
Circadian Disruption Primes Myofibroblasts for Accelerated Activation as a Mechanism Underpinning Fibrotic Progression in Non-Alcoholic Fatty Liver Disease
 , , , , , , ,
on behalf of the ID-LIVER Consortium
, , , , , , ,
on behalf of the ID-LIVER Consortium
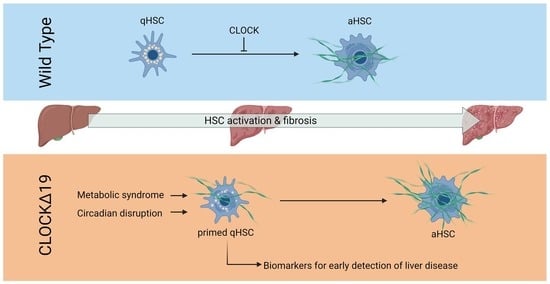
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Mice and In Vivo Fibrosis
2.2. Hepatic Stellate Cell Isolation and In Vitro Culture
2.3. Histology and Western Blot
2.4. Microarray Analysis
2.5. ATAC-Seq Integration
2.6. Human Serum Analysis
2.7. Statistical Analyses
3. Results
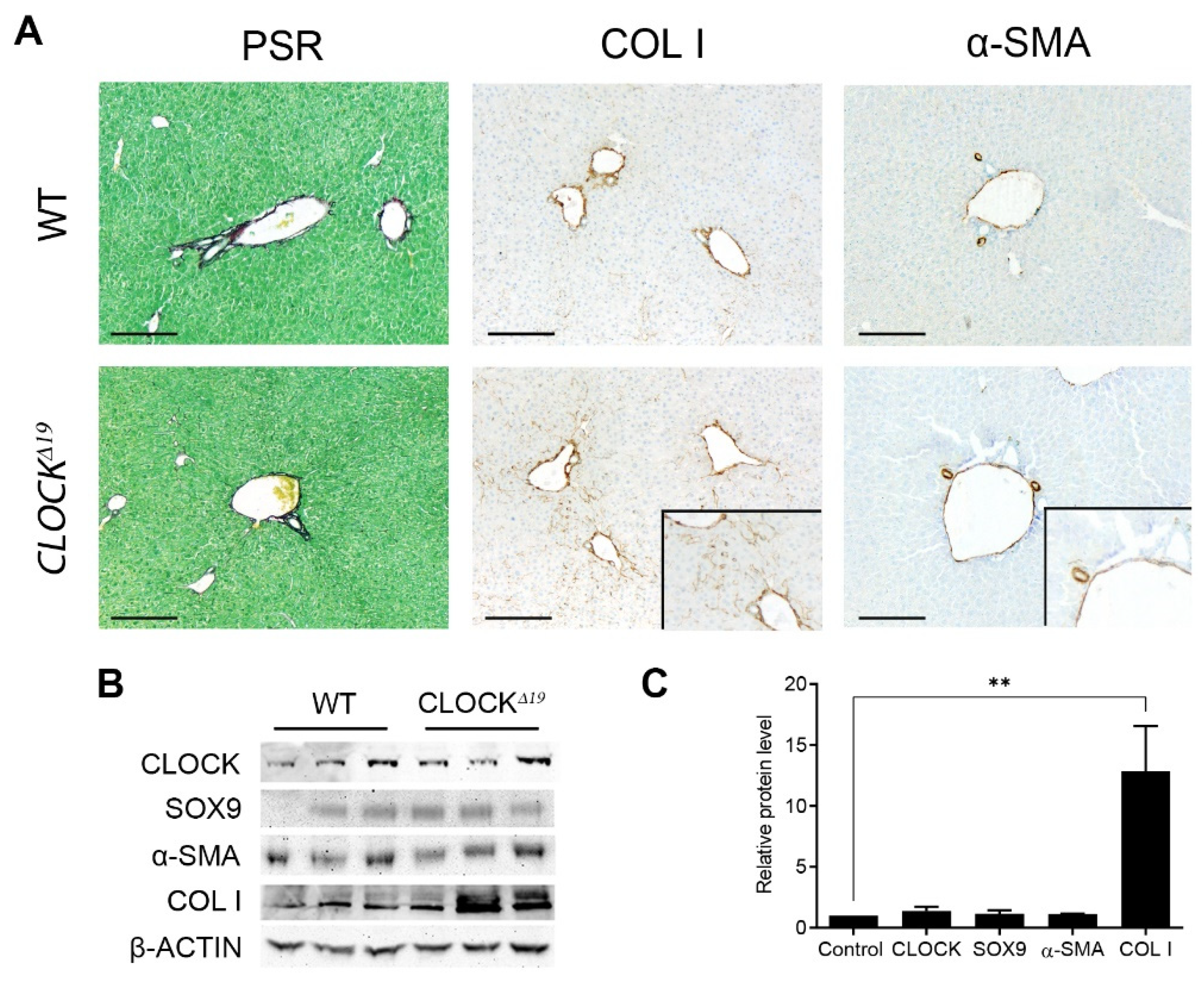
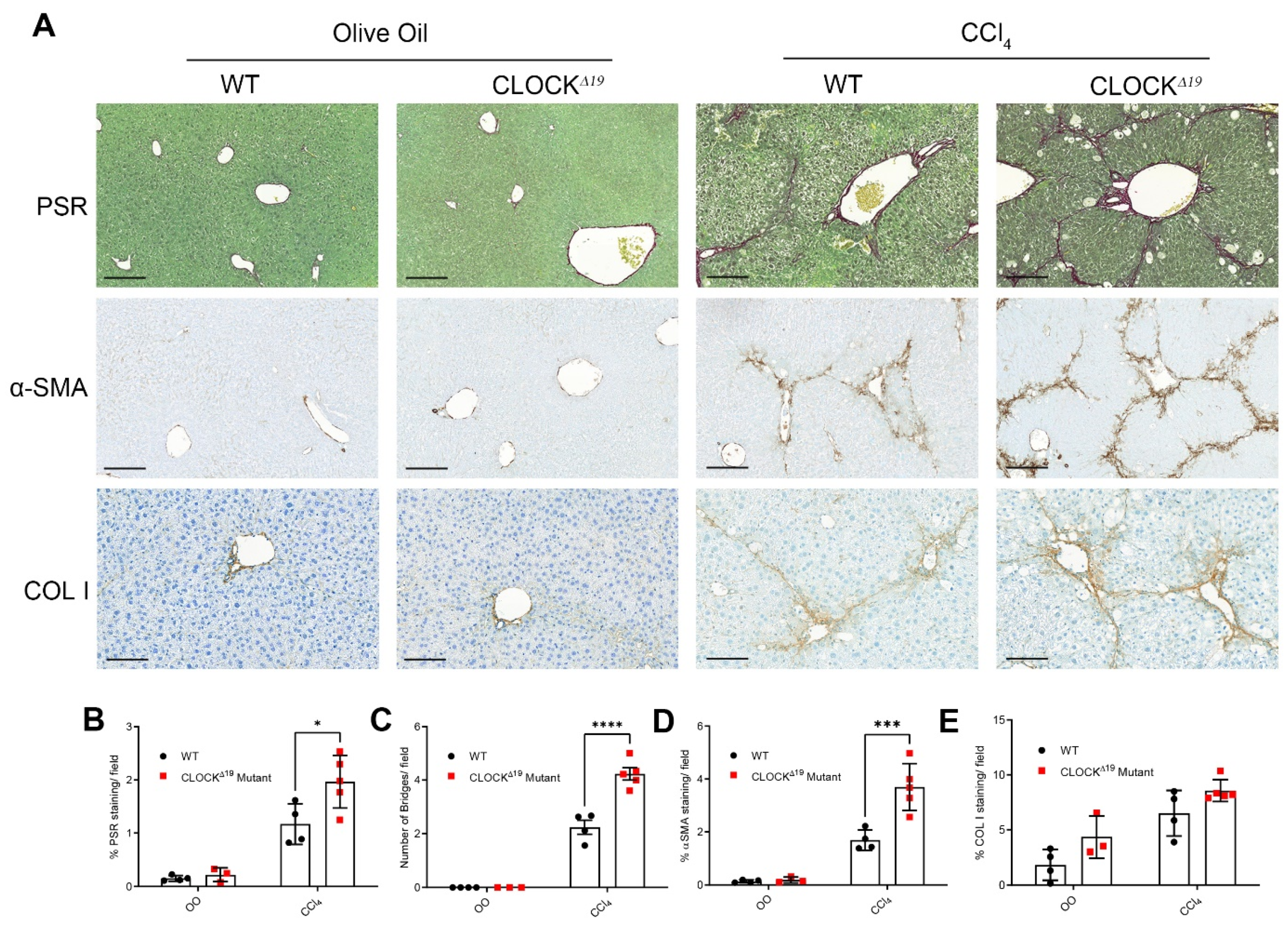
3.1. CLOCKΔ19 Mice Have an Enhanced Fibrotic Response to CCl4 Treatment
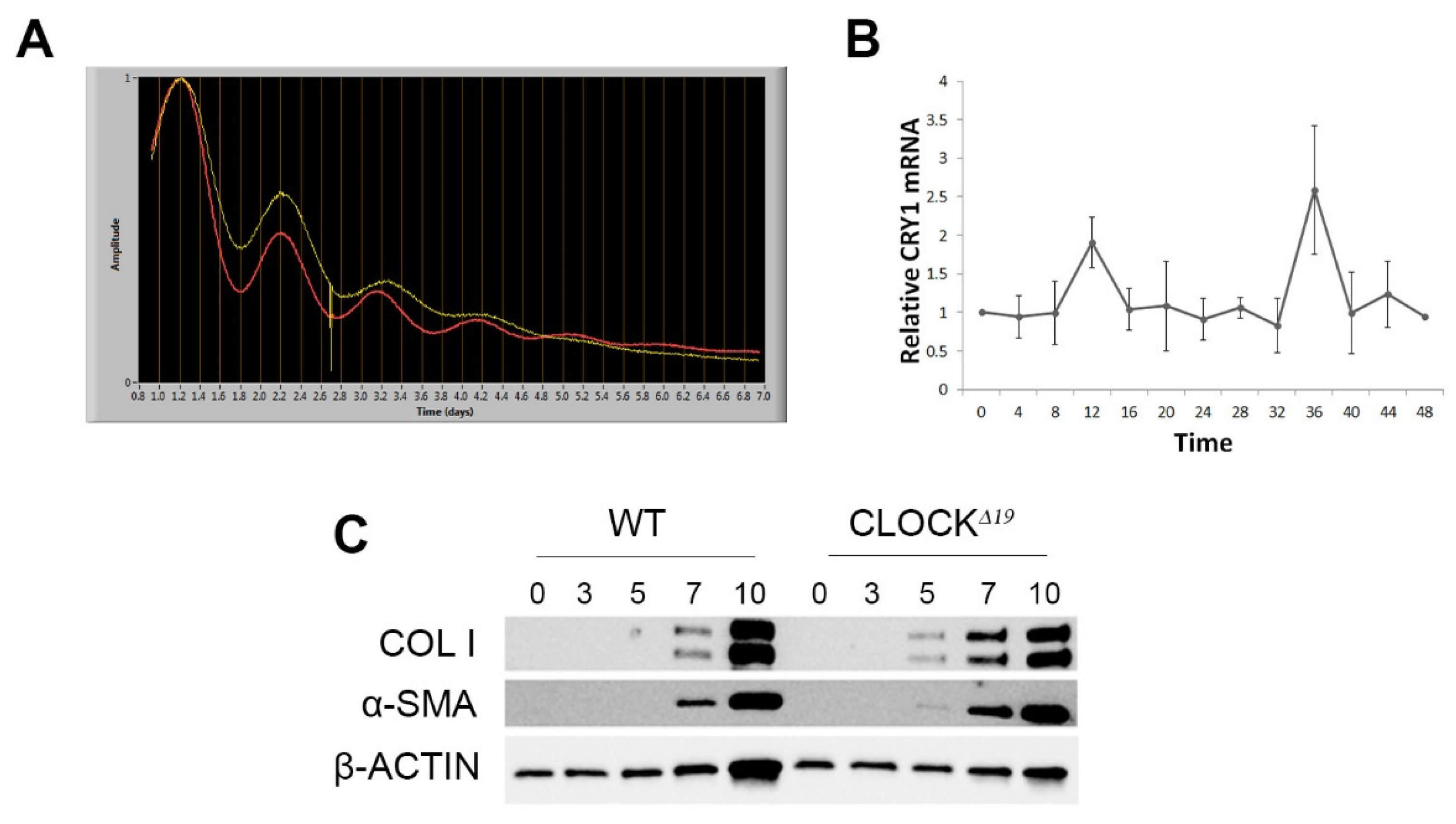
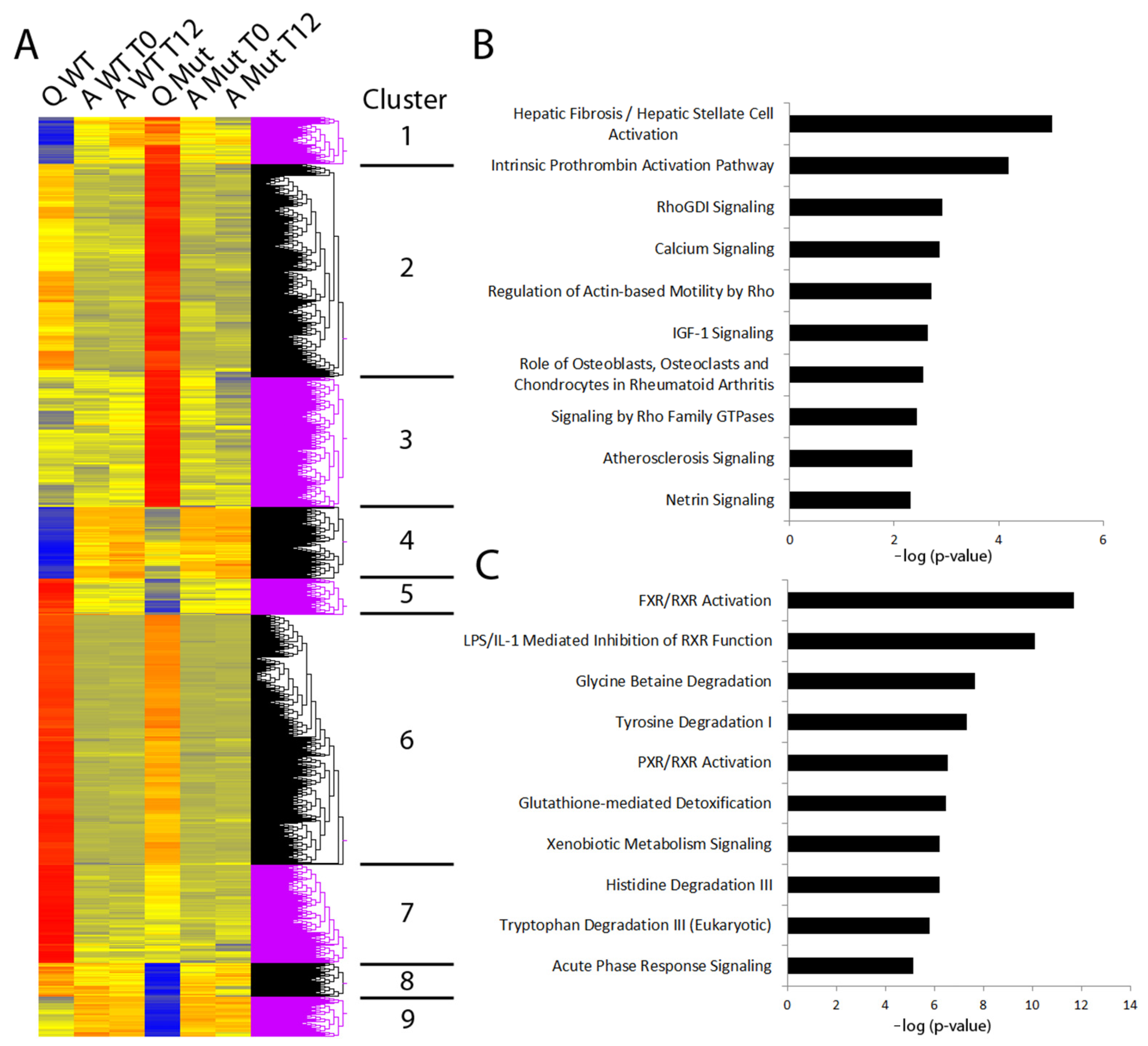
3.2. Disruption to Circadian Rhythm Accelerates HSC Activation
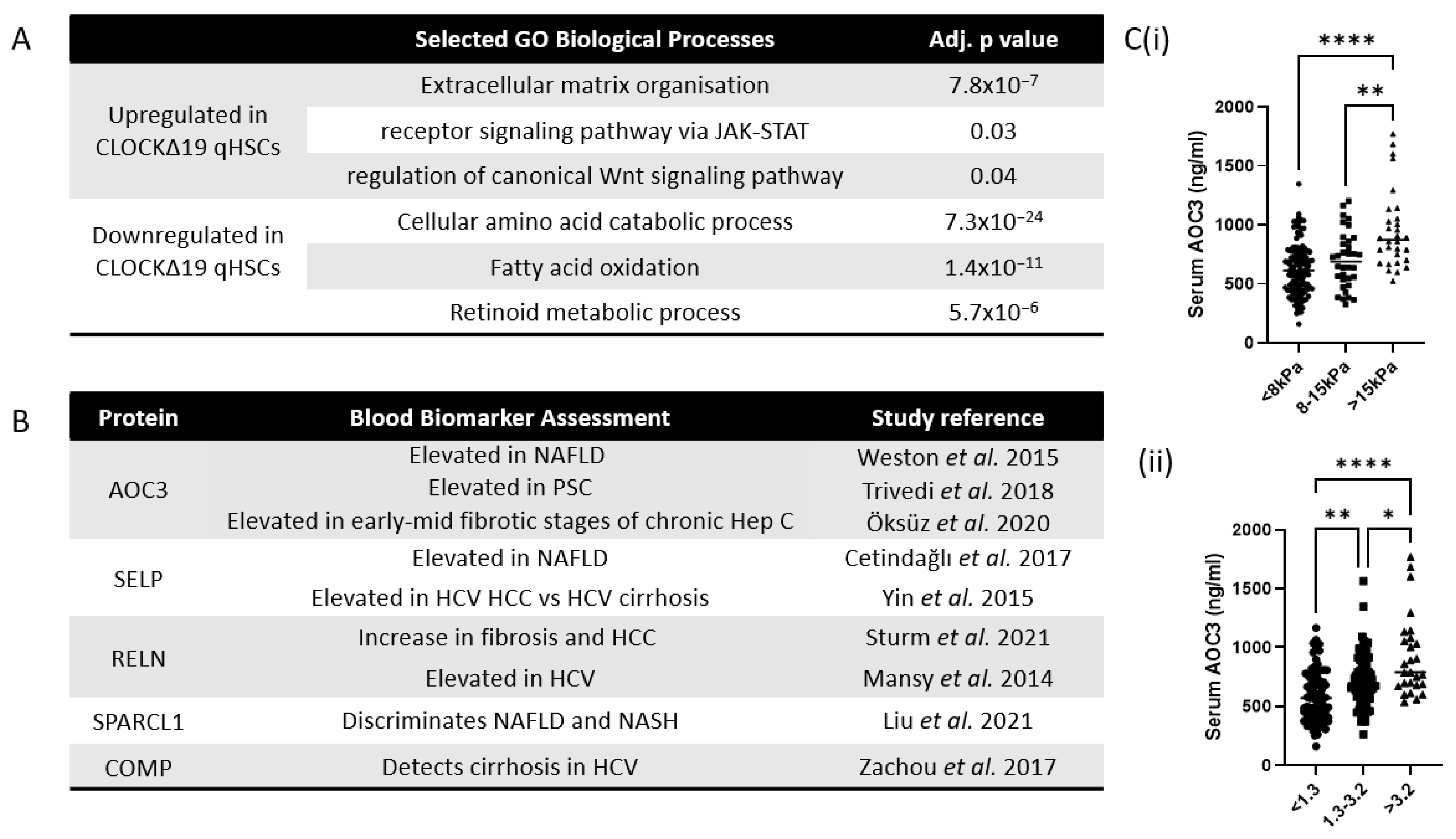
3.3. Factors Priming CLOCKΔ19 HSCs Include Liver Disease Biomarkers
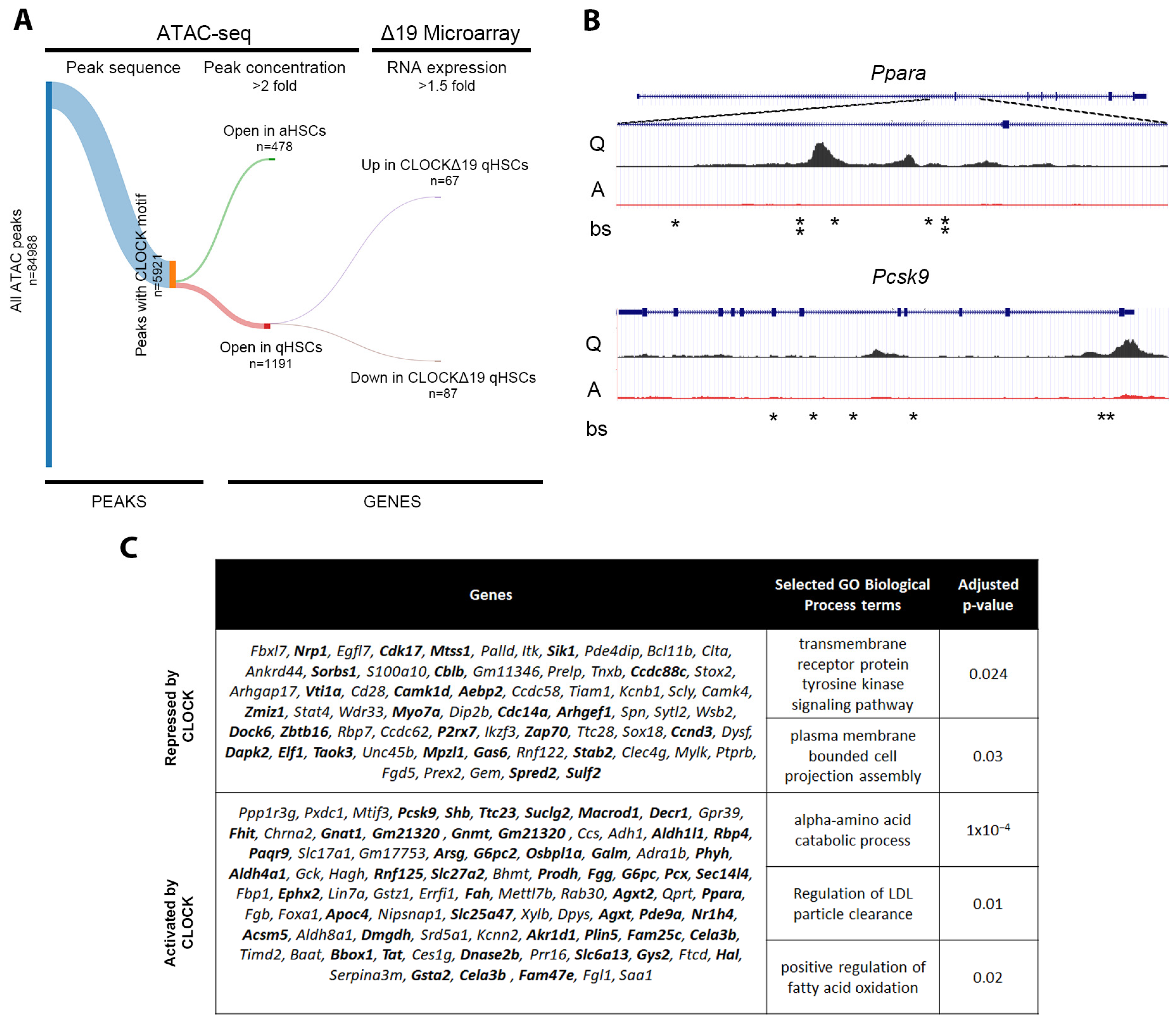
3.4. In Silico Analysis Predicts Direct Regulation of Quiescence Associated Genes by CLOCK
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Younossi, Z.; Henry, L. Contribution of Alcoholic and Nonalcoholic Fatty Liver Disease to the Burden of Liver-Related Morbidity and Mortality. Gastroenterology 2016, 150, 1778–1785. [Google Scholar] [CrossRef] [PubMed]
- Carr, R.M.; Oranu, A.; Khungar, V. Nonalcoholic Fatty Liver Disease: Pathophysiology and Management. Gastroenterol. Clin. N. Am. 2016, 45, 639–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherji, A.; Dachraoui, M.; Baumert, T.F. Perturbation of the circadian clock and pathogenesis of NAFLD. Metabolism 2020, 111, 154337. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Chen, J.; Wang, J.; Yao, J.; Huang, Y.; Zhang, G.; Bao, Z. Circadian Clock Genes in the Metabolism of Non-alcoholic Fatty Liver Disease. Front. Physiol. 2019, 10, 423. [Google Scholar] [CrossRef] [Green Version]
- Sookoian, S. Common genetic variations inCLOCKtranscription factor are associated with nonalcoholic fatty liver disease. World J. Gastroenterol. 2007, 13, 4242. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, Y.; Wang, Z.; Wang, H.; Xue, C.; Li, Q.; Guan, W.; Yuan, J. Rotating night shift work and non-alcoholic fatty liver disease among steelworkers in China: A cross-sectional survey. Occup. Environ. Med. 2020, 77, 333–339. [Google Scholar] [CrossRef]
- Naylor, E.; Bergmann, B.M.; Krauski, K.; Zee, P.C.; Takahashi, J.S.; Vitaterna, M.H.; Turek, F.W. The CircadianClockMutation Alters Sleep Homeostasis in the Mouse. J. Neurosci. 2000, 20, 8138–8143. [Google Scholar] [CrossRef] [Green Version]
- Turek, F.W. Obesity and Metabolic Syndrome in Circadian Clock Mutant Mice. Science 2005, 308, 1043–1045. [Google Scholar] [CrossRef] [Green Version]
- Vitaterna, M.; King, D.; Chang, A.; Kornhauser, J.; Lowrey, P.; McDonald, J.; Dove, W.; Pinto, L.; Turek, F.; Takahashi, J. Mutagenesis and mapping of a mouse gene, Clock, essential for circadian behavior. Science 1994, 264, 719–725. [Google Scholar] [CrossRef] [Green Version]
- Friedman, S.L. Mechanisms of Hepatic Fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar] [CrossRef] [Green Version]
- Akhtar, R.A.; Reddy, A.B.; Maywood, E.S.; Clayton, J.D.; King, V.M.; Smith, A.G.; Gant, T.W.; Hastings, M.H.; Kyriacou, C.P. Circadian Cycling of the Mouse Liver Transcriptome, as Revealed by cDNA Microarray, Is Driven by the Suprachiasmatic Nucleus. Curr. Biol. 2002, 12, 540–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, B.H.; McDearmon, E.L.; Panda, S.; Hayes, K.R.; Zhang, J.; Andrews, J.L.; Antoch, M.P.; Walker, J.R.; Esser, K.A.; Hogenesch, J.B.; et al. Circadian and CLOCK-controlled regulation of the mouse transcriptome and cell proliferation. Proc. Natl. Acad. Sci. USA 2007, 104, 3342–3347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storch, K.-F.; Lipan, O.; Leykin, I.; Viswanathan, N.; Davis, F.C.; Wong, W.H.; Weitz, C.J. Extensive and divergent circadian gene expression in liver and heart. Nature 2002, 417, 78–83. [Google Scholar] [CrossRef]
- Gekakis, N. Role of the CLOCK Protein in the Mammalian Circadian Mechanism. Science 1998, 280, 1564–1569. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.-N.; Malinin, N.; Yang, F.-C.; Staknis, D.; Gekakis, N.; Maier, B.; Reischl, S.; Kramer, A.; Weitz, C.J. CIPC is a mammalian circadian clock protein without invertebrate homologues. Nat. Cell Biol. 2007, 9, 268–275. [Google Scholar] [CrossRef]
- Mohawk, J.A.; Green, C.B.; Takahashi, J.S. Central and Peripheral Circadian Clocks in Mammals. Annu. Rev. Neurosci. 2012, 35, 445–462. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Yang, T.-Y.; Zhou, Y.-W.; Wu, M.-F.; Shen, J.; Cheng, J.-L.; Liu, Q.-X.; Cao, S.-Y.; Wang, J.-Q.; Zhang, L. Bmal1 inhibits phenotypic transformation of hepatic stellate cells in liver fibrosis via IDH1/α-KG-mediated glycolysis. Acta Pharmacol. Sin. 2022, 43, 316–329. [Google Scholar] [CrossRef]
- Pekovic-Vaughan, V.; Gibbs, J.; Yoshitane, H.; Yang, N.; Pathiranage, D.; Guo, B.; Sagami, A.; Taguchi, K.; Bechtold, D.; Loudon, A.; et al. The circadian clock regulates rhythmic activation of the NRF2/glutathione-mediated antioxidant defense pathway to modulate pulmonary fibrosis. Genes Dev. 2014, 28, 548–560. [Google Scholar] [CrossRef] [Green Version]
- Friedman, S.L.; Roll, F.J. Isolation and culture of hepatic lipocytes, Kupffer cells, and sinusoidal endothelial cells by density gradient centrifugation with Stractan. Anal Biochem. 1987, 161, 207–218. [Google Scholar] [CrossRef]
- Martin, K.; Pritchett, J.; Llewellyn, J.; Mullan, A.F.; Athwal, V.S.; Dobie, R.; Harvey, E.; Zeef, L.; Farrow, S.; Streuli, C.; et al. PAK proteins and YAP-1 signalling downstream of integrin beta-1 in myofibroblasts promote liver fibrosis. Nat. Commun. 2016, 7, 12502. [Google Scholar] [CrossRef] [Green Version]
- Yoo, S.H.; Yamazaki, S.; Lowrey, P.L.; Shimomura, K.; Ko, C.H.; Buhr, E.D.; Siepka, S.M.; Hong, H.K.; Oh, W.J.; Yoo, O.J.; et al. PERIOD2::LUCIFERASE real-time reporting of circadian dynamics reveals persistent circadian oscillations in mouse peripheral tissues. Proc. Natl. Acad. Sci. USA 2004, 101, 5339–5346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jokl, E.; Mullan, A.; Simpson, K.; Birchall, L.; Pearmain, L.; Martin, K.; Pritchett, J.; Shah, R.; Hodson, N.; Williams, C.; et al. PAK1-dependant mechanotransduction enables myofibroblast nuclear adaptation and chromatin organisation during fibrosis. bioRxiv 2023, in press. [CrossRef]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef] [PubMed]
- Sterling, R.K.; Lissen, E.; Clumeck, N.; Sola, R.; Correa, M.C.; Montaner, J.; Sulkowski, M.S.; Torriani, F.J.; Dieterich, D.T.; Thomas, D.L.; et al. Development of a simple noninvasive index to predict significant fibrosis in patients with HIV/HCV coinfection. Hepatology 2006, 43, 1317–1325. [Google Scholar] [CrossRef] [PubMed]
- Angulo, P.; Hui, J.M.; Marchesini, G.; Bugianesi, E.; George, J.; Farrell, G.C.; Enders, F.; Saksena, S.; Burt, A.D.; Bida, J.P.; et al. The NAFLD fibrosis score: A noninvasive system that identifies liver fibrosis in patients with NAFLD. Hepatology 2007, 45, 846–854. [Google Scholar] [CrossRef]
- Newsome, P.N.; Cramb, R.; Davison, S.M.; Dillon, J.F.; Foulerton, M.; Godfrey, E.M.; Hall, R.; Harrower, U.; Hudson, M.; Langford, A.; et al. Guidelines on the management of abnormal liver blood tests. Gut 2018, 67, 6–19. [Google Scholar] [CrossRef] [Green Version]
- Trivedi, P.J.; Tickle, J.; Vesterhus, M.N.; Eddowes, P.J.; Bruns, T.; Vainio, J.; Parker, R.; Smith, D.; Liaskou, E.; Thorbjørnsen, L.W.; et al. Vascular adhesion protein-1 is elevated in primary sclerosing cholangitis, is predictive of clinical outcome and facilitates recruitment of gut-tropic lymphocytes to liver in a substrate-dependent manner. Gut 2018, 67, 1135–1145. [Google Scholar] [CrossRef] [Green Version]
- Weston, C.J.; Shepherd, E.L.; Claridge, L.C.; Rantakari, P.; Curbishley, S.M.; Tomlinson, J.W.; Hubscher, S.G.; Reynolds, G.M.; Aalto, K.; Anstee, Q.M.; et al. Vascular adhesion protein-1 promotes liver inflammation and drives hepatic fibrosis. J. Clin. Investig. 2015, 125, 501–520. [Google Scholar] [CrossRef] [Green Version]
- Öksüz, Z.; Üçbilek, E.; Serin, M.S.; Yaraş, S.; Temel, G.O.; Sezgin, O. Circulating vascular adhesion protein-1(VAP-1): A possible biomarker for liver fibrosis associated with chronic hepatitis B and C. Braz. J. Microbiol. 2020, 51, 1757–1763. [Google Scholar] [CrossRef]
- Cetindağlı, I.; Kara, M.; Tanoglu, A.; Ozalper, V.; Aribal, S.; Hancerli, Y.; Unal, M.; Ozarı, O.; Hira, S.; Kaplan, M.; et al. Evaluation of endothelial dysfunction in patients with nonalcoholic fatty liver disease: Association of selenoprotein P with carotid intima-media thickness and endothelium-dependent vasodilation. Clin. Res. Hepatol. Gastroenterol. 2017, 41, 516–524. [Google Scholar] [CrossRef]
- Yin, H.; Tan, Z.; Wu, J.; Zhu, J.; Shedden, K.A.; Marrero, J.; Lubman, D.M. Mass-Selected Site-Specific Core-Fucosylation of Serum Proteins in Hepatocellular Carcinoma. J. Proteome Res. 2015, 14, 4876–4884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sturm, L.; Roth, L.; Zoldan, K.; Schultheiss, M.; Boettler, T.; Huber, J.P.; Kaeser, R.; Thimme, R.; Bettinger, D. Blood reelin in the progression of chronic liver disease. Adv. Med. Sci. 2021, 66, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Mansy, S.S.; Nosseir, M.M.; Zoheiry, M.A.; Hassanein, M.H.; Guda, M.F.; Othman, M.M.; AbuTalab, H. Value of reelin for assessing hepatic fibrogenesis in a group of Egyptian HCV infected patients. Clin. Chem. Lab. Med. 2014, 52, 1319–1328. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Xiang, L.; Ji, J.; Liu, W.; Chen, Y.; Xia, M.; Liu, Y.; Liu, W.; Zhu, P.; Jin, Y.; et al. Sparcl1 promotes nonalcoholic steatohepatitis progression in mice through upregulation of CCL2. J. Clin. Investig. 2021, 131, e144801. [Google Scholar] [CrossRef]
- Zachou, K.; Gabeta, S.; Shums, Z.; Gatselis, N.K.; Koukoulis, G.K.; Norman, G.L.; Dalekos, G.N. COMP serum levels: A new non-invasive biomarker of liver fibrosis in patients with chronic viral hepatitis. Eur. J. Intern. Med. 2017, 38, 83–88. [Google Scholar] [CrossRef]
- Yoshitane, H.; Ozaki, H.; Terajima, H.; Du, N.-H.; Suzuki, Y.; Fujimori, T.; Kosaka, N.; Shimba, S.; Sugano, S.; Takagi, T.; et al. CLOCK-Controlled Polyphonic Regulation of Circadian Rhythms through Canonical and Noncanonical E-Boxes. Mol. Cell. Biol. 2014, 34, 1776–1787. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Kakan, X.; Wang, S.; Dong, W.; Jia, A.; Cai, C.; Zhang, J. Deletion of clock gene Per2 exacerbates cholestatic liver injury and fibrosis in mice. Exp. Toxicol. Pathol. 2013, 65, 427–432. [Google Scholar] [CrossRef]
- Ota, T.; Maeda, M.; Okamoto, M.; Tatsuka, M. Positive regulation of Rho GTPase activity by RhoGDIs as a result of their direct interaction with GAPs. BMC Syst. Biol. 2015, 9, 3. [Google Scholar] [CrossRef] [Green Version]
- Golding, A.E.; Visco, I.; Bieling, P.; Bement, W.M. Extraction of active RhoGTPases by RhoGDI regulates spatiotemporal patterning of RhoGTPases. eLife 2019, 8, e50471. [Google Scholar] [CrossRef]
- Cho, H.J.; Kim, J.T.; Baek, K.E.; Kim, B.Y.; Lee, H.G. Regulation of Rho GTPases by RhoGDIs in Human Cancers. Cells 2019, 8, 1037. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, S.J.; Bernier, M.; Mattison, J.A.; Aon, M.A.; Kaiser, T.A.; Anson, R.M.; Ikeno, Y.; Anderson, R.M.; Ingram, D.K.; de Cabo, R. Daily Fasting Improves Health and Survival in Male Mice Independent of Diet Composition and Calories. Cell Metab. 2019, 29, 221–228.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jokl, E.; Llewellyn, J.; Simpson, K.; Adegboye, O.; Pritchett, J.; Zeef, L.; Donaldson, I.; Athwal, V.S.; Purssell, H.; Street, O.; et al. Circadian Disruption Primes Myofibroblasts for Accelerated Activation as a Mechanism Underpinning Fibrotic Progression in Non-Alcoholic Fatty Liver Disease. Cells 2023, 12, 1582. https://doi.org/10.3390/cells12121582
Jokl E, Llewellyn J, Simpson K, Adegboye O, Pritchett J, Zeef L, Donaldson I, Athwal VS, Purssell H, Street O, et al. Circadian Disruption Primes Myofibroblasts for Accelerated Activation as a Mechanism Underpinning Fibrotic Progression in Non-Alcoholic Fatty Liver Disease. Cells. 2023; 12(12):1582. https://doi.org/10.3390/cells12121582
Chicago/Turabian StyleJokl, Elliot, Jessica Llewellyn, Kara Simpson, Oluwatobi Adegboye, James Pritchett, Leo Zeef, Ian Donaldson, Varinder S. Athwal, Huw Purssell, Oliver Street, and et al. 2023. "Circadian Disruption Primes Myofibroblasts for Accelerated Activation as a Mechanism Underpinning Fibrotic Progression in Non-Alcoholic Fatty Liver Disease" Cells 12, no. 12: 1582. https://doi.org/10.3390/cells12121582