillères, O.; Corpechot, C.; et al. Aspirin may reduce liver fibrosis progression: Evidence from a multicenter retrospective study of recurrent hepatitis C after liver transplantation. Clin. Res. Hepatol. Gastroenterol. 2014, 38, 570–576. [Google Scholar] [CrossRef]
- Simon, T.G.; Duberg, A.S.; Aleman, S.; Chung, R.T.; Chan, A.T.; Ludvigsson, J.F. Association of Aspirin with Hepatocellular Carcinoma and Liver-Related Mortality. N. Engl. J. Med. 2020, 382, 1018–1028. [Google Scholar] [CrossRef]
- Mohanty, A.; Tate, J.P.; Garcia-Tsao, G. Statins Are Associated With a Decreased Risk of Decompensation and Death in Veterans With Hepatitis C-Related Compensated Cirrhosis. Gastroenterology 2016, 150, 430–440.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, T.G.; King, L.Y.; Zheng, H.; Chung, R.T. Statin use is associated with a reduced risk of fibrosis progression in chronic hepatitis C. J. Hepatol. 2015, 62, 18–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bitto, N.; Salerno, F.; Tripodi, A.; La Mura, V. Coagulation and fibrosis: A potential non-negligible target of statins in chronic hepatitis. J. Hepatol. 2015, 63, 277–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraldes, J.G.; Albillos, A.; Bañares, R.; Turnes, J.; González, R.; García-Pagán, J.C.; Bosch, J. Simvastatin lowers portal pressure in patients with cirrhosis and portal hypertension: A randomized controlled trial. Gastroenterology 2009, 136, 1651–1658. [Google Scholar] [CrossRef]
- Trebicka, J.; Hennenberg, M.; Laleman, W.; Shelest, N.; Biecker, E.; Schepke, M.; Nevens, F.; Sauerbruch, T.; Heller, J. Atorvastatin lowers portal pressure in cirrhotic rats by inhibition of RhoA/Rho-kinase and activation of endothelial nitric oxide synthase. Hepatology 2007, 46, 242–253. [Google Scholar] [CrossRef]
- Trebicka, J.; Hennenberg, M.; Odenthal, M.; Shir, K.; Klein, S.; Granzow, M.; Vogt, A.; Dienes, H.P.; Lammert, F.; Reichen, J.; et al. Atorvastatin attenuates hepatic fibrosis in rats after bile duct ligation via decreased turnover of hepatic stellate cells. J. Hepatol. 2010, 53, 702–712. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, D.M.; Vilaseca, M.; Lafoz, E.; Garcia-Calderó, H.; Viegas Haute, G.; Fernández-Iglesias, A.; Rodrigues de Oliveira, J.; García-Pagán, J.C.; Bosch, J.; Gracia-Sancho, J. Simvastatin Prevents Progression of Acute on Chronic Liver Failure in Rats With Cirrhosis and Portal Hypertension. Gastroenterology 2018, 155, 1564–1577. [Google Scholar] [CrossRef] [Green Version]
- La Mura, V.; Gagliano, N.; Arnaboldi, F.; Sartori, P.; Procacci, P.; Denti, L.; Liguori, E.; Bitto, N.; Ristagno, G.; Latini, R.; et al. Simvastatin Prevents Liver Microthrombosis and Sepsis Induced Coagulopathy in a Rat Model of Endotoxemia. Cells 2022, 11, 1148. [Google Scholar] [CrossRef]

| Study | Design | Clinical Setting | Marker | Outcome |
|---|---|---|---|---|
| Kalambosis et al. [124] | Observational | Liver cirrhosis with thrombocytopenia | FVIII/PC and VWF Ag | ↑ liver-related death (VWF Ag cut-off 321% FVIII/PC cut-off 2.36%) ↑ variceal bleeding (VWF Ag cut-off 466% FVIII/PC cut-off 3.29%) ↑ new-onset ascites (VWF Ag cut-off 213% FVIII/PC cut-off 1.99%) ↑ portal vein thrombosis |
| Schneiner et al. [125] | Observational | Liver cirrhosis with portal hypertension | FVIII/PC | ↑ MELD score ↑ HVPG ↑ ACLF (FVIII/PC cut-off > 4.46) |
| Ponziani et al. [126] | Observational | Liver cirrhosis | ↓ ADAMTS-13/VWF and ↑ FVIII/PC | ↑ decompensation rate and risk of liver-related death (FVIII/PC cut-off > 2.6 ADAMTS-13/VWF cut-off < 0.26) |
| Matsuyama et al. [119] | Observational | Alcoholic hepatitis | ↓ ADAMTS-13 activity and ↑ VWF Ag | ↑ risk of severe alcoholic hepatitis ↓ survival |
| Takaya et al. [127] | Observational | ACLF | ↓ VWF Ag, ↑ ADAMTS-13 activity | ↑ survival (VWF/ADAMTS-13 cut-off < 9) |
| Driever et al. [132] | Observational | Drug-induced acute liver injury | ↑ VWF Ag, ↓ ADAMTS-13 activity | ↑ hepatic encephalopathy ↑ bleeding complications ↑ acute kidney injury predictors of the need for liver transplantation |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Airola, C.; Pallozzi, M.; Cerrito, L.; Santopaolo, F.; Stella, L.; Gasbarrini, A.; Ponziani, F.R. Microvascular Thrombosis and Liver Fibrosis Progression: Mechanisms and Clinical Applications. Cells 2023, 12, 1712. https://doi.org/10.3390/cells12131712
Airola C, Pallozzi M, Cerrito L, Santopaolo F, Stella L, Gasbarrini A, Ponziani FR. Microvascular Thrombosis and Liver Fibrosis Progression: Mechanisms and Clinical Applications. Cells. 2023; 12(13):1712. https://doi.org/10.3390/cells12131712
Chicago/Turabian StyleAirola, Carlo, Maria Pallozzi, Lucia Cerrito, Francesco Santopaolo, Leonardo Stella, Antonio Gasbarrini, and Francesca Romana Ponziani. 2023. "Microvascular Thrombosis and Liver Fibrosis Progression: Mechanisms and Clinical Applications" Cells 12, no. 13: 1712. https://doi.org/10.3390/cells12131712