
HIV–Host Cell Interactions

 , , ,
, , , 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Structure of HIV
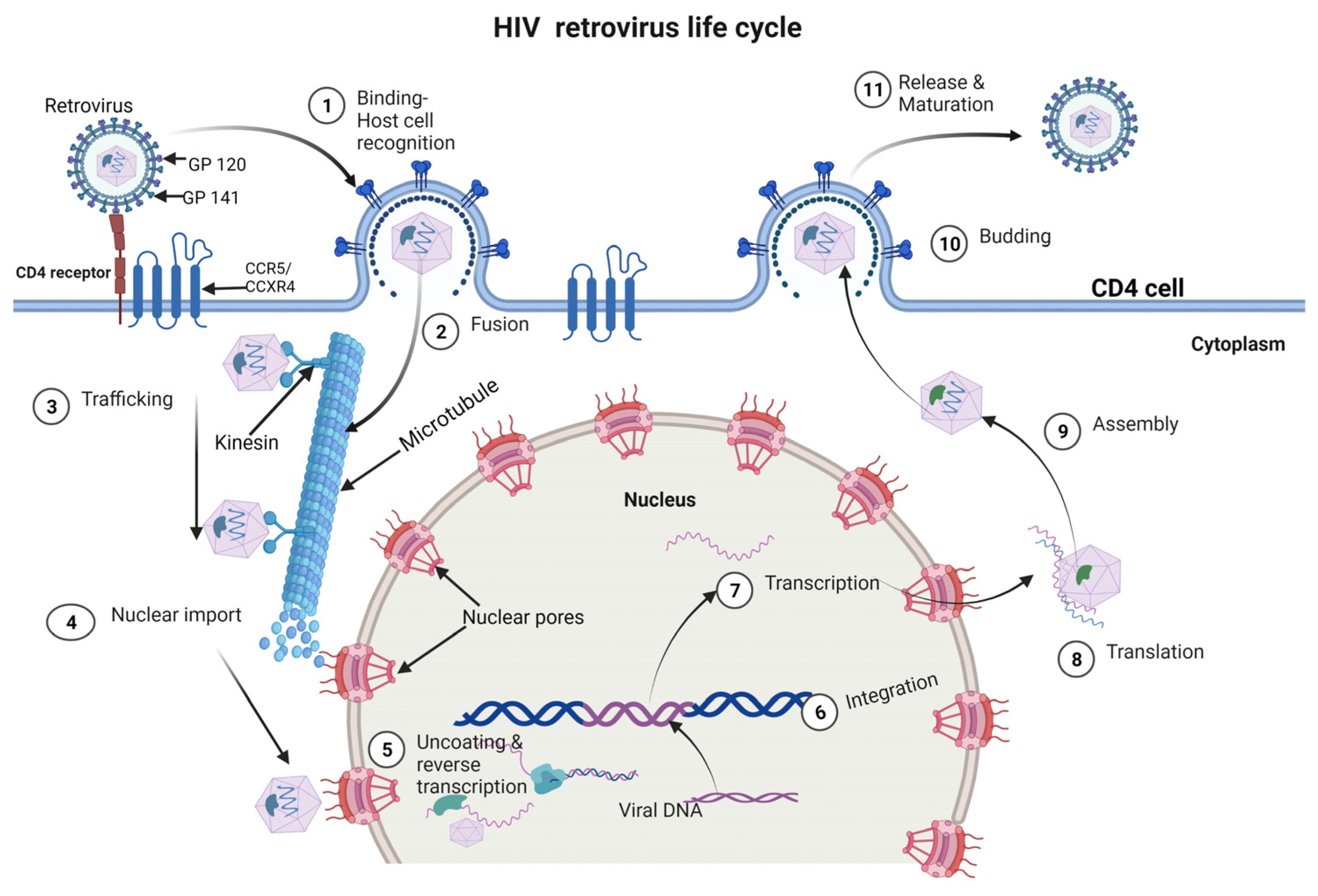
3. HIV Life Cycle
HIV Receptors, Fusion and Uncoating Mechanism
4. HIV-Related Factors Promoting Infection and Immune Evasion
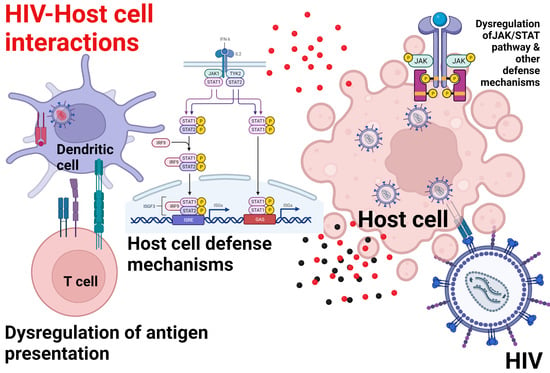
4.1. Downregulation of MHC Class I and II
4.2. Production of Non-Neutralizing Antibodies
4.3. Induction of Immune Exhaustion
4.4. Destruction of Virus-Specific T Helper Cells
4.5. The Emergence of Antigenic Escape Variants
4.6. Expression of an Envelope Complex That Minimizes Antibody Access
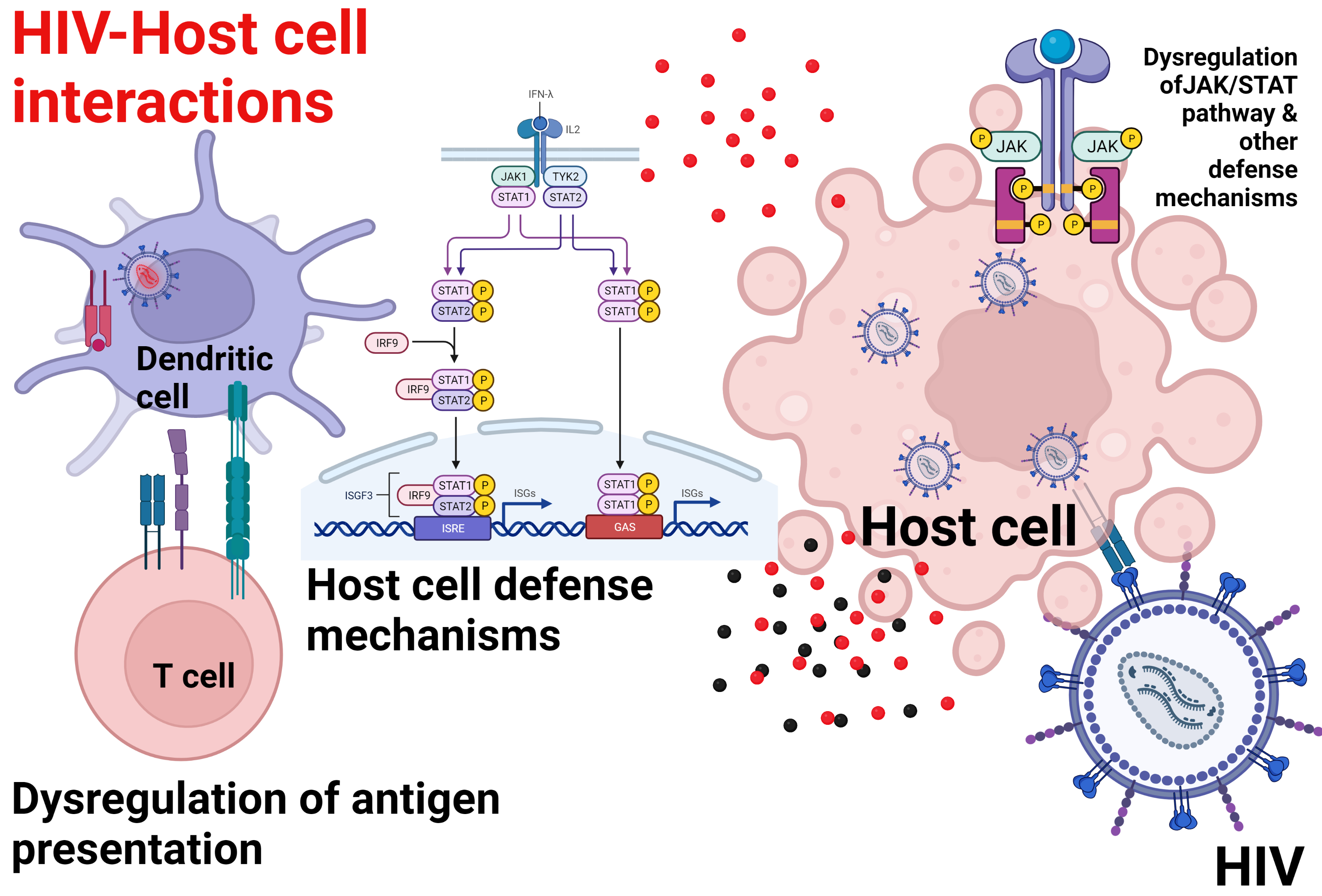
4.7. Dysregulation of the JAK/STAT Pathway
4.8. Other Factors That Promote HIV Infection
5. Host Cell Mechanisms That Control Infection and Replication
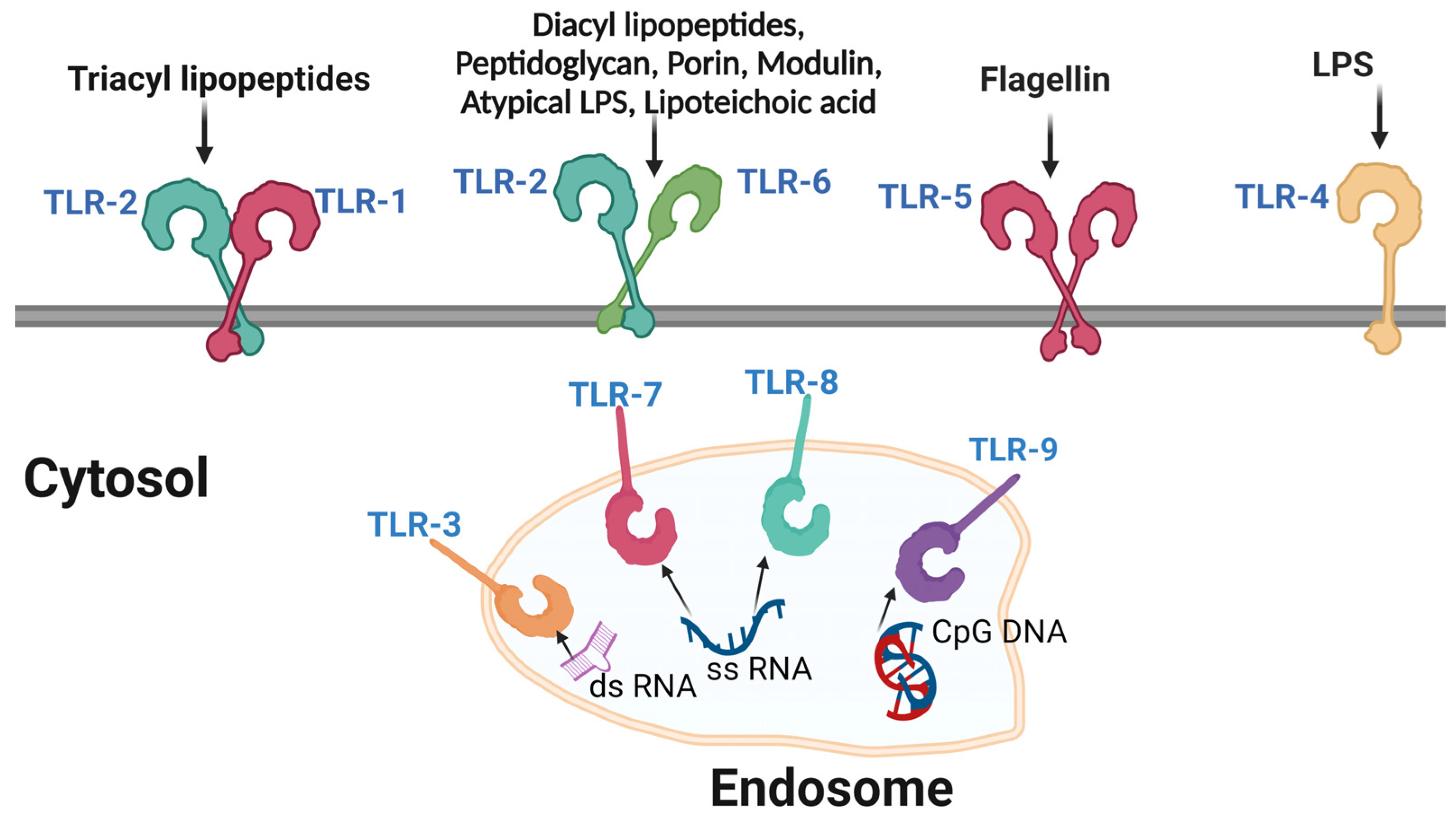
5.1. Pathogen Recognition Receptors (PRRs)
5.2. Dendritic Cells
5.2.1. Plasmacytoid Dendritic Cells (pDCs)
5.2.2. Conventional Dendritic Cells (cDCs)
5.3. Macrophages
5.4. CD4+ T Cells
5.5. CD8+ T Cells
6. Influence of Sex on HIV Transmission and Immune Responses
7. Strengths and Limitations
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- German Advisory Committee Blood. Human Immunodeficiency Virus (HIV). Transfus. Med. Hemotherapy 2016, 43, 203–222. [Google Scholar] [CrossRef]
- Moss, J.A. HIV/AIDS Review. Radiol. Technol. 2013, 84, 247–267. [Google Scholar] [PubMed]
- Faria, N.R.; Rambaut, A.; Suchard, M.A.; Baele, G.; Bedford, T.; Ward, M.J.; Tatem, A.J.; Sousa, J.D.; Arinaminpathy, N.; Pépin, J.; et al. HIV Epidemiology. The Early Spread and Epidemic Ignition of HIV-1 in Human Populations. Science 2014, 346, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Rife, B.; Salemi, M. On the Early Dynamics and Spread of HIV-1. Trends Microbiol. 2015, 23, 3–4. [Google Scholar] [CrossRef]
- Visseaux, B.; Le Hingrat, Q.; Damond, F.; Charpentier, C.; Descamps, D. [Physiopathology of HIV-2 infection]. Virol. Montrouge Fr. 2019, 23, 277–291. [Google Scholar] [CrossRef] [PubMed]
- Kalinichenko, S.; Komkov, D.; Mazurov, D. HIV-1 and HTLV-1 Transmission Modes: Mechanisms and Importance for Virus Spread. Viruses 2022, 14, 152. [Google Scholar] [CrossRef]
- Visseaux, B.; Damond, F.; Matheron, S.; Descamps, D.; Charpentier, C. Hiv-2 Molecular Epidemiology. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2016, 46, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Boswell, M.T.; Rowland-Jones, S.L. Delayed Disease Progression in HIV-2: The Importance of TRIM5α and the Retroviral Capsid. Clin. Exp. Immunol. 2019, 196, 305–317. [Google Scholar] [CrossRef]
- Vidya Vijayan, K.K.; Karthigeyan, K.P.; Tripathi, S.P.; Hanna, L.E. Pathophysiology of CD4+ T-Cell Depletion in HIV-1 and HIV-2 Infections. Front. Immunol. 2017, 8, 580. [Google Scholar] [CrossRef]
- Joseph, S.B.; Arrildt, K.T.; Sturdevant, C.B.; Swanstrom, R. HIV-1 Target Cells in the CNS. J. Neurovirol. 2015, 21, 276–289. [Google Scholar] [CrossRef]
- Woodham, A.W.; Skeate, J.G.; Sanna, A.M.; Taylor, J.R.; Da Silva, D.M.; Cannon, P.M.; Kast, W.M. Human Immunodeficiency Virus Immune Cell Receptors, Coreceptors, and Cofactors: Implications for Prevention and Treatment. AIDS Patient Care STDs 2016, 30, 291–306. [Google Scholar] [CrossRef]
- Fenwick, C.; Joo, V.; Jacquier, P.; Noto, A.; Banga, R.; Perreau, M.; Pantaleo, G. T-Cell Exhaustion in HIV Infection. Immunol. Rev. 2019, 292, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Luo, X.; George, A.F.; Mukherjee, G.; Sen, N.; Spitzer, T.L.; Giudice, L.C.; Greene, W.C.; Roan, N.R. HIV Efficiently Infects T Cells from the Endometrium and Remodels Them to Promote Systemic Viral Spread. eLife 2020, 9, e55487. [Google Scholar] [CrossRef]
- Renault, C.; Veyrenche, N.; Mennechet, F.; Bedin, A.-S.; Routy, J.-P.; Van de Perre, P.; Reynes, J.; Tuaillon, E. Th17 CD4+ T-Cell as a Preferential Target for HIV Reservoirs. Front. Immunol. 2022, 13, 822576. [Google Scholar] [CrossRef] [PubMed]
- Ng’eno, B.N.; Kellogg, T.A.; Kim, A.A.; Mwangi, A.; Mwangi, M.; Wamicwe, J.; Rutherford, G.W. Modes of HIV Transmission among Adolescents and Young Adults Aged 10-24 Years in Kenya. Int. J. STD AIDS 2018, 29, 800–805. [Google Scholar] [CrossRef]
- del Rio, C. The Global HIV Epidemic: What the Pathologist Needs to Know. Semin. Diagn. Pathol. 2017, 34, 314–317. [Google Scholar] [CrossRef] [PubMed]
- Kassa, G.M. Mother-to-Child Transmission of HIV Infection and Its Associated Factors in Ethiopia: A Systematic Review and Meta-Analysis. BMC Infect. Dis. 2018, 18, 216. [Google Scholar] [CrossRef] [PubMed]
- Osório, D.; Munyangaju, I.; Nacarapa, E.; Muhiwa, A.; Nhangave, A.V.; Ramos, J.M. Mother-to-Child Transmission of HIV Infection and Its Associated Factors in the District of Bilene, Gaza Province-Mozambique. PLoS ONE 2021, 16, e0260941. [Google Scholar] [CrossRef]
- Morar, M.M.; Pitman, J.P.; McFarland, W.; Bloch, E.M. The Contribution of Unsafe Blood Transfusion to Human Immunodeficiency Virus Incidence in Sub-Saharan Africa: Reexamination of the 5% to 10% Convention. Transfusion 2016, 56, 3121–3132. [Google Scholar] [CrossRef] [PubMed]
- Belov, A.; Yang, H.; Forshee, R.A.; Whitaker, B.I.; Eder, A.F.; Chancey, C.; Anderson, S.A. Modeling the Risk of HIV Transfusion Transmission. J. Acquir. Immune Defic. Syndr. 1999 2023, 92, 173–179. [Google Scholar] [CrossRef]
- Ball, L.J.; Puka, K.; Speechley, M.; Wong, R.; Hallam, B.; Wiener, J.C.; Koivu, S.; Silverman, M.S. Sharing of Injection Drug Preparation Equipment Is Associated With HIV Infection: A Cross-Sectional Study. J. Acquir. Immune Defic. Syndr. 1999 2019, 81, e99–e103. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Aridoss, S.; Mathiyazhakan, M.; Balasubramanian, G.; Jaganathasamy, N.; Natesan, M.; Padmapriya, V.M.; David, J.K.; Rajan, S.; Adhikary, R.; et al. Substance Use and Risk of HIV Infection among Men Who Have Sex with Men in India. Medicine 2020, 99, e21360. [Google Scholar] [CrossRef]
- Scheim, A.I.; Nosova, E.; Knight, R.; Hayashi, K.; Kerr, T. HIV Incidence Among Men Who Have Sex with Men and Inject Drugs in a Canadian Setting. AIDS Behav. 2018, 22, 3957–3961. [Google Scholar] [CrossRef]
- El-Bassel, N.; Shaw, S.A.; Dasgupta, A.; Strathdee, S.A. Drug Use as a Driver of HIV Risks: Re-Emerging and Emerging Issues. Curr. Opin. HIV AIDS 2014, 9, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Edeza, A.; Bazzi, A.; Salhaney, P.; Biancarelli, D.; Childs, E.; Mimiaga, M.J.; Drainoni, M.-L.; Biello, K. HIV Pre-Exposure Prophylaxis for People Who Inject Drugs: The Context of Co-Occurring Injection- and Sexual-Related HIV Risk in the U.S. Northeast. Subst. Use Misuse 2020, 55, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Bruxelle, J.-F.; Trattnig, N.; Mureithi, M.W.; Landais, E.; Pantophlet, R. HIV-1 Entry and Prospects for Protecting against Infection. Microorganisms 2021, 9, 228. [Google Scholar] [CrossRef]
- Jewanraj, J.; Ngcapu, S.; Liebenberg, L.J.P. Semen: A Modulator of Female Genital Tract Inflammation and a Vector for HIV-1 Transmission. Am. J. Reprod. Immunol. 2021, 86, e13478. [Google Scholar] [CrossRef]
- Cavarelli, M.; Le Grand, R. The Importance of Semen Leukocytes in HIV-1 Transmission and the Development of Prevention Strategies. Hum. Vaccines Immunother. 2020, 16, 2018–2032. [Google Scholar] [CrossRef] [PubMed]
- Kaul, R.; Liu, C.M.; Park, D.E.; Galiwango, R.M.; Tobian, A.A.R.; Prodger, J.L. The Penis, the Vagina and HIV Risk: Key Differences (Aside from the Obvious). Viruses 2022, 14, 1164. [Google Scholar] [CrossRef]
- WHO The Global Health Observatory. HIV. Available online: https://www.who.int/data/gho/data/themes/hiv-aids (accessed on 19 February 2023).
- UNAIDS Global HIV & AIDS Statistics—Fact Sheet. Available online: https://www.unaids.org/en/resources/fact-sheet (accessed on 19 February 2023).
- Gupta, S.; Granich, R.; Williams, B.G. Update on Treatment as Prevention of HIV Illness, Death, and Transmission: Sub-Saharan Africa HIV Financing and Progress towards the 95-95-95 Target. Curr. Opin. HIV AIDS 2022, 17, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Kharsany, A.B.M.; Karim, Q.A. HIV Infection and AIDS in Sub-Saharan Africa: Current Status, Challenges and Opportunities. Open. AIDS J. 2016, 10, 34–48. [Google Scholar] [CrossRef]
- Jones, J.; Sullivan, P.S.; Curran, J.W. Progress in the HIV Epidemic: Identifying Goals and Measuring Success. PLoS Med. 2019, 16, e1002729. [Google Scholar] [CrossRef] [PubMed]
- Shcherbatova, O.; Grebennikov, D.; Sazonov, I.; Meyerhans, A.; Bocharov, G. Modeling of the HIV-1 Life Cycle in Productively Infected Cells to Predict Novel Therapeutic Targets. Pathogens 2020, 9, 255. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.G.; Zhang, X.; Ganapathi, U.; Szekely, Z.; Flexner, C.W.; Owen, A.; Sinko, P.J. Drug Delivery Strategies and Systems for HIV/AIDS Pre-Exposure Prophylaxis and Treatment. J. Control. Release 2015, 219, 669–680. [Google Scholar] [CrossRef]
- Sankaranantham, M. HIV—Is a Cure Possible? Indian J. Sex. Transm. Dis. AIDS 2019, 40, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Bailon, L.; Mothe, B.; Berman, L.; Brander, C. Novel Approaches Towards a Functional Cure of HIV/AIDS. Drugs 2020, 80, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Sakuragi, J. Morphogenesis of the Infectious HIV-1 Virion. Front. Microbiol. 2011, 2, 242. [Google Scholar] [CrossRef]
- McGettigan, J.P.; Naper, K.; Orenstein, J.; Koser, M.; McKenna, P.M.; Schnell, M.J. Functional Human Immunodeficiency Virus Type 1 (HIV-1) Gag-Pol or HIV-1 Gag-Pol and Env Expressed from a Single Rhabdovirus-Based Vaccine Vector Genome. J. Virol. 2003, 77, 10889–10899. [Google Scholar] [CrossRef]
- Hill, M.; Tachedjian, G.; Mak, J. The Packaging and Maturation of the HIV-1 Pol Proteins. Curr. HIV Res. 2005, 3, 73–85. [Google Scholar] [CrossRef]
- Staudt, R.P.; Alvarado, J.J.; Emert-Sedlak, L.A.; Shi, H.; Shu, S.T.; Wales, T.E.; Engen, J.R.; Smithgall, T.E. Structure, Function, and Inhibitor Targeting of HIV-1 Nef-Effector Kinase Complexes. J. Biol. Chem. 2020, 295, 15158–15171. [Google Scholar] [CrossRef]
- NIH HIV Replication Cycle|NIH: National Institute of Allergy and Infectious Diseases. Available online: https://www.niaid.nih.gov/diseases-conditions/hiv-replication-cycle (accessed on 23 February 2023).
- Greta Hughson HIV Lifecycle. Available online: https://www.aidsmap.com/about-hiv/hiv-lifecycle (accessed on 3 March 2023).
- Xiao, T.; Cai, Y.; Chen, B. HIV-1 Entry and Membrane Fusion Inhibitors. Viruses 2021, 13, 735. [Google Scholar] [CrossRef]
- Wilen, C.B.; Tilton, J.C.; Doms, W. HIV: Cell Binding and Entry. Cold Spring Harb. Perspect. Med. 2012, 2, a006866. [Google Scholar] [CrossRef] [PubMed]
- Negi, G.; Sharma, A.; Dey, M.; Dhanawat, G.; Parveen, N. Membrane Attachment and Fusion of HIV-1, Influenza A, and SARS-CoV-2: Resolving the Mechanisms with Biophysical Methods. Biophys. Rev. 2022, 14, 1109–1140. [Google Scholar] [CrossRef] [PubMed]
- Doms, R.W.; Moore, J.P. HIV-1 Membrane Fusion. J. Cell. Biol. 2000, 151, f9–f14. [Google Scholar] [CrossRef]
- Sapp, N.; Burge, N.; Cox, K.; Prakash, P.; Balasubramaniam, M.; Thapa, S.; Christensen, D.; Li, M.; Linderberger, J.; Kvaratskhelia, M.; et al. HIV-1 Preintegration Complex Preferentially Integrates the Viral DNA into Nucleosomes Containing Trimethylated Histone 3-Lysine 36 Modification and Flanking Linker DNA. J. Virol. 2022, 96, e0101122. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Rodriguez, G.; Gazi, A.; Monel, B.; Frabetti, S.; Scoca, V.; Mueller, F.; Schwartz, O.; Krijnse-Locker, J.; Charneau, P.; Di Nunzio, F. Remodeling of the Core Leads HIV-1 Preintegration Complex into the Nucleus of Human Lymphocytes. J. Virol. 2020, 94, e00135-20. [Google Scholar] [CrossRef]
- Rozina, A.; Anisenko, A.; Kikhai, T.; Silkina, M.; Gottikh, M. Complex Relationships between HIV-1 Integrase and Its Cellular Partners. Int. J. Mol. Sci. 2022, 23, 12341. [Google Scholar] [CrossRef]
- Elliott, J.L.; Kutluay, S.B. Going beyond Integration: The Emerging Role of HIV-1 Integrase in Virion Morphogenesis. Viruses 2020, 12, 1005. [Google Scholar] [CrossRef]
- PubChem HIV Life Cycle. Available online: https://pubchem.ncbi.nlm.nih.gov/pathway/Reactome:R-HSA-162587 (accessed on 3 March 2023).
- Murray, J.M.; Kelleher, A.D.; Cooper, D.A. Timing of the Components of the HIV Life Cycle in Productively Infected CD4+ T Cells in a Population of HIV-Infected Individuals. J. Virol. 2011, 85, 10798–10805. [Google Scholar] [CrossRef]
- Roebuck, K.A.; Saifuddin, M. Regulation of HIV-1 Transcription. Gene Expr. 1999, 8, 67–84. [Google Scholar]
- NIH. The HIV Life Cycle|NIH. Available online: https://hivinfo.nih.gov/understanding-hiv/fact-sheets/hiv-life-cycle (accessed on 3 March 2023).
- Gary Kaiser 10.6C: The Life Cycle of HIV. Available online: https://bio.libretexts.org/Bookshelves/Microbiology/Microbiology_(Kaiser)/Unit_4%3A_Eukaryotic_Microorganisms_and_Viruses/10%3A_Viruses/10.06%3A_Animal_Virus_Life_Cycles%3A_An_Overview/10.6C%3A_The_Life_Cycle_of_HIV (accessed on 3 March 2023).
- Sundquist, W.I.; Kräusslich, H.-G. HIV-1 Assembly, Budding, and Maturation. Cold Spring Harb. Perspect. Med. 2012, 2, a006924. [Google Scholar] [CrossRef]
- Freed, E.O. Freed HIV-1 Assembly, Release and Maturation. Nat. Rev. Microbiol. 2015, 13, 484–496. [Google Scholar] [CrossRef]
- Ganser-Pornillos, B.; Yeager, M.; Sundquist, W.I. The Structural Biology of HIV Assembly. Curr. Opin. Struct. Biol. 2008, 18, 203. [Google Scholar] [CrossRef] [PubMed]
- Yandrapally, S.; Mohareer, K.; Arekuti, G.; Vadankula, G.R.; Banerjee, S. HIV Co-Receptor-Tropism: Cellular and Molecular Events behind the Enigmatic Co-Receptor Switching. Crit. Rev. Microbiol. 2021, 47, 499–516. [Google Scholar] [CrossRef]
- Simon, V.; Ho, D.D.; Karim, Q.A. HIV/AIDS Epidemiology, Pathogenesis, Prevention, and Treatment. Lancet 2006, 368, 489–504. [Google Scholar] [CrossRef]
- Naif, H.M. Pathogenesis of HIV Infection. Infect. Dis. Rep. 2013, 5, e6. [Google Scholar] [CrossRef] [PubMed]
- White, J.M.; Delos, S.E.; Brecher, M.; Schornberg, K. Structures and Mechanisms of Viral Membrane Fusion Proteins. Crit. Rev. Biochem. Mol. Biol. 2008, 43, 189–219. [Google Scholar] [CrossRef]
- Dimitrov, D.S. Virus Entry: Molecular Mechanisms and Biomedical Applications. Nat. Rev. Microbiol. 2004, 2, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Belouzard, S.; Millet, J.K.; Licitra, B.N.; Whittaker, G.R. Mechanisms of Coronavirus Cell Entry Mediated by the Viral Spike Protein. Viruses 2012, 4, 1011–1033. [Google Scholar] [CrossRef]
- Leroy, H.; Han, M.; Woottum, M.; Bracq, L.; Bouchet, J.; Xie, M.; Benichou, S. Virus-Mediated Cell-Cell Fusion. Int. J. Mol. Sci. 2020, 21, 9644. [Google Scholar] [CrossRef]
- Zhou, H.; Møhlenberg, M.; Thakor, J.C.; Tuli, H.S.; Wang, P.; Assaraf, Y.G.; Dhama, K.; Jiang, S. Sensitivity to Vaccines, Therapeutic Antibodies, and Viral Entry Inhibitors and Advances To Counter the SARS-CoV-2 Omicron Variant. Clin. Microbiol. Rev. 2022, 35, e00014-22. [Google Scholar] [CrossRef] [PubMed]
- Burdick, R.C.; Li, C.; Munshi, M.; Rawson, J.M.O.; Nagashima, K.; Hu, W.-S.; Pathak, V.K. HIV-1 Uncoats in the Nucleus near Sites of Integration. Proc. Natl. Acad. Sci. USA 2020, 117, 5486–5493. [Google Scholar] [CrossRef]
- Mamede, J.I.; Cianci, G.C.; Anderson, M.R.; Hope, T.J. Early Cytoplasmic Uncoating Is Associated with Infectivity of HIV-1. Proc. Natl. Acad. Sci. USA 2017, 114, E7169–E7178. [Google Scholar] [CrossRef] [PubMed]
- Rossi, E.; Meuser, M.E.; Cunanan, C.J.; Cocklin, S. Structure, Function, and Interactions of the HIV-1 Capsid Protein. Life Basel Switz. 2021, 11, 100. [Google Scholar] [CrossRef] [PubMed]
- Henning, M.S.; Dubose, B.N.; Burse, M.J.; Aiken, C.; Yamashita, M. In Vivo Functions of CPSF6 for HIV-1 as Revealed by HIV-1 Capsid Evolution in HLA-B27-Positive Subjects. PLoS Pathog. 2014, 10, e1003868. [Google Scholar] [CrossRef]
- Kane, M.; Rebensburg, S.V.; Takata, M.A.; Zang, T.M.; Yamashita, M.; Kvaratskhelia, M.; Bieniasz, P.D. Nuclear Pore Heterogeneity Influences HIV-1 Infection and the Antiviral Activity of MX2. eLife 2018, 7, e35738. [Google Scholar] [CrossRef] [PubMed]
- Buffone, C.; Martinez-Lopez, A.; Fricke, T.; Opp, S.; Severgnini, M.; Cifola, I.; Petiti, L.; Frabetti, S.; Skorupka, K.; Zadrozny, K.K.; et al. Nup153 Unlocks the Nuclear Pore Complex for HIV-1 Nuclear Translocation in Nondividing Cells. J. Virol. 2018, 92, e00648-18. [Google Scholar] [CrossRef]
- De Iaco, A.; Santoni, F.; Vannier, A.; Guipponi, M.; Antonarakis, S.; Luban, J. TNPO3 Protects HIV-1 Replication from CPSF6-Mediated Capsid Stabilization in the Host Cell Cytoplasm. Retrovirology 2013, 10, 20. [Google Scholar] [CrossRef]
- Zhong, Z.; Ning, J.; Boggs, E.A.; Jang, S.; Wallace, C.; Telmer, C.; Bruchez, M.P.; Ahn, J.; Engelman, A.N.; Zhang, P.; et al. Cytoplasmic CPSF6 Regulates HIV-1 Capsid Trafficking and Infection in a Cyclophilin A-Dependent Manner. mBio 2021, 12, e03142-20. [Google Scholar] [CrossRef]
- Hendricks, C.M.; Cordeiro, T.; Gomes, A.P.; Stevenson, M. The Interplay of HIV-1 and Macrophages in Viral Persistence. Front. Microbiol. 2021, 12, 646447. [Google Scholar] [CrossRef]
- Delannoy, A.; Poirier, M.; Bell, B. Cat and Mouse: HIV Transcription in Latency, Immune Evasion and Cure/Remission Strategies. Viruses 2019, 11, 269. [Google Scholar] [CrossRef] [PubMed]
- Balasubramaniam, M.; Pandhare, J.; Dash, C. Immune Control of HIV. J. Life Sci. Westlake Village Calif. 2019, 1, 4–37. [Google Scholar] [CrossRef]
- Boasso, A.; Shearer, G.M.; Chougnet, C. Immune Dysregulation in Human Immunodeficiency Virus Infection: Know It, Fix It, Prevent It? J. Intern. Med. 2009, 265, 78–96. [Google Scholar] [CrossRef]
- Okoye, A.A.; Picker, L.J. CD4+ T Cell Depletion in HIV Infection: Mechanisms of Immunological Failure. Immunol. Rev. 2013, 254, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Février, M.; Dorgham, K.; Rebollo, A. CD4+ T Cell Depletion in Human Immunodeficiency Virus (HIV) Infection: Role of Apoptosis. Viruses 2011, 3, 586–612. [Google Scholar] [CrossRef]
- Guha, D.; Ayyavoo, V. Innate Immune Evasion Strategies by Human Immunodeficiency Virus Type 1. ISRN AIDS 2013, 2013, 954806. [Google Scholar] [CrossRef] [PubMed]
- Saez-Cirion, A.; Jacquelin, B.; Barré-Sinoussi, F.; Müller-Trutwin, M. Immune Responses during Spontaneous Control of HIV and AIDS: What Is the Hope for a Cure? Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130436. [Google Scholar] [CrossRef] [PubMed]
- Goulder, P.J.R.; Watkins, D.I. Impact of MHC Class I Diversity on Immune Control of Immunodeficiency Virus Replication. Nat. Rev. Immunol. 2008, 8, 619–630. [Google Scholar] [CrossRef]
- Swigut, T.; Alexander, L.; Morgan, J.; Lifson, J.; Mansfield, K.G.; Lang, S.; Johnson, R.P.; Skowronski, J.; Desrosiers, R. Impact of Nef-Mediated Downregulation of Major Histocompatibility Complex Class I on Immune Response to Simian Immunodeficiency Virus. J. Virol. 2004, 78, 13335–13344. [Google Scholar] [CrossRef] [PubMed]
- Kerkau, T.; Bacik, I.; Bennink, J.R.; Yewdell, J.W.; Hünig, T.; Schimpl, A.; Schubert, U. The Human Immunodeficiency Virus Type 1 (HIV-1) Vpu Protein Interferes with an Early Step in the Biosynthesis of Major Histocompatibility Complex (MHC) Class I Molecules. J. Exp. Med. 1997, 185, 1295–1306. [Google Scholar] [CrossRef]
- Koyanagi, N.; Kawaguchi, Y. Evasion of the Cell-Mediated Immune Response by Alphaherpesviruses. Viruses 2020, 12, 1354. [Google Scholar] [CrossRef]
- Stumptner-Cuvelette, P.; Morchoisne, S.; Dugast, M.; Le Gall, S.; Raposo, G.; Schwartz, O.; Benaroch, P. HIV-1 Nef Impairs MHC Class II Antigen Presentation and Surface Expression. Proc. Natl. Acad. Sci. USA 2001, 98, 12144–12149. [Google Scholar] [CrossRef] [PubMed]
- Wonderlich, E.R.; Leonard, J.A.; Collins, K.L. HIV Immune Evasion: Disruption of Antigen Presentation by the HIV Nef Protein. Adv. Virus Res. 2011, 80, 103–127. [Google Scholar] [CrossRef] [PubMed]
- Roeth, J.F.; Collins, K.L. Human Immunodeficiency Virus Type 1 Nef: Adapting to Intracellular Trafficking Pathways. Microbiol. Mol. Biol. Rev. 2006, 70, 548–563. [Google Scholar] [CrossRef]
- Blagoveshchenskaya, A.D.; Thomas, L.; Feliciangeli, S.F.; Hung, C.-H.; Thomas, G. HIV-1 Nef Downregulates MHC-I by a PACS-1- and PI3K-Regulated ARF6 Endocytic Pathway. Cell 2002, 111, 853–866. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Geiger, J.D. Role of Viral Protein U (Vpu) in HIV-1 Infection and Pathogenesis. Viruses 2021, 13, 1466. [Google Scholar] [CrossRef] [PubMed]
- Dubé, M.; Bego, M.G.; Paquay, C.; Cohen, É.A. Modulation of HIV-1-Host Interaction: Role of the Vpu Accessory Protein. Retrovirology 2010, 7, 114. [Google Scholar] [CrossRef]
- Carrington, M.; Alter, G. Innate Immune Control of HIV. Cold Spring Harb. Perspect. Med. 2012, 2, a007070. [Google Scholar] [CrossRef]
- Tomescu, C.; Law, W.K.; Kedes, D.H. Surface Downregulation of Major Histocompatibility Complex Class I, PE-CAM, and ICAM-1 Following De Novo Infection of Endothelial Cells with Kaposi’s Sarcoma-Associated Herpesvirus. J. Virol. 2003, 77, 9669–9684. [Google Scholar] [CrossRef]
- Dirk, B.S.; Pawlak, E.N.; Johnson, A.L.; Van Nynatten, L.R.; Jacob, R.A.; Heit, B.; Dikeakos, J.D. HIV-1 Nef Sequesters MHC-I Intracellularly by Targeting Early Stages of Endocytosis and Recycling. Sci. Rep. 2016, 6, 37021. [Google Scholar] [CrossRef]
- Mitchell, R.S.; Katsura, C.; Skasko, M.A.; Fitzpatrick, K.; Lau, D.; Ruiz, A.; Stephens, E.B.; Margottin-Goguet, F.; Benarous, R.; Guatelli, J.C. Vpu Antagonizes BST-2–Mediated Restriction of HIV-1 Release via β-TrCP and Endo-Lysosomal Trafficking. PLOS Pathog. 2009, 5, e1000450. [Google Scholar] [CrossRef] [PubMed]
- Le Tortorec, A.; Willey, S.; Neil, S.J.D. Antiviral Inhibition of Enveloped Virus Release by Tetherin/BST-2: Action and Counteraction. Viruses 2011, 3, 520–540. [Google Scholar] [CrossRef]
- Miyagi, E.; Andrew, A.J.; Kao, S.; Strebel, K. Vpu Enhances HIV-1 Virus Release in the Absence of Bst-2 Cell Surface down-Modulation and Intracellular Depletion. Proc. Natl. Acad. Sci. USA 2009, 106, 2868–2873. [Google Scholar] [CrossRef] [PubMed]
- Arias, J.F.; Heyer, L.N.; von Bredow, B.; Weisgrau, K.L.; Moldt, B.; Burton, D.R.; Rakasz, E.G.; Evans, D.T. Tetherin Antagonism by Vpu Protects HIV-Infected Cells from Antibody-Dependent Cell-Mediated Cytotoxicity. Proc. Natl. Acad. Sci. USA 2014, 111, 6425–6430. [Google Scholar] [CrossRef] [PubMed]
- Waheed, A.A.; Swiderski, M.; Khan, A.; Gitzen, A.; Majadly, A.; Freed, E.O. The Viral Protein U (Vpu)-Interacting Host Protein ATP6V0C down-Regulates Cell-Surface Expression of Tetherin and Thereby Contributes to HIV-1 Release. J. Biol. Chem. 2020, 295, 7327–7340. [Google Scholar] [CrossRef]
- Donaldson, J.G.; Williams, D.B. Intracellular Assembly and Trafficking of MHC Class I Molecules. Traffic 2009, 10, 1745–1752. [Google Scholar] [CrossRef] [PubMed]
- Mack, K.; Starz, K.; Sauter, D.; Langer, S.; Bibollet-Ruche, F.; Learn, G.H.; Stürzel, C.M.; Leoz, M.; Plantier, J.-C.; Geyer, M.; et al. Efficient Vpu-Mediated Tetherin Antagonism by an HIV-1 Group O Strain. J. Virol. 2017, 91, e02177-16. [Google Scholar] [CrossRef]
- Tokarev, A.; Skasko, M.; Fitzpatrick, K.; Guatelli, J. Antiviral Activity of the Interferon-Induced Cellular Protein BST-2/Tetherin. AIDS Res. Hum. Retrovir. 2009, 25, 1197–1210. [Google Scholar] [CrossRef] [PubMed]
- Mielke, D.; Bandawe, G.; Zheng, J.; Jones, J.; Abrahams, M.-R.; Bekker, V.; Ochsenbauer, C.; Garrett, N.; Abdool Karim, S.; Moore, P.L.; et al. ADCC-Mediating Non-Neutralizing Antibodies Can Exert Immune Pressure in Early HIV-1 Infection. PLoS Pathog. 2021, 17, e1010046. [Google Scholar] [CrossRef]
- Martín, J.; LaBranche, C.C.; González-Scarano, F. Differential CD4/CCR5 Utilization, Gp120 Conformation, and Neutralization Sensitivity between Envelopes from a Microglia-Adapted Human Immunodeficiency Virus Type 1 and Its Parental Isolate. J. Virol. 2001, 75, 3568–3580. [Google Scholar] [CrossRef]
- Raja, A.; Venturi, M.; Kwong, P.; Sodroski, J. CD4 Binding Site Antibodies Inhibit Human Immunodeficiency Virus Gp120 Envelope Glycoprotein Interaction with CCR5. J. Virol. 2003, 77, 713–718. [Google Scholar] [CrossRef]
- Chen, B. Molecular Mechanism of HIV-1 Entry. Trends Microbiol. 2019, 27, 878–891. [Google Scholar] [CrossRef] [PubMed]
- Klasse, P.J. The Molecular Basis of HIV Entry. Cell. Microbiol. 2012, 14, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Sachdeva, M.; Fischl, M.A.; Pahwa, R.; Sachdeva, N.; Pahwa, S. Immune Exhaustion Occurs Concomitantly with Immune Activation and Decrease in Regulatory T Cells in Viremic Chronically HIV-1 Infected Patients. J. Acquir. Immune Defic. Syndr. 1999 2010, 54, 447–454. [Google Scholar] [CrossRef]
- Khaitan, A.; Unutmaz, D. Revisiting Immune Exhaustion During HIV Infection. Curr. HIV/AIDS Rep. 2011, 8, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.; McMichael, A. The T-Cell Response to HIV. Cold Spring Harb. Perspect. Med. 2012, 2, a007054. [Google Scholar] [CrossRef] [PubMed]
- Cummins, N.W.; Badley, A.D. Mechanisms of HIV-Associated Lymphocyte Apoptosis: 2010. Cell. Death Dis. 2010, 1, e99. [Google Scholar] [CrossRef]
- Oyaizu, N.; McCloskey, T.; Than, S.; Hu, R.; Kalyanaraman, V.; Pahwa, S. Cross-Linking of CD4 Molecules Upregulates Fas Antigen Expression in Lymphocytes by Inducing Interferon-Gamma and Tumor Necrosis Factor- Alpha Secretion. Blood 1994, 84, 2622–2631. [Google Scholar] [CrossRef]
- Chandrasekar, A.P.; Cummins, N.W.; Badley, A.D. The Role of the BCL-2 Family of Proteins in HIV-1 Pathogenesis and Persistence. Clin. Microbiol. Rev. 2019, 33, e00107-19. [Google Scholar] [CrossRef]
- Xu, H.; Wang, X.; Veazey, R.S. Mucosal Immunology of HIV Infection. Immunol. Rev. 2013, 254, 10–33. [Google Scholar] [CrossRef]
- McMichael, A.J.; Borrow, P.; Tomaras, G.D.; Goonetilleke, N.; Haynes, B.F. The Immune Response during Acute HIV-1 Infection: Clues for Vaccine Development. Nat. Rev. Immunol. 2010, 10, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Batorsky, R.; Sergeev, R.A.; Rouzine, I.M. The Route of HIV Escape from Immune Response Targeting Multiple Sites Is Determined by the Cost-Benefit Tradeoff of Escape Mutations. PLoS Comput. Biol. 2014, 10, e1003878. [Google Scholar] [CrossRef]
- Cuevas, J.M.; Geller, R.; Garijo, R.; López-Aldeguer, J.; Sanjuán, R. Extremely High Mutation Rate of HIV-1 In Vivo. PLoS Biol. 2015, 13, e1002251. [Google Scholar] [CrossRef]
- Abram, M.E.; Ferris, A.L.; Das, K. Mutations in HIV-1 Reverse Transcriptase Affect the Errors Made in a Single Cycle of Viral Replication. J. Virol. 2014, 88, 7589–7601. [Google Scholar] [CrossRef] [PubMed]
- Balasubramaniam, S.D.; Balakrishnan, V.; Oon, C.E.; Kaur, G. Key Molecular Events in Cervical Cancer Development. Medicina 2019, 55, 384. [Google Scholar] [CrossRef]
- Burton, D.R.; Mascola, J.R. Antibody Responses to Envelope Glycoproteins in HIV-1 Infection. Nat. Immunol. 2015, 16, 571–576. [Google Scholar] [CrossRef]
- Prabakaran, P.; Dimitrov, A.S.; Fouts, T.R.; Dimitrov, D.S. Structure and Function of the HIV Envelope Glycoprotein as Entry Mediator, Vaccine Immunogen, and Target for Inhibitors. Adv. Pharmacol. San. Diego Calif. 2007, 55, 33–97. [Google Scholar] [CrossRef]
- Anand, S.P.; Grover, J.R.; Tolbert, W.D.; Prévost, J.; Richard, J.; Ding, S.; Baril, S.; Medjahed, H.; Evans, D.T.; Pazgier, M.; et al. Antibody-Induced Internalization of HIV-1 Env Proteins Limits Surface Expression of the Closed Conformation of Env. J. Virol. 2019, 93, e00293-19. [Google Scholar] [CrossRef]
- Swiecki, M.; Colonna, M. Type I Interferons: Diversity of Sources, Production Pathways and Effects on Immune Responses. Curr. Opin. Virol. 2011, 1, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Marziali, F.; Delpeuch, M.; Kumar, A.; Appourchaux, R.; Dufloo, J.; Tartour, K.; Etienne, L.; Cimarelli, A. Functional Heterogeneity of Mammalian IFITM Proteins against HIV-1. J. Virol. 2021, 95, e0043921. [Google Scholar] [CrossRef]
- Ali, S.; Mann-Nüttel, R.; Schulze, A.; Richter, L.; Alferink, J.; Scheu, S. Sources of Type I Interferons in Infectious Immunity: Plasmacytoid Dendritic Cells Not Always in the Driver’s Seat. Front. Immunol. 2019, 10, 778. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of Type I Interferon Responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Hermant, P.; Michiels, T. Interferon-λ in the Context of Viral Infections: Production, Response and Therapeutic Implications. J. Innate Immun. 2014, 6, 563–574. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Tran, J.T.; Sanchez, D.J. Manipulation of Type I Interferon Signaling by HIV and AIDS-Associated Viruses. J. Immunol. Res. 2019, 2019, 8685312. [Google Scholar] [CrossRef]
- Nguyen, N.V.; Tran, J.T.; Sanchez, D.J. HIV Blocks Type I IFN Signaling through Disruption of STAT1 Phosphorylation. Innate Immun. 2018, 24, 490–500. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Qian, G.; Zhu, L.; Zhao, Z.; Liu, Y.; Han, W.; Zhang, X.; Zhang, Y.; Xiong, T.; Zeng, H.; et al. HIV-1 Vif Suppresses Antiviral Immunity by Targeting STING. Cell. Mol. Immunol. 2022, 19, 108–121. [Google Scholar] [CrossRef]
- Ezeonwumelu, I.J.; Garcia-Vidal, E.; Ballana, E. JAK-STAT Pathway: A Novel Target to Tackle Viral Infections. Viruses 2021, 13, 2379. [Google Scholar] [CrossRef]
- Khanal, S.; Schank, M.; El Gazzar, M.; Moorman, J.P.; Yao, Z.Q. HIV-1 Latency and Viral Reservoirs: Existing Reversal Approaches and Potential Technologies, Targets, and Pathways Involved in HIV Latency Studies. Cells 2021, 10, 475. [Google Scholar] [CrossRef] [PubMed]
- Lackner, A.A.; Lederman, M.M.; Rodriguez, B. HIV Pathogenesis: The Host. Cold Spring Harb. Perspect. Med. 2012, 2, a007005. [Google Scholar] [CrossRef]
- Azimi, F.C.; Lee, J.E. Structural Perspectives on HIV-1 Vif and APOBEC3 Restriction Factor Interactions. Protein Sci. Publ. Protein Soc. 2020, 29, 391–406. [Google Scholar] [CrossRef]
- Kung, W.-W.; Ramachandran, S.; Makukhin, N.; Bruno, E.; Ciulli, A. Structural Insights into Substrate Recognition by the SOCS2 E3 Ubiquitin Ligase. Nat. Commun. 2019, 10, 2534. [Google Scholar] [CrossRef]
- Stanley, B.J.; Ehrlich, E.S.; Short, L.; Yu, Y.; Xiao, Z.; Yu, X.-F.; Xiong, Y. Structural Insight into the Human Immunodeficiency Virus Vif SOCS Box and Its Role in Human E3 Ubiquitin Ligase Assembly. J. Virol. 2008, 82, 8656. [Google Scholar] [CrossRef]
- Lata, S.; Mishra, R.; Banerjea, A.C. Proteasomal Degradation Machinery: Favorite Target of HIV-1 Proteins. Front. Microbiol. 2018, 9, 2738. [Google Scholar] [CrossRef]
- Du, Y.; Hu, Z.; Luo, Y.; Wang, H.Y.; Yu, X.; Wang, R.-F. Function and Regulation of CGAS-STING Signaling in Infectious Diseases. Front. Immunol. 2023, 14, 1130423. [Google Scholar] [CrossRef] [PubMed]
- Maelfait, J.; Bridgeman, A.; Benlahrech, A.; Cursi, C.; Rehwinkel, J. Restriction by SAMHD1 Limits CGAS/STING-Dependent Innate and Adaptive Immune Responses to HIV-1. Cell. Rep. 2016, 16, 1492–1501. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; König, R.; Deng, M.; Riess, M.; Mo, J.; Zhang, L.; Petrucelli, A.; Yoh, S.M.; Barefoot, B.; Samo, M.; et al. NLRX1 Sequesters STING to Negatively Regulate the Interferon Response, Thereby Facilitating the Replication of HIV-1 and DNA Viruses. Cell Host Microbe 2016, 19, 515–528. [Google Scholar] [CrossRef]
- Sivro, A.; Su, R.-C.; Plummer, F.A.; Ball, T.B. Interferon Responses in HIV Infection: From Protection to Disease. AIDS Rev. 2014, 16, 43–51. [Google Scholar] [PubMed]
- Ananworanich, J.; Sacdalan, C.P.; Pinyakorn, S.; Chomont, N.; Souza, M.; Luekasemsuk, T.; Schuetz, A.; Krebs, S.J.; Dewar, R.; Jagodzinski, L.; et al. Virological and Immunological Characteristics of HIV-Infected Individuals at the Earliest Stage of Infection. J. Virus Erad. 2016, 2, 43–48. [Google Scholar] [CrossRef]
- Robb, M.L.; Eller, L.A.; Kibuuka, H.; Rono, K.; Maganga, L.; Nitayaphan, S.; Kroon, E.; Sawe, F.K.; Sinei, S.; Sriplienchan, S.; et al. Prospective Study of Acute HIV-1 Infection in Adults in East Africa and Thailand. N. Engl. J. Med. 2016, 374, 2120–2130. [Google Scholar] [CrossRef]
- Martín-Moreno, A.; Muñoz-Fernández, M.A. Dendritic Cells, the Double Agent in the War Against HIV-1. Front. Immunol. 2019, 10, 2485. [Google Scholar] [CrossRef]
- Lee, H.-C.; Chathuranga, K.; Lee, J.-S. Intracellular Sensing of Viral Genomes and Viral Evasion. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Su, J.; Chen, R.; Wei, W.; Yuan, Z.; Chen, X.; Wang, X.; Liang, H.; Ye, L.; Jiang, J. The Role of Innate Immunity in Natural Elite Controllers of HIV-1 Infection. Front. Immunol. 2022, 13, 780922. [Google Scholar] [CrossRef]
- Chintala, K.; Mohareer, K.; Banerjee, S. Dodging the Host Interferon-Stimulated Gene Mediated Innate Immunity by HIV-1: A Brief Update on Intrinsic Mechanisms and Counter-Mechanisms. Front. Immunol. 2021, 12, 716927. [Google Scholar] [CrossRef]
- Mogensen, T.H.; Melchjorsen, J.; Larsen, C.S.; Paludan, S.R. Innate Immune Recognition and Activation during HIV Infection. Retrovirology 2010, 7, 54. [Google Scholar] [CrossRef]
- Stunnenberg, M.; Sprokholt, J.K.; van Hamme, J.L.; Kaptein, T.M.; Zijlstra-Willems, E.M.; Gringhuis, S.I.; Geijtenbeek, T.B.H. Synthetic Abortive HIV-1 RNAs Induce Potent Antiviral Immunity. Front. Immunol. 2020, 11, 8. [Google Scholar] [CrossRef]
- Chow, K.T.; Gale, M.; Loo, Y.-M. RIG-I and Other RNA Sensors in Antiviral Immunity. Annu. Rev. Immunol. 2018, 36, 667–694. [Google Scholar] [CrossRef]
- Kell, A.M.; Gale, M. RIG-I in RNA Virus Recognition. Virology 2015, 479–480, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Radoshevich, L.; Dussurget, O. Cytosolic Innate Immune Sensing and Signaling upon Infection. Front. Microbiol. 2016, 7, 313. [Google Scholar] [CrossRef]
- Goubau, D.; Deddouche, S.; Reis e Sousa, C. Cytosolic Sensing of Viruses. Immunity 2013, 38, 855–869. [Google Scholar] [CrossRef] [PubMed]
- Abbas, F.; Cenac, C.; Youness, A.; Azar, P.; Delobel, P.; Guéry, J.-C. HIV-1 Infection Enhances Innate Function and TLR7 Expression in Female Plasmacytoid Dendritic Cells. Life Sci. Alliance 2022, 5, e202201452. [Google Scholar] [CrossRef] [PubMed]
- Scagnolari, C.; Antonelli, G. Type I Interferon and HIV: Subtle Balance between Antiviral Activity, Immunopathogenesis and the Microbiome. Cytokine Growth Factor. Rev. 2018, 40, 19. [Google Scholar] [CrossRef]
- Li, D.; Wu, M. Pattern Recognition Receptors in Health and Diseases. Signal Transduct. Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef] [PubMed]
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.C.; Weinlich, R.; Bortoluci, K.R. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front. Immunol. 2018, 9, 2379. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The Role of Pattern-Recognition Receptors in Innate Immunity: Update on Toll-like Receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.-H.; Shin, H.W.; Lee, J.M.; Lee, H.-W.; Kim, E.-C.; Park, S.H. An Overview of Pathogen Recognition Receptors for Innate Immunity in Dental Pulp. Mediat. Inflamm. 2015, 2015, 794143. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, T.H. Pathogen Recognition and Inflammatory Signaling in Innate Immune Defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [PubMed]
- Schoenemeyer, A.; Barnes, B.J.; Mancl, M.E.; Latz, E.; Goutagny, N.; Pitha, P.M.; Fitzgerald, K.A.; Golenbock, D.T. The Interferon Regulatory Factor, IRF5, Is a Central Mediator of Toll-like Receptor 7 Signaling. J. Biol. Chem. 2005, 280, 17005–17012. [Google Scholar] [CrossRef] [PubMed]
- Kwa, M.Q.; Nguyen, T.; Huynh, J.; Ramnath, D.; De Nardo, D.; Lam, P.Y.; Reynolds, E.C.; Hamilton, J.A.; Sweet, M.J.; Scholz, G.M. Interferon Regulatory Factor 6 Differentially Regulates Toll-like Receptor 2-Dependent Chemokine Gene Expression in Epithelial Cells. J. Biol. Chem. 2014, 289, 19758–19768. [Google Scholar] [CrossRef]
- Bergantz, L.; Subra, F.; Deprez, E.; Delelis, O.; Richetta, C. Interplay between Intrinsic and Innate Immunity during HIV Infection. Cells 2019, 8, 922. [Google Scholar] [CrossRef]
- Meås, H.Z.; Haug, M.; Beckwith, M.S.; Louet, C.; Ryan, L.; Hu, Z.; Landskron, J.; Nordbø, S.A.; Taskén, K.; Yin, H.; et al. Sensing of HIV-1 by TLR8 Activates Human T Cells and Reverses Latency. Nat. Commun. 2020, 11, 147. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, E.N.; Dirk, B.S.; Jacob, R.A.; Johnson, A.L.; Dikeakos, J.D. The HIV-1 Accessory Proteins Nef and Vpu Downregulate Total and Cell Surface CD28 in CD4+ T Cells. Retrovirology 2018, 15, 6. [Google Scholar] [CrossRef] [PubMed]
- Manches, O.; Frleta, D.; Bhardwaj, N. Dendritic Cells in Progression and Pathology of HIV Infection. Trends Immunol. 2014, 35, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Chathuranga, K.; Weerawardhana, A.; Dodantenna, N.; Lee, J.-S. Regulation of Antiviral Innate Immune Signaling and Viral Evasion Following Viral Genome Sensing. Exp. Mol. Med. 2021, 53, 1647–1668. [Google Scholar] [CrossRef] [PubMed]
- Collin, M.; Bigley, V. Human Dendritic Cell Subsets: An Update. Immunology 2018, 154, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.; Bhardwaj, N. Dendritic Cell Dysregulation during HIV-1 Infection. Immunol. Rev. 2013, 254, 170–189. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.L.; Takata, H.; Muir, R.; Colby, D.J.; Kroon, E.; Crowell, T.A.; Sacdalan, C.; Pinyakorn, S.; Puttamaswin, S.; Benjapornpong, K.; et al. Plasmacytoid Dendritic Cells Sense HIV Replication before Detectable Viremia Following Treatment Interruption. J. Clin. Invest. 2020, 130, 2845–2858. [Google Scholar] [CrossRef]
- Borrow, P. Innate Immunity in Acute HIV-1 Infection. Curr. Opin. HIV AIDS 2011, 6, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Mbita, Z.; Hull, R.; Dlamini, Z. Human Immunodeficiency Virus-1 (HIV-1)-Mediated Apoptosis: New Therapeutic Targets. Viruses 2014, 6, 3181–3227. [Google Scholar] [CrossRef] [PubMed]
- Lester, R.T.; Yao, X.-D.; Ball, T.B.; McKinnon, L.R.; Kaul, R.; Wachihi, C.; Jaoko, W.; Plummer, F.A.; Rosenthal, K.L. Toll-like Receptor Expression and Responsiveness Are Increased in Viraemic HIV-1 Infection. AIDS Lond. Engl. 2008, 22, 685–694. [Google Scholar] [CrossRef]
- Falschlehner, C.; Schaefer, U.; Walczak, H. Following TRAIL’s Path in the Immune System. Immunology 2009, 127, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.E.; El-Deiry, W.S. Regulation of the Human TRAIL Gene. Cancer Biol. Ther. 2012, 13, 1143–1151. [Google Scholar] [CrossRef]
- Svanberg, C.; Nyström, S.; Govender, M.; Bhattacharya, P.; Che, K.F.; Ellegård, R.; Shankar, E.M.; Larsson, M. HIV-1 Induction of Tolerogenic Dendritic Cells Is Mediated by Cellular Interaction with Suppressive T Cells. Front. Immunol. 2022, 13, 790276. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Baban, B.; Johnson, B.A.; Mellor, A.L. Dendritic Cells, Indoleamine 2,3 Dioxygenase and Acquired Immune Privilege. Int. Rev. Immunol. 2010, 29, 133–155. [Google Scholar] [CrossRef] [PubMed]
- Acovic, A.; Simovic Markovic, B.; Gazdic, M.; Arsenijevic, A.; Jovicic, N.; Gajovic, N.; Jovanovic, M.; Zdravkovic, N.; Kanjevac, T.; Harrell, C.R.; et al. Indoleamine 2,3-Dioxygenase-Dependent Expansion of T-Regulatory Cells Maintains Mucosal Healing in Ulcerative Colitis. Ther. Adv. Gastroenterol. 2018, 11, 1756284818793558. [Google Scholar] [CrossRef]
- Hirayama, D.; Iida, T.; Nakase, H. The Phagocytic Function of Macrophage-Enforcing Innate Immunity and Tissue Homeostasis. Int. J. Mol. Sci. 2017, 19, 92. [Google Scholar] [CrossRef]
- Parihar, A.; Eubank, T.D.; Doseff, A.I. Monocytes and Macrophages Regulate Immunity through Dynamic Networks of Survival and Cell Death. J. Innate Immun. 2010, 2, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Mass, E.; Nimmerjahn, F.; Kierdorf, K.; Schlitzer, A. Tissue-Specific Macrophages: How They Develop and Choreograph Tissue Biology. Nat. Rev. Immunol. 2023, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Schulz, C.; Perdiguero, E.G.; Chorro, L.; Szabo-Rogers, H.; Cagnard, N.; Kierdorf, K.; Prinz, M.; Wu, B.; Jacobsen, S.E.W.; Pollard, J.W.; et al. A Lineage of Myeloid Cells Independent of Myb and Hematopoietic Stem Cells. Science 2012, 336, 86–90. [Google Scholar] [CrossRef]
- Hashimoto, D.; Chow, A.; Noizat, C.; Teo, P.; Beasley, M.B.; Leboeuf, M.; Becker, C.D.; See, P.; Price, J.; Lucas, D.; et al. Tissue-Resident Macrophages Self-Maintain Locally throughout Adult Life with Minimal Contribution from Circulating Monocytes. Immunity 2013, 38, 792–804. [Google Scholar] [CrossRef]
- Bajgar, A.; Krejčová, G. On the Origin of the Functional Versatility of Macrophages. Front. Physiol. 2023, 14, 246. [Google Scholar] [CrossRef] [PubMed]
- Koppensteiner, H.; Brack-Werner, R.; Schindler, M. Macrophages and Their Relevance in Human Immunodeficiency Virus Type I Infection. Retrovirology 2012, 9, 82. [Google Scholar] [CrossRef] [PubMed]
- Wu, L. Biology of HIV Mucosal Transmission. Curr. Opin. HIV AIDS 2008, 3, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Shen, R.; Richter, H.E.; Smith, P.D. Interactions between HIV-1 and Mucosal Cells in the Female Reproductive Tract. Am. J. Reprod. Immunol. 2014, 71, 608–617. [Google Scholar] [CrossRef]
- Teer, E.; Joseph, D.E.; Glashoff, R.H.; Faadiel Essop, M. Monocyte/Macrophage-Mediated Innate Immunity in HIV-1 Infection: From Early Response to Late Dysregulation and Links to Cardiovascular Diseases Onset. Virol. Sin. 2021, 36, 565–576. [Google Scholar] [CrossRef]
- Bertram, K.M.; Tong, O.; Royle, C.; Turville, S.G.; Nasr, N.; Cunningham, A.L.; Harman, A.N. Manipulation of Mononuclear Phagocytes by HIV: Implications for Early Transmission Events. Front. Immunol. 2019, 10, 2263. [Google Scholar] [CrossRef] [PubMed]
- Vijay, K. Toll-like Receptors in Immunity and Inflammatory Diseases: Past, Present, and Future. Int. Immunopharmacol. 2018, 59, 391–412. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-Like Receptor Signaling Pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [PubMed]
- Duan, T.; Du, Y.; Xing, C.; Wang, H.Y.; Wang, R.-F. Toll-Like Receptor Signaling and Its Role in Cell-Mediated Immunity. Front. Immunol. 2022, 13, 812774. [Google Scholar] [CrossRef]
- Sefik, E.; Qu, R.; Junqueira, C.; Kaffe, E.; Mirza, H.; Zhao, J.; Brewer, J.R.; Han, A.; Steach, H.R.; Israelow, B.; et al. Inflammasome Activation in Infected Macrophages Drives COVID-19 Pathology. Nature 2022, 606, 585–593. [Google Scholar] [CrossRef]
- Roh, J.S.; Sohn, D.H. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune Netw. 2018, 18, e27. [Google Scholar] [CrossRef] [PubMed]
- Nardacci, R.; Amendola, A.; Ciccosanti, F.; Corazzari, M.; Esposito, V.; Vlassi, C.; Taibi, C.; Fimia, G.M.; Del Nonno, F.; Ippolito, G.; et al. Autophagy Plays an Important Role in the Containment of HIV-1 in Nonprogressor-Infected Patients. Autophagy 2014, 10, 1167–1178. [Google Scholar] [CrossRef] [PubMed]
- Dinkins, C.; Arko-Mensah, J.; Deretic, V. Autophagy and HIV. Semin. Cell. Dev. Biol. 2010, 21, 712–718. [Google Scholar] [CrossRef]
- Meulendyke, K.A.; Croteau, J.D.; Zink, M.C. HIV Life Cycle, Innate Immunity, and Autophagy in the Central Nervous System. Curr. Opin. HIV AIDS 2014, 9, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Zhen, A.; Krutzik, S.R.; Levin, B.R.; Kasparian, S.; Zack, J.A.; Kitchen, S.G. CD4 Ligation on Human Blood Monocytes Triggers Macrophage Differentiation and Enhances HIV Infection. J. Virol. 2014, 88, 9934–9946. [Google Scholar] [CrossRef] [PubMed]
- Richards, K.H.; Aasa-Chapman, M.M.; McKnight, Á.; Clapham, P.R. Modulation of HIV-1 Macrophage-Tropism among R5 Envelopes Occurs before Detection of Neutralizing Antibodies. Retrovirology 2010, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Kruize, Z.; Kootstra, N.A. The Role of Macrophages in HIV-1 Persistence and Pathogenesis. Front. Microbiol. 2019, 10, 2828. [Google Scholar] [CrossRef] [PubMed]
- Lopez, P.; Koh, W.H.; Hnatiuk, R.; Murooka, T.T. HIV Infection Stabilizes Macrophage-T Cell Interactions To Promote Cell-Cell HIV Spread. J. Virol. 2019, 93, e00805-19. [Google Scholar] [CrossRef] [PubMed]
- Clayton, K.L.; Collins, D.R.; Lengieza, J.; Ghebremichael, M.; Dotiwala, F.; Lieberman, J.; Walker, B.D. Resistance of HIV-Infected Macrophages to CD8 T Lymphocyte-Mediated Killing Drives Immune Activation. Nat. Immunol. 2018, 19, 475–486. [Google Scholar] [CrossRef]
- Devanathan, A.S.; Pirone, J.R.; Akkina, R.; Remling-Mulder, L.; Luciw, P.; Adamson, L.; Garcia, J.V.; Kovarova, M.; White, N.R.; Schauer, A.P.; et al. Antiretroviral Penetration across Three Preclinical Animal Models and Humans in Eight Putative HIV Viral Reservoirs. Antimicrob. Agents Chemother. 2019, 64, e01639-19. [Google Scholar] [CrossRef]
- Colino, C.I.; Lanao, J.M.; Gutierrez-Millan, C. Targeting of Hepatic Macrophages by Therapeutic Nanoparticles. Front. Immunol. 2020, 11, 218. [Google Scholar] [CrossRef] [PubMed]
- Hammonds, J.E.; Beeman, N.; Ding, L.; Takushi, S.; Francis, A.C.; Wang, J.-J.; Melikyan, G.B.; Spearman, P. Siglec-1 Initiates Formation of the Virus-Containing Compartment and Enhances Macrophage-to-T Cell Transmission of HIV-1. PLoS Pathog. 2017, 13, e1006181. [Google Scholar] [CrossRef]
- Chu, H.; Wang, J.-J.; Qi, M.; Yoon, J.-J.; Wen, X.; Chen, X.; Ding, L.; Spearman, P. The Intracellular Virus-Containing Compartments in Primary Human Macrophages Are Largely Inaccessible to Antibodies and Small Molecules. PLoS ONE 2012, 7, e35297. [Google Scholar] [CrossRef]
- Abbas, W.; Herbein, G. T-Cell Signaling in HIV-1 Infection. Open Virol. J. 2013, 7, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Courtney, A.H.; Lo, W.-L.; Weiss, A. TCR signaling: Mechanisms of initiation and propagation. Trends Biochem. Sci. 2018, 43, 108–123. [Google Scholar] [CrossRef] [PubMed]
- Pino, S.C.; O’Sullivan-Murphy, B.; Lidstone, E.A.; Thornley, T.B.; Jurczyk, A.; Urano, F.; Greiner, D.L.; Mordes, J.P.; Rossini, A.A.; Bortell, R. Protein Kinase C Signaling during T Cell Activation Induces the Endoplasmic Reticulum Stress Response. Cell. Stress. Chaperones 2008, 13, 421–434. [Google Scholar] [CrossRef]
- Oh, H.; Ghosh, S. NF-ΚB: Roles and Regulation In Different CD4+ T Cell Subsets. Immunol. Rev. 2013, 252, 41–51. [Google Scholar] [CrossRef]
- Perdomo-Celis, F.; Taborda, N.A.; Rugeles, M.T. CD8+ T-Cell Response to HIV Infection in the Era of Antiretroviral Therapy. Front. Immunol. 2019, 10, 1896. [Google Scholar] [CrossRef]
- McBrien, J.B.; Kumar, N.A.; Silvestri, G. Mechanisms of CD8+ T Cell-Mediated Suppression of HIV/SIV Replication. Eur. J. Immunol. 2018, 48, 898–914. [Google Scholar] [CrossRef] [PubMed]
- Juno, J.A.; van Bockel, D.; Kent, S.J.; Kelleher, A.D.; Zaunders, J.J.; Munier, C.M.L. Cytotoxic CD4 T Cells—Friend or Foe during Viral Infection? Front. Immunol. 2017, 8, 19. [Google Scholar] [CrossRef]
- Bhat, P.; Leggatt, G.; Waterhouse, N.; Frazer, I.H. Interferon-γ Derived from Cytotoxic Lymphocytes Directly Enhances Their Motility and Cytotoxicity. Cell. Death Dis. 2017, 8, e2836. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Brown, H.M.; Hwang, S. Direct Antiviral Mechanisms of Interferon-Gamma. Immune Netw. 2018, 18, e33. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.; Wu, L. Different Subsets of T Cells, Memory, Effector Functions, and CAR-T Immunotherapy. Cancers 2016, 8, 36. [Google Scholar] [CrossRef]
- Yuzefpolskiy, Y.; Baumann, F.M.; Kalia, V.; Sarkar, S. Early CD8 T-Cell Memory Precursors and Terminal Effectors Exhibit Equipotent in Vivo Degranulation. Cell. Mol. Immunol. 2015, 12, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Pennock, N.D.; White, J.T.; Cross, E.W.; Cheney, E.E.; Tamburini, B.A.; Kedl, R.M. T Cell Responses: Naïve to Memory and Everything in Between. Adv. Physiol. Educ. 2013, 37, 273–283. [Google Scholar] [CrossRef]
- Remakus, S.; Sigal, L.J. Memory CD8+ T Cell Protection. Adv. Exp. Med. Biol. 2013, 785, 77–86. [Google Scholar] [CrossRef]
- Vajpayee, M.; Negi, N.; Kurapati, S. The Enduring Tale of T Cells in HIV Immunopathogenesis. Indian J. Med. Res. 2013, 138, 682–699. [Google Scholar] [PubMed]
- Subra, C.; Trautmann, L. Role of T Lymphocytes in HIV Neuropathogenesis. Curr. HIV/AIDS Rep. 2019, 16, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.S.; Cox, M.A.; Zajac, A.J. T-Cell Exhaustion: Characteristics, Causes and Conversion. Immunology 2010, 129, 474–481. [Google Scholar] [CrossRef]
- Bucks, C.M.; Norton, J.A.; Boesteanu, A.C.; Mueller, Y.M.; Katsikis, P.D. Chronic Antigen Stimulation Alone Is Sufficient to Drive CD8+ T Cell Exhaustion. J. Immunol. 2009, 182, 6697–6708. [Google Scholar] [CrossRef] [PubMed]
- Mbongue, J.C.; Nicholas, D.A.; Torrez, T.W.; Kim, N.-S.; Firek, A.F.; Langridge, W.H.R. The Role of Indoleamine 2, 3-Dioxygenase in Immune Suppression and Autoimmunity. Vaccines 2015, 3, 703–729. [Google Scholar] [CrossRef] [PubMed]
- Boasso, A.; Herbeuval, J.-P.; Hardy, A.W.; Anderson, S.A.; Dolan, M.J.; Fuchs, D.; Shearer, G.M. HIV Inhibits CD4+ T-Cell Proliferation by Inducing Indoleamine 2,3-Dioxygenase in Plasmacytoid Dendritic Cells. Blood 2007, 109, 3351–3359. [Google Scholar] [CrossRef]
- Ziegler, S.; Altfeld, M. Sex Differences in HIV-1-Mediated Immunopathology. Curr. Opin. HIV AIDS 2016, 11, 209. [Google Scholar] [CrossRef] [PubMed]
- Santinelli, L.; Ceccarelli, G.; Borrazzo, C.; Innocenti, G.P.; Frasca, F.; Cavallari, E.N.; Celani, L.; Nonne, C.; Mastroianni, C.M.; d’Ettorre, G. Sex-Related Differences in Markers of Immune Activation in Virologically Suppressed HIV-Infected Patients. Biol. Sex. Differ. 2020, 11, 23. [Google Scholar] [CrossRef] [PubMed]
- Anastos, K.; Gange, S.J.; Lau, B.; Weiser, B.; Detels, R.; Giorgi, J.V.; Margolick, J.B.; Cohen, M.; Phair, J.; Melnick, S.; et al. Association of Race and Gender with HIV-1 RNA Levels and Immunologic Progression. J. Acquir. Immune Defic. Syndr. 2000, 24, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Portales, P.; Clot, J.; Corbeau, P. Sex Differences in HIV-1 Viral Load Due to Sex Difference in CCR5 Expression. Ann. Intern. Med. 2001, 134, 81–82. [Google Scholar] [CrossRef]
- Prodger, J.L.; Gray, R.; Kigozi, G.; Nalugoda, F.; Galiwango, R.; Hirbod, T.; Wawer, M.; Hofer, S.O.P.; Sewankambo, N.; Serwadda, D.; et al. Foreskin T Cell Subsets Differ Substantially from Blood with Respect to HIV Co-Receptor Expression, Inflammatory Profile and Memory Status. Mucosal Immunol. 2012, 5, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Ballweber, L.; Robinson, B.; Kreger, A.; Fialkow, M.; Lentz, G.; McElrath, M.J.; Hladik, F. Vaginal Langerhans Cells Nonproductively Transporting HIV-1 Mediate Infection of T Cells. J. Virol. 2011, 85, 13443–13447. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, Z. Male Circumcision and Human Immunodeficiency Virus Infection: An Update on Randomized Controlled Trials and Molecular Evidences. Int. J. Health Sci. 2018, 12, 1–3. [Google Scholar]
- Prodger, J.L.; Kaul, R. The Biology of How Circumcision Reduces HIV Susceptibility: Broader Implications for the Prevention Field. AIDS Res. Ther. 2017, 14, 49. [Google Scholar] [CrossRef] [PubMed]
- Weiss, H.A.; Dickson, K.E.; Agot, K.; Hankins, C.A. Male Circumcision for HIV Prevention: Current Research and Programmatic Issues. AIDS Lond. Engl. 2010, 24 (Suppl. 4), S61–S69. [Google Scholar] [CrossRef]
- Plesniarski, A.; Siddik, A.B.; Su, R.-C. The Microbiome as a Key Regulator of Female Genital Tract Barrier Function. Front. Cell. Infect. Microbiol. 2021, 11, 790627. [Google Scholar] [CrossRef] [PubMed]
- Ochiel, D.O.; Fahey, J.V.; Ghosh, M.; Haddad, S.N.; Wira, C.R. Innate Immunity in the Female Reproductive Tract: Role of Sex Hormones in Regulating Uterine Epithelial Cell Protection Against Pathogens. Curr. Womens Health Rev. 2008, 4, 102–117. [Google Scholar] [CrossRef] [PubMed]
- Tasker, C.; Ding, J.; Schmolke, M.; Rivera-Medina, A.; García-Sastre, A.; Chang, T.L. 17β-Estradiol Protects Primary Macrophages Against HIV Infection Through Induction of Interferon-Alpha. Viral Immunol. 2014, 27, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Raghupathy, R.; Szekeres-Bartho, J. Progesterone: A Unique Hormone with Immunomodulatory Roles in Pregnancy. Int. J. Mol. Sci. 2022, 23, 1333. [Google Scholar] [CrossRef] [PubMed]
- Hall, O.J.; Klein, S.L. Progesterone-Based Compounds Affect Immune Responses and Susceptibility to Infections at Diverse Mucosal Sites. Mucosal Immunol. 2017, 10, 1097–1107. [Google Scholar] [CrossRef]
- Abbai, N.S.; Wand, H.; Ramjee, G. Biological Factors That Place Women at Risk for HIV: Evidence from a Large-Scale Clinical Trial in Durban. BMC Womens Health 2016, 16, 19. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.L.; Flanagan, K.L. Sex Differences in Immune Responses. Nat. Rev. Immunol. 2016, 16, 626–638. [Google Scholar] [CrossRef] [PubMed]
- Moran, J.A.; Turner, S.R.; Marsden, M.D. Contribution of Sex Differences to HIV Immunology, Pathogenesis, and Cure Approaches. Front. Immunol. 2022, 13, 905773. [Google Scholar] [CrossRef] [PubMed]
- Addo, M.M.; Altfeld, M. Sex-Based Differences in HIV Type 1 Pathogenesis. J. Infect. Dis. 2014, 209, S86–S92. [Google Scholar] [CrossRef] [PubMed]
- Youness, A.; Miquel, C.-H.; Guéry, J.-C. Escape from X Chromosome Inactivation and the Female Predominance in Autoimmune Diseases. Int. J. Mol. Sci. 2021, 22, 1114. [Google Scholar] [CrossRef] [PubMed]
- Mousavi, M.J.; Mahmoudi, M.; Ghotloo, S. Escape from X Chromosome Inactivation and Female Bias of Autoimmune Diseases. Mol. Med. 2020, 26, 127. [Google Scholar] [CrossRef] [PubMed]
- Arnold, A.P. A General Theory of Sexual Differentiation. J. Neurosci. Res. 2017, 95, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Dhanoa, J.K.; Mukhopadhyay, C.S.; Arora, J.S. Y-Chromosomal Genes Affecting Male Fertility: A Review. Vet. World 2016, 9, 783–791. [Google Scholar] [CrossRef]
- Young, N.A.; Wu, L.-C.; Burd, C.J.; Friedman, A.K.; Kaffenberger, B.H.; Rajaram, M.V.S.; Schlesinger, L.S.; James, H.; Shupnik, M.A.; Jarjour, W.N. Estrogen Modulation of Endosome-Associated Toll-like Receptor 8: An IFNα-Independent Mechanism of Sex-Bias in Systemic Lupus Erythematosus. Clin. Immunol. 2014, 151, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Justina, V.D.; Giachini, F.R.; Sullivan, J.C.; Webb, R.C. Toll-Like Receptors Contribute to Sex Differences in Blood Pressure Regulation. J. Cardiovasc. Pharmacol. 2020, 76, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Zandieh, Z.; Amjadi, F.; Ashrafi, M.; Aflatoonian, A.; Fazeli, A.; Aflatoonian, R. The Effect of Estradiol and Progesterone on Toll Like Receptor Gene Expression in A Human Fallopian Tube Epithelial Cell Line. Cell. J. Yakhteh 2016, 17, 678–691. [Google Scholar]
- Cunningham, M.A.; Naga, O.S.; Eudaly, J.G.; Scott, J.L.; Gilkeson, G.S. Estrogen Receptor Alpha Modulates Toll-like Receptor Signaling in Murine Lupus. Clin. Immunol. 2012, 144, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Khan, D.; Ansar Ahmed, S. The Immune System Is a Natural Target for Estrogen Action: Opposing Effects of Estrogen in Two Prototypical Autoimmune Diseases. Front. Immunol. 2016, 6, 635. [Google Scholar] [CrossRef]
- Young, N.A.; Valiente, G.R.; Hampton, J.M.; Wu, L.-C.; Burd, C.J.; Willis, W.L.; Bruss, M.; Steigelman, H.; Gotsatsenko, M.; Amici, S.A.; et al. Estrogen-Regulated STAT1 Activation Promotes TLR8 Expression to Facilitate Signaling via MicroRNA-21 in Systemic Lupus Erythematosus. Clin. Immunol. 2017, 176, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Rettew, J.A.; Huet-Hudson, Y.M.; Marriott, I. Testosterone Reduces Macrophage Expression in the Mouse of Toll-Like Receptor 4, a Trigger for Inflammation and Innate Immunity. Biol. Reprod. 2008, 78, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Ben-Batalla, I.; Vargas-Delgado, M.E.; von Amsberg, G.; Janning, M.; Loges, S. Influence of Androgens on Immunity to Self and Foreign: Effects on Immunity and Cancer. Front. Immunol. 2020, 11, 1184. [Google Scholar] [CrossRef] [PubMed]
- Harding, A.T.; Heaton, N.S. The Impact of Estrogens and Their Receptors on Immunity and Inflammation during Infection. Cancers 2022, 14, 909. [Google Scholar] [CrossRef] [PubMed]
- Alesci, A.; Nicosia, N.; Fumia, A.; Giorgianni, F.; Santini, A.; Cicero, N. Resveratrol and Immune Cells: A Link to Improve Human Health. Molecules 2022, 27, 424. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.H.; Huang, T.; Xiao, J.W.; Gu, R.C.; Ouyang, J.; Wu, G.; Liao, H. Estrogen Signaling Effects on Muscle-Specific Immune Responses through Controlling the Recruitment and Function of Macrophages and T Cells. Skelet. Muscle 2019, 9, 20. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in Inflammation, Immunity, and Disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Zhou, J.; Webb, D.C. Estrogen Stimulates Th2 Cytokine Production and Regulates the Compartmentalisation of Eosinophils during Allergen Challenge in a Mouse Model of Asthma. Int. Arch. Allergy Immunol. 2012, 158, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Joseph, K.; Tholanikunnel, B.G.; Kaplan, A.P. Cytokine and Estrogen Stimulation of Endothelial Cells Augments Activation of the Prekallikrein-High Molecular Weight Kininogen Complex: Implications for Hereditary Angioedema. J. Allergy Clin. Immunol. 2017, 140, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Shivers, K.-Y.; Amador, N.; Abrams, L.; Hunter, D.; Jenab, S.; Quiñones-Jenab, V. Estrogen Alters Baseline and Inflammatory-Induced Cytokine Levels Independent from Hypothalamic–Pituitary–Adrenal Axis Activity. Cytokine 2015, 72, 121–129. [Google Scholar] [CrossRef]
- Cutolo, M.; Sulli, A.; Straub, R.H. Estrogen Metabolism and Autoimmunity. Autoimmun. Rev. 2012, 11, A460–A464. [Google Scholar] [CrossRef] [PubMed]
- Javadian, A.; Salehi, E.; Bidad, K.; Sahraian, M.A.; Izad, M. Effect of Estrogen on Th1, Th2 and Th17 Cytokines Production by Proteolipid Protein and PHA Activated Peripheral Blood Mononuclear Cells Isolated from Multiple Sclerosis Patients. Arch. Med. Res. 2014, 45, 177–182. [Google Scholar] [CrossRef]
- Gao, H.; Liu, L.; Zhao, Y.; Hara, H.; Chen, P.; Xu, J.; Tang, J.; Wei, L.; Li, Z.; Cooper, D.K.C.; et al. Human IL-6, IL-17, IL-1β, and TNF-α Differently Regulate the Expression of pro-Inflammatory Related Genes, Tissue Factor, and Swine Leukocyte Antigen Class I in Porcine Aortic Endothelial Cells. Xenotransplantation 2017, 24, e12291. [Google Scholar] [CrossRef]
- Hagman, S.; Mäkinen, A.; Ylä-Outinen, L.; Huhtala, H.; Elovaara, I.; Narkilahti, S. Effects of Inflammatory Cytokines IFN-γ, TNF-α and IL-6 on the Viability and Functionality of Human Pluripotent Stem Cell-Derived Neural Cells. J. Neuroimmunol. 2019, 331, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Theofilopoulos, A.N.; Koundouris, S.; Kono, D.H.; Lawson, B.R. The Role of IFN-Gamma in Systemic Lupus Erythematosus: A Challenge to the Th1/Th2 Paradigm in Autoimmunity. Arthritis Res. Ther. 2001, 3, 136. [Google Scholar] [CrossRef] [PubMed]
- Chaouali, M.; Azaiez, M.B.; Tezeghdenti, A.; Yacoubi-Oueslati, B.; Ghazouani, E.; Kochkar, R. High Levels of Proinflammatory Cytokines IL-6, IL-8, TNF-A, IL-23, and IFN-γ in Tunisian Patients with Type 1 Autoimmune Hepatitis. Eur. Cytokine Netw. 2020, 31, 94–103. [Google Scholar] [CrossRef]
- Paterni, I.; Granchi, C.; Katzenellenbogen, J.A.; Minutolo, F. Estrogen Receptors Alpha (ERα) and Beta (ERβ): Subtype-Selective Ligands and Clinical Potential. Steroids 2014, 0, 13–29. [Google Scholar] [CrossRef]
- Fuentes, N.; Silveyra, P. Estrogen Receptor Signaling Mechanisms. Adv. Protein Chem. Struct. Biol. 2019, 116, 135–170. [Google Scholar] [CrossRef]
- Moulton, V.R. Sex Hormones in Acquired Immunity and Autoimmune Disease. Front. Immunol. 2018, 9, 2279. [Google Scholar] [CrossRef] [PubMed]
- Atanaskova, N.; Keshamouni, V.G.; Krueger, J.S.; Schwartz, J.A.; Miller, F.; Reddy, K.B. MAP Kinase/Estrogen Receptor Cross-Talk Enhances Estrogen-Mediated Signaling and Tumor Growth but Does Not Confer Tamoxifen Resistance. Oncogene 2002, 21, 4000–4008. [Google Scholar] [CrossRef]
- Liu, A.; Zhang, D.; Yang, X.; Song, Y. Estrogen Receptor Alpha Activates MAPK Signaling Pathway to Promote the Development of Endometrial Cancer. J. Cell. Biochem. 2019, 120, 17593–17601. [Google Scholar] [CrossRef] [PubMed]
- Filardo, E.J.; Quinn, J.A.; Frackelton, A.R.; Bland, K.I. Estrogen Action via the G Protein-Coupled Receptor, GPR30: Stimulation of Adenylyl Cyclase and CAMP-Mediated Attenuation of the Epidermal Growth Factor Receptor-to-MAPK Signaling Axis. Mol. Endocrinol. 2002, 16, 70–84. [Google Scholar] [CrossRef] [PubMed]
- Petrova, T.; Pesic, J.; Pardali, K.; Gaestel, M.; Arthur, J.S.C. P38 MAPK Signalling Regulates Cytokine Production in IL-33 Stimulated Type 2 Innate Lymphoid Cells. Sci. Rep. 2020, 10, 3479. [Google Scholar] [CrossRef] [PubMed]
- Morrison, D.K. MAP Kinase Pathways. Cold Spring Harb. Perspect. Biol. 2012, 4, a011254. [Google Scholar] [CrossRef] [PubMed]
- Atsaves, V.; Leventaki, V.; Rassidakis, G.Z.; Claret, F.X. AP-1 Transcription Factors as Regulators of Immune Responses in Cancer. Cancers 2019, 11, 1037. [Google Scholar] [CrossRef] [PubMed]
- Guma, M.; Firestein, G.S. C-Jun N-Terminal Kinase in Inflammation and Rheumatic Diseases. Open. Rheumatol. J. 2012, 6, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Kyriakis, J.M. Activation of the AP-1 Transcription Factor by Inflammatory Cytokines of the TNF Family. Gene Expr. 2018, 7, 217–231. [Google Scholar]
- Mebratu, Y.; Tesfaigzi, Y. How ERK1/2 Activation Controls Cell Proliferation and Cell Death Is Subcellular Localization the Answer? Cell. Cycle Georget. Tex. 2009, 8, 1168–1175. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.Y.; Kim, C.G.; Lim, Y.; Lee, Y.H. The ETS Family Transcription Factor ELK-1 Regulates Induction of the Cell Cycle-Regulatory Gene P21Waf1/Cip1 and the BAX Gene in Sodium Arsenite-Exposed Human Keratinocyte HaCaT Cells. J. Biol. Chem. 2011, 286, 26860–26872. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, J.D.; Ammit, A.J.; Clark, A.R. MAPK P38 Regulates Inflammatory Gene Expression via Tristetraprolin: Doing Good by Stealth. Int. J. Biochem. Cell. Biol. 2018, 94, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Rao, S. Sex Differences in HIV-1 Persistence and the Implications for a Cure. Front. Glob. Womens Health 2022, 3, 942345. [Google Scholar] [CrossRef] [PubMed]
- Gianella, S.; Rawlings, S.A.; Dobrowolski, C.; Nakazawa, M.; Chaillon, A.; Strain, M.; Layman, L.; Caballero, G.; Scully, E.; Scott, B.; et al. Sex Differences in Human Immunodeficiency Virus Persistence and Reservoir Size During Aging. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2021, 75, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Kissick, H.T.; Sanda, M.G.; Dunn, L.K.; Pellegrini, K.L.; On, S.T.; Noel, J.K.; Arredouani, M.S. Androgens Alter T-Cell Immunity by Inhibiting T-Helper 1 Differentiation. Proc. Natl. Acad. Sci. USA 2014, 111, 9887–9892. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, V.D.; Cephus, J.-Y.; Norlander, A.E.; Chowdhury, N.U.; Zhang, J.; Ceneviva, Z.J.; Tannous, E.; Polosukhin, V.V.; Putz, N.D.; Wickersham, N.; et al. Androgen Receptor Signaling Promotes Treg Suppressive Function during Allergic Airway Inflammation. J. Clin. Invest. 2022, 132, e153397. [Google Scholar] [CrossRef] [PubMed]
- Henze, L.; Schwinge, D.; Schramm, C. The Effects of Androgens on T Cells: Clues to Female Predominance in Autoimmune Liver Diseases? Front. Immunol. 2020, 11, 1567. [Google Scholar] [CrossRef]
- Schmidt, S.V.; Nino-Castro, A.C.; Schultze, J.L. Regulatory Dendritic Cells: There Is More than Just Immune Activation. Front. Immunol. 2012, 3, 274. [Google Scholar] [CrossRef] [PubMed]
- Becerra-Diaz, M.; Song, M.; Heller, N. Androgen and Androgen Receptors as Regulators of Monocyte and Macrophage Biology in the Healthy and Diseased Lung. Front. Immunol. 2020, 11, 1698. [Google Scholar] [CrossRef] [PubMed]
- Trigunaite, A.; Dimo, J.; Jørgensen, T.N. Suppressive Effects of Androgens on the Immune System. Cell. Immunol. 2015, 294, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Sun, Y.; Huang, C.-P.; Chen, L.; Liu, B.; You, B.; Wang, Z.; Chang, C. High Dose Androgen Suppresses Natural Killer Cytotoxicity of Castration-Resistant Prostate Cancer Cells via Altering AR/CircFKBP5/MiRNA-513a-5p/PD-L1 Signals. Cell. Death Dis. 2022, 13, 746. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Lin, H.; Li, G.; Jin, R.-A.; Xu, J.; Sun, Y.; Ma, W.-L.; Yeh, S.; Cai, X.; Chang, C. Targeting Androgen Receptor (AR)→IL12A Signal Enhances Efficacy of Sorafenib Plus NK Cells-Immunotherapy to Better Suppress HCC Progression. Mol. Cancer Ther. 2016, 15, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Griesbeck, M.; Scully, E.; Altfeld, M. Sex and Gender Differences in HIV-1 Infection. Clin. Sci. Lond. Engl. 1979 2016, 130, 1435–1451. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, A.; Silvestri, G. Host-Pathogen Interaction in HIV Infection. Curr. Opin. Immunol. 2013, 25, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Shukla, E.; Chauhan, R. Host-HIV-1 Interactome: A Quest for Novel Therapeutic Intervention. Cells 2019, 8, 1155. [Google Scholar] [CrossRef] [PubMed]
- Herskovitz, J.; Gendelman, H.E. HIV and the Macrophage: From Cell Reservoirs to Drug Delivery to Viral Eradication. J. Neuroimmune Pharmacol. Off. J. Soc. NeuroImmune Pharmacol. 2019, 14, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.; Mayuresh Patkar, M.; Momin, M.; Omri, A. Macrophage Targeted Nanocarrier Delivery Systems in HIV Therapeutics. Expert. Opin. Drug. Deliv. 2020, 17, 903–918. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masenga, S.K.; Mweene, B.C.; Luwaya, E.; Muchaili, L.; Chona, M.; Kirabo, A. HIV–Host Cell Interactions. Cells 2023, 12, 1351. https://doi.org/10.3390/cells12101351
Masenga SK, Mweene BC, Luwaya E, Muchaili L, Chona M, Kirabo A. HIV–Host Cell Interactions. Cells. 2023; 12(10):1351. https://doi.org/10.3390/cells12101351
Chicago/Turabian StyleMasenga, Sepiso K., Bislom C. Mweene, Emmanuel Luwaya, Lweendo Muchaili, Makondo Chona, and Annet Kirabo. 2023. "HIV–Host Cell Interactions" Cells 12, no. 10: 1351. https://doi.org/10.3390/cells12101351