
Platelet Versus Megakaryocyte: Who Is the Real Bandleader of Thromboinflammation in Sepsis?

 , ,
, ,
Abstract
:1. Introduction
2. Immunothrombosis or Thromboinflammation?

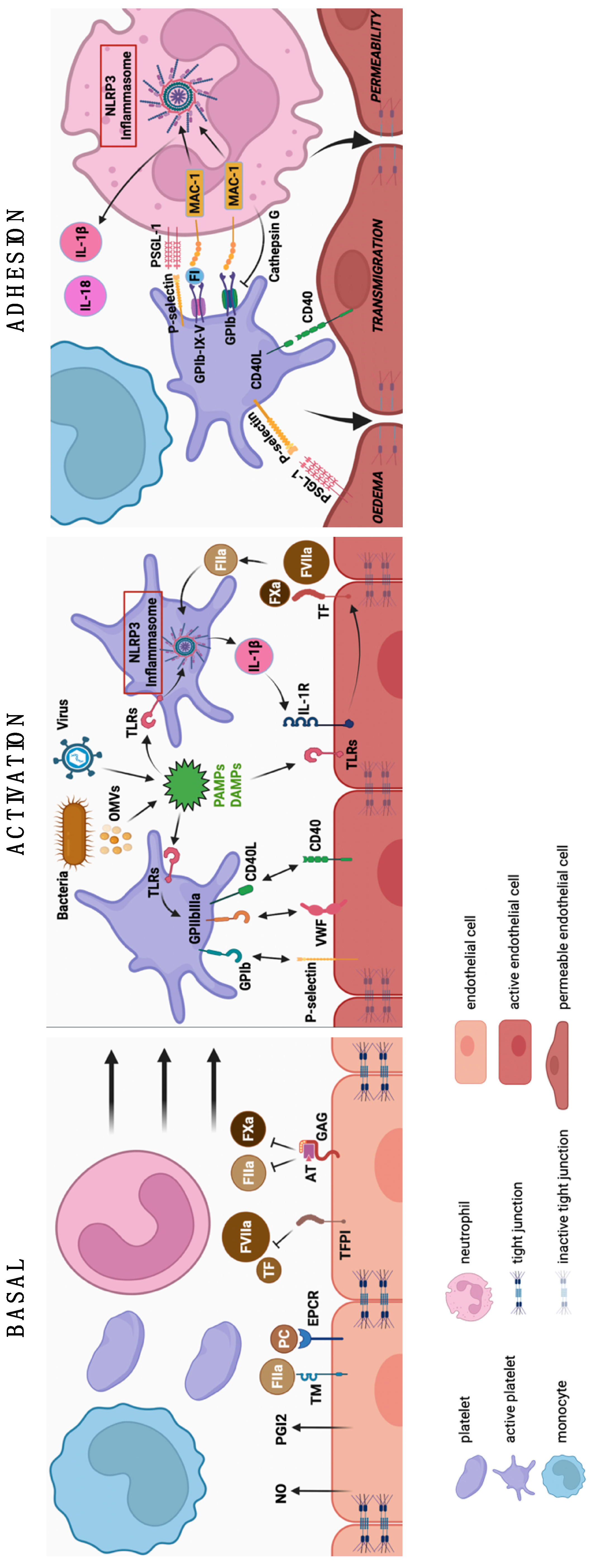{kind=link}
{kind=link}
| POSITIVE EFFECTS | REFERENCES | NEGATIVE EFFECTS | REFERENCES |
|---|---|---|---|
| Promote leukocyte transmigration by interacting with endothelial cells | [14,15,16,17,18,19] | Neutrophil activation by aggregating with them | [20,21,22,23,24,25,26] |
| Fill endothelial fenestration with TLT-1 to prevent edema and microhemorrhage | [27,28] | Soluble TLT-1 precipitates plasma fibrinogen | [27,29] |
| Catch pathogens thanks to their integrins and membrane receptors | [30,31,32,33,34,35,36] | Triggers NETs and acute thrombosis | [37,38,39,40,41,42,43,44,45] |
| Phagocyte pathogens | [46,47,48,49,50,51] | Procoagulant activity of their membrane Production of microparticles | [52,53,54,55,56] |
| Production of interleukin (IL)1β and IL18 in response to PAMPs and DAMPs | [19,57,58,59] | Exacerbation of leukocyte inflammasome | [38,60,61] |
| Tissue repair and regeneration by the local release of the α-granule content | [62,63,64,65,66,67] | Endothelial barrier disruption and organ damage caused by hemorrhage and microthrombosis | [5,7,20,68,69,70,71,72,73,74] |
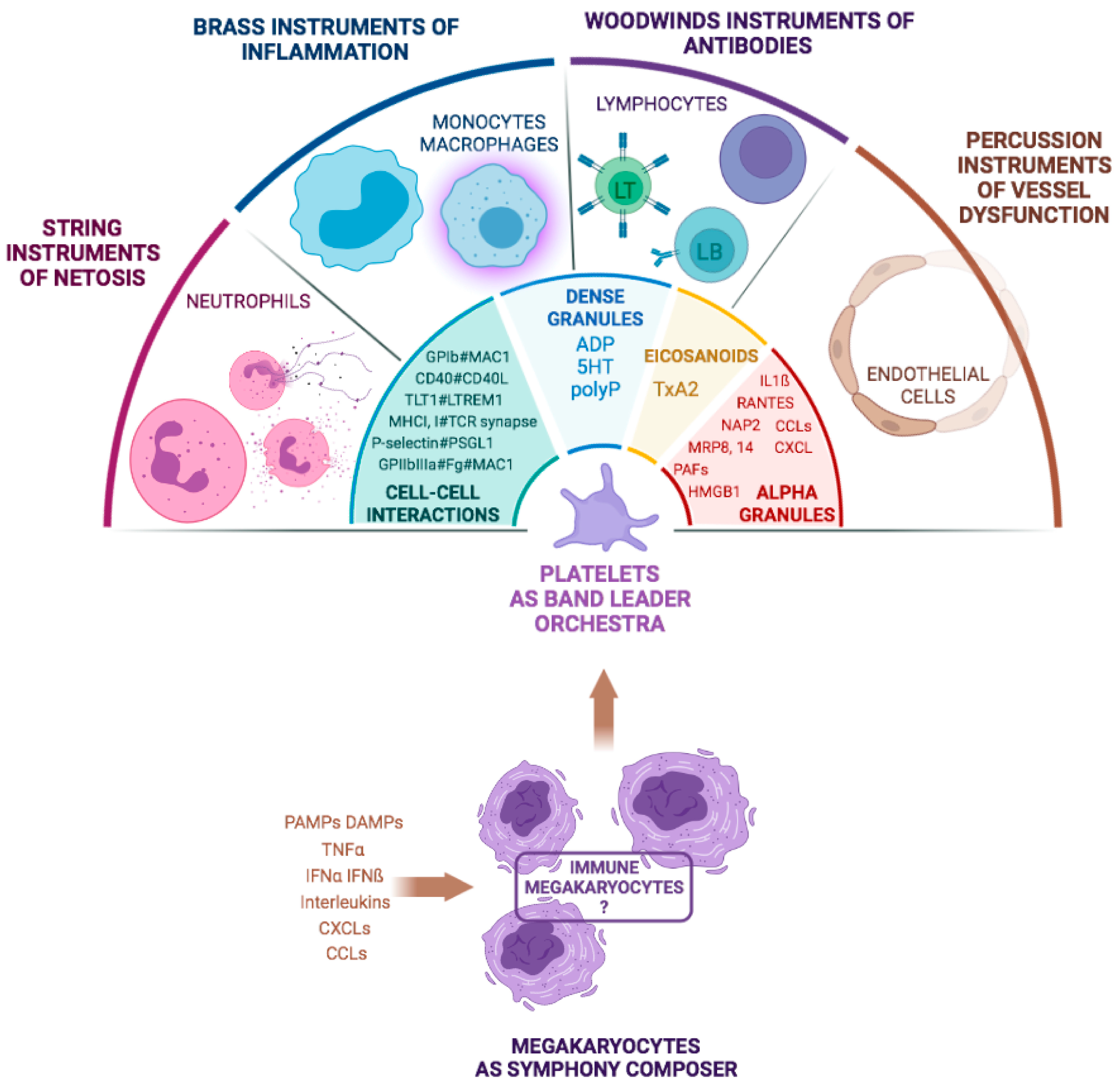
3. Stakeholders in the Septic Scene
3.1. From Leuko–Platelet Interactions to the Involvement in NETosis
3.2. Platelet and Endothelial Cell (EC) Crosstalk during Sepsis
3.3. A Platelet Inflammasome during Sepsis?
3.4. Effects of Antiplatelet Drugs in Sepsis
4. Vaccine-Induced Immune Thrombotic Thrombocytopenia: An Example of Immunothrombosis Involving Platelets
5. Relevance of Platelet-Derived Extracellular Vesicles
6. A Role for Specialized Megakaryocytes during Sepsis
6.1. Occurrence of a Specific MK Population in Sepsis?
6.2. Differential Protein Content in Megakaryocytes and Platelets in Viral and Bacterial Sepsis
7. Conclusions
Funding
Conflicts of Interest
References
- Palacios-Acedo, A.L.; Mège, D.; Crescence, L.; Dignat-George, F.; Dubois, C.; Panicot-Dubois, L. Platelets, Thrombo-Inflammation, and Cancer: Collaborating With the Enemy. Front. Immunol. 2019, 10, 1805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oikonomou, E.; Leopoulou, M.; Theofilis, P.; Antonopoulos, A.; Siasos, G.; Latsios, G.; Mystakidi, V.C.; Antoniades, C.; Tousoulis, D. A link between inflammation and thrombosis in atherosclerotic cardiovascular diseases: Clinical and therapeutic implications. Atherosclerosis 2020, 309, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Bültmann, A.; Li, Z.; Wagner, S.; Peluso, M.; Schönberger, T.; Weis, C.; Konrad, I.; Stellos, K.; Massberg, S.; Nieswandt, B.; et al. Impact of glycoprotein VI and platelet adhesion on atherosclerosis—A possible role of fibronectin. J. Mol. Cell. Cardiol. 2010, 49, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Burkard, P.; Vögtle, T.; Nieswandt, B. Platelets in Thrombo-Inflammation: Concepts, Mechanisms, and Therapeutic Strategies for Ischemic Stroke. Hamostaseologie 2020, 40, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Semeraro, N.; Ammollo, C.T.; Semeraro, F.; Colucci, M. Sepsis-associated disseminated intravascular coagulation and thromboembolic disease. Mediterr. J. Hematol. Infect. Dis. 2010, 2, e2010024. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.C. Disseminated intravascular coagulation: New identity as endotheliopathy-associated vascular microthrombotic disease based on in vivo hemostasis and endothelial molecular pathogenesis. Thromb. J. 2020, 18, 25. [Google Scholar] [CrossRef] [PubMed]
- Martinod, K.; Deppermann, C. Immunothrombosis and thromboinflammation in host defense and disease. Platelets 2020, 32, 314–324. [Google Scholar] [CrossRef]
- Guo, L.; Rondina, M.T. The Era of Thromboinflammation: Platelets Are Dynamic Sensors and Effector Cells During Infectious Diseases. Front. Immunol. 2019, 10, 2204. [Google Scholar] [CrossRef] [Green Version]
- Engelmann, B.; Massberg, S. Thrombosis as an intravascular effector of innate immunity. Nat. Rev. Immunol. 2013, 13, 34–45. [Google Scholar] [CrossRef]
- Tanguay, J.-F.; Geoffroy, P.; Sirois, M.G.; Libersan, D.; Kumar, A.; Schaub, R.G.; Merhi, Y. Prevention of in-stent restenosis via reduction of thrombo-inflammatory reactions with recombinant P-selectin glycoprotein ligand-1. Thromb. Haemost. 2004, 91, 1186–1193. [Google Scholar] [CrossRef]
- Blair, P.; Rex, S.; Vitseva, O.; Beaulieu, L.; Tanriverdi, K.; Chakrabarti, S.; Hayashi, C.; Genco, C.; Iafrati, M.; Freedman, J.E. Stimulation of Toll-Like Receptor 2 in Human Platelets Induces a Thromboinflammatory Response Through Activation of Phosphoinositide 3-Kinase. Circ. Res. 2009, 104, 346–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeaman, M.R. Platelets in defense against bacterial pathogens. Cell Mol. Life Sci. 2009, 67, 525–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szaba, F.M.; Smiley, S.T. Roles for thrombin and fibrin(ogen) in cytokine/chemokine production and macrophage adhesion in vivo. Blood 2002, 99, 1053–1059. [Google Scholar] [CrossRef] [Green Version]
- Gardiner, E.E.; De Luca, M.; McNally, T.; Michelson, A.D.; Andrews, R.K.; Berndt, M.C. Regulation of P-selectin binding to the neutrophil P-selectin counter-receptor P-selectin glycoprotein ligand-1 by neutrophil elastase and cathepsin G. Blood 2001, 98, 1440–1447. [Google Scholar] [CrossRef] [PubMed]
- Gorina, R.; Lyck, R.; Vestweber, D.; Engelhardt, B. β2 integrin-mediated crawling on endothelial ICAM-1 and ICAM-2 is a prerequisite for transcellular neutrophil diapedesis across the inflamed blood-brain barrier. J. Immunol. 2014, 192, 324–337. [Google Scholar] [CrossRef] [Green Version]
- Vestweber, D. How leukocytes cross the vascular endothelium. Nat. Rev. Immunol. 2015, 15, 692–704. [Google Scholar] [CrossRef]
- Wilkins, P.P.; Moore, K.L.; McEver, R.P.; Cummings, R.D. Tyrosine Sulfation of P-selectin Glycoprotein Ligand-1 Is Required for High Affinity Binding to P-selectin. J. Biol. Chem. 1995, 270, 22677–22680. [Google Scholar] [CrossRef] [Green Version]
- Dole, V.S.; Bergmeier, W.; Mitchell, H.A.; Eichenberger, S.C.; Wagner, D.D. Activated platelets induce Weibel-Palade–body secretion and leukocyte rolling in vivo: Role of P-selectin. Blood 2005, 106, 2334–2339. [Google Scholar] [CrossRef] [Green Version]
- Cornelius, D.C.; Baik, C.H.; Travis, O.K.; White, D.L.; Young, C.M.; Pierce, W.A.; Shields, C.A.; Poudel, B.; Williams, J.M. NLRP3 inflammasome activation in platelets in response to sepsis. Physiol. Rep. 2019, 7, e14073. [Google Scholar] [CrossRef]
- Massberg, S.; Grahl, L.; von Bruehl, M.-L.; Manukyan, D.; Pfeiler, S.; Goosmann, C.; Brinkmann, V.; Lorenz, M.; Bidzhekov, K.; Khandagale, A.B.; et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat. Med. 2010, 16, 887–896. [Google Scholar] [CrossRef]
- Joffre, J.; Hellman, J.; Ince, C.; Ait-Oufella, H. Endothelial Responses in Sepsis. Am. J. Respir. Crit. Care Med. 2020, 202, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Van der Poll, T.; Parker, R.I. Platelet Activation and Endothelial Cell Dysfunction. Crit. Care Clin. 2020, 36, 233–253. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Carestia, A.; Kaufman, T.; Schattner, M. Platelets: New Bricks in the Building of Neutrophil Extracellular Traps. Front. Immunol. 2016, 7, 271. [Google Scholar] [CrossRef] [Green Version]
- Maugeri, N.; Campana, L.; Gavina, M.; Covino, C.; De Metrio, M.; Panciroli, C.; Maiuri, L.; Maseri, A.; D’Angelo, A.; Bianchi, M.E.; et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J. Thromb. Haemost. 2014, 12, 2074–2088. [Google Scholar] [CrossRef]
- Caudrillier, A.; Kessenbrock, K.; Gilliss, B.M.; Nguyen, J.X.; Marques, M.B.; Monestier, M.; Toy, P.; Werb, Z.; Looney, M.R. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J. Clin. Investig. 2012, 122, 2661–2671. [Google Scholar] [CrossRef]
- Washington, A.V.; Schubert, R.L.; Quigley, L.; Disipio, T.; Feltz, R.; Cho, E.H.; McVicar, D.W. A TREM family member, TLT-1, is found exclusively in the alpha-granules of megakaryocytes and platelets. Blood 2004, 104, 1042–1047. [Google Scholar] [CrossRef] [Green Version]
- Diacovo, T.G.; Roth, S.J.; Buccola, J.M.; Bainton, D.F.; Springer, T.A. Neutrophil rolling, arrest, and transmigration across activated, surface-adherent platelets via sequential action of P-selectin and the beta 2-integrin CD11b/CD18. Blood 1996, 88, 146–157. [Google Scholar] [CrossRef] [Green Version]
- Morales-Ortíz, J.; Deal, V.; Reyes, F.; Maldonado-Martínez, G.; Ledesma, N.; Staback, F.; Croft, C.; Pacheco, A.; Ortiz-Zuazaga, H.; Yost, C.C.; et al. Platelet-derived TLT-1 is a prognostic indicator in ALI/ARDS and prevents tissue damage in the lungs in a mouse model. Blood 2018, 132, 2495–2505. [Google Scholar] [CrossRef] [Green Version]
- Assinger, A.; Laky, M.; Schabbauer, G.; Hirschl, A.M.; Buchberger, E.; Binder, B.R.; Volf, I. Efficient phagocytosis of periodontopathogens by neutrophils requires plasma factors, platelets and TLR2. J. Thromb. Haemost. 2011, 9, 799–809. [Google Scholar] [CrossRef]
- Miyabe, K.; Sakamoto, N.; Wu, Y.H.; Mori, N.; Sakamoto, H. Effects of platelet release products on neutrophilic phagocytosis and complement receptors. Thromb. Res. 2004, 114, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, B.F.; Campbell, R.A.; Schwertz, H.; Cody, M.J.; Franks, Z.; Tolley, N.D.; Kahr, W.H.A.; Lindemann, S.; Seizer, P.; Yost, C.C.; et al. Novel Anti-bacterial Activities of β-defensin 1 in Human Platelets: Suppression of Pathogen Growth and Signaling of Neutrophil Extracellular Trap Formation. PLoS Pathog. 2011, 7, e1002355. [Google Scholar] [CrossRef] [PubMed]
- Plummer, C.; Wu, H.; Kerrigan, S.W.; Meade, G.; Cox, D.; Douglas, C.W.I. A serine-rich glycoprotein of Streptococcus sanguis mediates adhesion to platelets via GPIb. Br. J. Haematol. 2005, 129, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Ohta, T.; Takatsuka, H.; Yamaguchi, K. High-field magnetic resonance image of a huge calcified chronic subdural haematoma, so-called “armoured brain”. Acta Neurochir. 1992, 114, 151–153. [Google Scholar] [CrossRef]
- Coburn, J.; Leong, J.M.; Erban, J.K. Integrin alpha IIb beta 3 mediates binding of the Lyme disease agent Borrelia burgdorferi to human platelets. Proc. Natl. Acad. Sci. USA 1993, 90, 7059–7063. [Google Scholar] [CrossRef] [Green Version]
- Siboo, I.R.; Cheung, A.L.; Bayer, A.S.; Sullam, P.M. Clumping Factor A Mediates Binding of Staphylococcus aureus to Human Platelets. Infect. Immun. 2001, 69, 3120–3127. [Google Scholar] [CrossRef] [Green Version]
- Campos, J.; Ponomaryov, T.; De Prendergast, A.; Whitworth, K.; Smith, C.W.; Khan, A.O.; Kavanagh, D.; Brill, A. Neutrophil extracellular traps and inflammasomes cooperatively promote venous thrombosis in mice. Blood Adv. 2021, 5, 2319–2324. [Google Scholar] [CrossRef]
- Rolfes, V.; Ribeiro, L.S.; Hawwari, I.; Böttcher, L.; Rosero, N.; Maasewerd, S.; Santos, M.L.S.; Próchnicki, T.; Silva, C.M.D.S.; Wanderley, C.W.D.S.; et al. Platelets Fuel the Inflammasome Activation of Innate Immune Cells. Cell Rep. 2020, 31, 107615. [Google Scholar] [CrossRef]
- Pilsczek, F.H.; Salina, D.; Poon, K.K.H.; Fahey, C.; Yipp, B.G.; Sibley, C.D.; Robbins, S.M.; Green, F.H.Y.; Surette, M.G.; Sugai, M.; et al. A Novel Mechanism of Rapid Nuclear Neutrophil Extracellular Trap Formation in Response to Staphylococcus aureus. J. Immunol. 2010, 185, 7413–7425. [Google Scholar] [CrossRef] [Green Version]
- Yipp, B.G.; Petri, B.; Salina, D.; Jenne, C.N.; Scott, B.N.V.; Zbytnuik, L.D.; Pittman, K.; Asaduzzaman, M.; Wu, K.; Meijndert, H.C.; et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat. Med. 2012, 18, 1386–1393. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, M.; Stadler, S.C.; Correll, S.; Li, P.; Wang, D.; Hayama, R.; Leonelli, L.; Han, H.; Grigoryev, S.A.; et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J. Cell Biol. 2009, 184, 205–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leshner, M.; Wang, S.; Lewis, C.; Zheng, H.; Chen, X.A.; Santy, L.; Wang, Y. PAD4 mediated histone hypercitrullination induces heterochromatin decondensation and chromatin unfolding to form neutrophil extracellular trap-like structures. Front. Immunol. 2012, 3, 307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papayannopoulos, V.; Metzle, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skendros, P.; Mitsios, A.; Chrysanthopoulou, A.; Mastellos, D.C.; Metallidis, S.; Rafailidis, P.; Ntinopoulou, M.; Sertaridou, E.; Tsironidou, V.; Tsigalou, C.; et al. Complement and tissue factor–enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J. Clin. Investig. 2020, 130, 6151–6157. [Google Scholar] [CrossRef]
- Semeraro, F.; Ammollo, C.T.; Morrissey, J.H.; Dale, G.L.; Friese, P.; Esmon, N.L.; Esmon, C.T. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: Involvement of platelet TLR2 and TLR4. Blood 2011, 118, 1952–1961. [Google Scholar] [CrossRef] [Green Version]
- Nagasawa, T.; Nakayasu, C.; Rieger, A.M.; Barreda, D.; Somamoto, T.; Nakao, M. Phagocytosis by Thrombocytes is a Conserved Innate Immune Mechanism in Lower Vertebrates. Front. Immunol. 2014, 5, 445. [Google Scholar] [CrossRef] [Green Version]
- Koupenova, M.; Corkrey, H.A.; Vitseva, O.; Manni, G.; Pang, C.J.; Clancy, L.; Yao, C.; Rade, J.; Levy, D.; Wang, J.P.; et al. The role of platelets in mediating a response to human influenza infection. Nat. Commun. 2019, 10, 1780. [Google Scholar] [CrossRef] [Green Version]
- Onlamoon, N.; Noisakran, S.; Hsiao, H.-M.; Duncan, A.; Villinger, F.; Ansari, A.A.; Perng, O. Dengue virus–induced hemorrhage in a nonhuman primate model. Blood 2010, 115, 1823–1834. [Google Scholar] [CrossRef] [Green Version]
- Cruz-Oliveira, C.; Freire, J.M.; Conceição, T.M.; Higa, L.M.; Castanho, M.A.; Da Poian, A.T. Receptors and routes of dengue virus entry into the host cells. FEMS Microbiol. Rev. 2015, 39, 155–170. [Google Scholar] [CrossRef] [Green Version]
- Simon, A.Y.; Sutherland, M.R.; Pryzdial, E.L.G. Dengue virus binding and replication by platelets. Blood 2015, 126, 378–385. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, M.; Huang, Y.; Joshi, S.; Popa, G.J.; Mendenhall, M.D.; Wang, Q.J.; Garvy, B.A.; Myint, T.; Whiteheart, S.W. Platelets Endocytose Viral Particles and Are Activated via TLR (Toll-Like Receptor) Signaling. Arter. Thromb. Vasc. Biol. 2020, 40, 1635–1650. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Maruyama, I. Thrombomodulin: Protectorate God of the vasculature in thrombosis and inflammation. J. Thromb. Haemost. 2011, 9, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Zeerleder, S.; Schroeder, V.; Hack, C.E.; Kohler, H.P.; Wuillemin, W.A. TAFI and PAI-1 levels in human sepsis. Thromb. Res. 2006, 118, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Binette, T.M.; Taylor, F.B.; Peer, G.; Bajzar, L. Thrombin-thrombomodulin connects coagulation and fibrinolysis: More than an in vitro phenomenon. Blood 2007, 110, 3168–3175. [Google Scholar] [CrossRef]
- Fourrier, F. Severe sepsis, coagulation, and fibrinolysis: Dead end or one way? Crit. Care Med. 2012, 40, 2704–2708. [Google Scholar] [CrossRef]
- Boilard, E.; Nigrovic, P.A.; Larabee, K.; Watts, G.F.M.; Coblyn, J.S.; Weinblatt, M.E.; Massarotti, E.M.; Remold-O’Donnell, E.; Farndale, R.W.; Ware, J.; et al. Platelets Amplify Inflammation in Arthritis via Collagen-Dependent Microparticle Production. Science 2010, 327, 580–583. [Google Scholar] [CrossRef] [Green Version]
- Tourkochristou, E.; Aggeletopoulou, I.; Konstantakis, C.; Triantos, C. Role of NLRP3 inflammasome in inflammatory bowel diseases. World J. Gastroenterol. 2019, 25, 4796–4804. [Google Scholar] [CrossRef]
- Lien, T.-S.; Chan, H.; Sun, D.-S.; Wu, J.-C.; Lin, Y.-Y.; Lin, G.-L.; Chang, H.-H. Exposure of Platelets to Dengue Virus and Envelope Protein Domain III Induces Nlrp3 Inflammasome-Dependent Platelet Cell Death and Thrombocytopenia in Mice. Front. Immunol. 2021, 12, 616394. [Google Scholar] [CrossRef]
- Xue, Y.; Chen, H.; Zhang, S.; Bao, L.; Chen, B.; Gong, H.; Zhao, Y.; Qi, R. Resveratrol Confers Vascular Protection by Suppressing TLR4/Syk/NLRP3 Signaling in Oxidized Low-Density Lipoprotein-Activated Platelets. Oxid. Med. Cell. Longev. 2021, 2021, 8819231. [Google Scholar] [CrossRef]
- Middleton, E.A.; Rowley, J.W.; Campbell, R.A.; Grissom, C.K.; Brown, S.; Beesley, S.J.; Schwertz, H.; Kosaka, Y.; Manne, B.K.; Krauel, K.; et al. Sepsis alters the transcriptional and translational landscape of human and murine platelets. Blood 2019, 134, 911–923. [Google Scholar] [CrossRef]
- Sinauridze, E.I.; Kireev, D.A.; Popenko, N.Y.; Pichugin, A.; Panteleev, M.; Krymskaya, O.V.; Ataullakhanov, F.I. Platelet microparticle membranes have 50- to 100-fold higher specific procoagulant activity than activated platelets. Thromb. Haemost. 2007, 97, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Semple, J.W.; Freedman, J. Platelets and innate immunity. Experientia 2009, 67, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Gros, A.; Ollivier, V.; Ho-Tin-Noã, B. Platelets in Inflammation: Regulation of Leukocyte Activities and Vascular Repair. Front. Immunol. 2015, 5, 678. [Google Scholar] [CrossRef]
- Bhattacharya, J.; Matthay, M.A. Regulation and Repair of the Alveolar-Capillary Barrier in Acute Lung Injury. Annu. Rev. Physiol. 2013, 75, 593–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gawaz, M.P.; Vogel, S. Platelets in tissue repair: Control of apoptosis and interactions with regenerative cells. Blood 2013, 122, 2550–2554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafii, S.; Cao, Z.; Lis, R.; Siempos, I.I.; Chavez, D.; Shido, K.; Rabbany, S.Y.; Ding, B.-S. Platelet-derived SDF-1 primes the pulmonary capillary vascular niche to drive lung alveolar regeneration. Nat. Cell Biol. 2015, 17, 123–136. [Google Scholar] [CrossRef]
- Mazzucco, L.; Borzini, P.; Gope, R. Platelet-Derived Factors Involved in Tissue Repair—From Signal to Function. Transfus. Med. Rev. 2010, 24, 218–234. [Google Scholar] [CrossRef] [PubMed]
- McDonald, B.; Davis, R.P.; Kim, S.-J.; Tse, M.; Esmon, C.T.; Kolaczkowska, E.; Jenne, C.N. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood 2017, 129, 1357–1367. [Google Scholar] [CrossRef] [Green Version]
- Etulain, J.; Martinod, K.; Wong, S.L.; Cifuni, S.M.; Schattner, M.; Wagner, D.D. P-selectin promotes neutrophil extracellular trap formation in mice. Blood 2015, 126, 242–246. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, E.P.; Yang, Y.; Janssen, W.J.; Gandjeva, A.; Perez, M.J.; Barthel, L.; Zemans, R.L.; Bowman, J.C.; Koyanagi, D.E.; Yunt, Z.X.; et al. The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nat. Med. 2012, 18, 1217–1223. [Google Scholar] [CrossRef] [Green Version]
- Wiesinger, A.; Peters, W.; Chappell, D.; Kentrup, D.; Reuter, S.; Pavenstädt, H.; Oberleithner, H.; Kümpers, P. Nanomechanics of the endothelial glycocalyx in experimental sepsis. PLoS ONE 2013, 8, e80905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, H.; Takemura, G.; Suzuki, K.; Oda, K.; Takada, C.; Hotta, Y.; Miyazaki, N.; Tsujimoto, A.; Muraki, I.; Ando, Y.; et al. Three-dimensional ultrastructure of capillary endothelial glycocalyx under normal and experimental endotoxemic conditions. Crit. Care 2017, 21, 261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipowsky, H.H.; Gao, L.; Lescanic, A. Shedding of the endothelial glycocalyx in arterioles, capillaries, and venules and its effect on capillary hemodynamics during inflammation. Am. J. Physiol. Circ. Physiol. 2011, 301, H2235–H2245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deutschman, C.S.; Tracey, K.J. Sepsis: Current Dogma and New Perspectives. Immunity 2014, 40, 463–475. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, O.; Akira, S. Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Kang, R.; Coyne, C.B.; Zeh, H.J.; Lotze, M.T. PAMPs and DAMPs: Signal 0s that spur autophagy and immunity. Immunol. Rev. 2012, 249, 158–175. [Google Scholar] [CrossRef]
- Guo, R.-F.; Ward, P.A. Role of C5a in inflammatory responses. Annu. Rev. Immunol. 2005, 23, 821–852. [Google Scholar] [CrossRef]
- Bierhaus, A.; Nawroth, P.P. Modulation of the vascular endothelium during infection—The role of NF-kappa B activation. Contrib. Microbiol. 2003, 10, 86–105. [Google Scholar]
- Lam, F.W.; Brown, C.A.; Valladolid, C.; Emebo, D.C.; Palzkill, T.G.; Cruz, M.A. The vimentin rod domain blocks P-selectin-P-selectin glycoprotein ligand 1 interactions to attenuate leukocyte adhesion to inflamed endothelium. PLoS ONE 2020, 15, e0240164. [Google Scholar] [CrossRef]
- Parikh, S.M. Dysregulation of the angiopoietin–Tie-2 axis in sepsis and ARDS. Virulence 2013, 4, 517–524. [Google Scholar] [CrossRef] [Green Version]
- Palmer, R.M.J.; Ashton, D.S.; Moncada, S. Vascular endothelial cells synthesize nitric oxide from L-arginine. Nature 1988, 333, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Noris, M.; Morigi, M.; Donadelli, R.; Aiello, S.; Foppolo, M.; Todeschini, M.; Orisio, S.; Remuzzi, G.; Remuzzi, A. Nitric Oxide Synthesis by Cultured Endothelial Cells Is Modulated by Flow Conditions. Circ. Res. 1995, 76, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Mohan Rao, L.V.; Esmon, C.T.; Pendurthi, U.R. Endothelial cell protein C receptor: A multiliganded and multifunctional receptor. Blood 2014, 124, 1553–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stearns-Kurosawa, D.J.; Kurosawa, S.; Mollica, J.S.; Ferrell, G.L.; Esmon, C.T. The endothelial cell protein C receptor augments protein C activation by the thrombin-thrombomodulin complex. Proc. Natl. Acad. Sci. USA 1996, 93, 10212–10216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catieau, B.; Devos, V.; Chtourou, S.; Borgel, D.; Plantier, J.-L. Endothelial cell surface limits coagulation without modulating the antithrombin potency. Thromb. Res. 2018, 167, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Lupu, C.; Goodwin, C.A.; Westmuckett, A.D.; Emeis, J.J.; Scully, M.F.; Kakkar, V.V.; Lupu, F. Tissue Factor Pathway Inhibitor in Endothelial Cells Colocalizes With Glycolipid Microdomains/Caveolae. Arter. Thromb. Vasc. Biol. 1997, 17, 2964–2974. [Google Scholar] [CrossRef]
- Ali, M.M.; Mahmoud, A.M.; Le Master, E.; Levitan, I.; Phillips, S.A. Role of matrix metalloproteinases and histone deacetylase in oxidative stress-induced degradation of the endothelial glycocalyx. Am. J. Physiol. Circ. Physiol. 2019, 316, H647–H663. [Google Scholar] [CrossRef]
- Angus, D.C.; van der Poll, T. Severe sepsis and septic shock. N. Engl. J. Med. 2013, 369, 840–851. [Google Scholar] [CrossRef]
- Bevilacqua, M.P.; Pober, J.S.; Wheeler, M.E.; Cotran, R.S.; Gimbrone, M.A. Interleukin-1 activation of vascular endothelium. Effects on procoagulant activity and leukocyte adhesion. Am. J. Pathol. 1985, 121, 394–403. [Google Scholar]
- Rahman, M.; Zhang, S.; Chew, M.; Syk, I.; Jeppsson, B.; Thorlacius, H. Platelet shedding of CD40L is regulated by matrix metalloproteinase-9 in abdominal sepsis. J. Thromb. Haemost. 2013, 11, 1385–1398. [Google Scholar] [CrossRef]
- Brunn, G.J.; Saadi, S.; Platt, J.L. Differential Regulation of Endothelial Cell Activation by Complement and Interleukin 1α. Circ. Res. 2006, 98, 793–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sreeramkumar, V.; Adrover, J.M.; Ballesteros, I.; Cuartero, M.I.; Rossaint, J.; Bilbao, I.; Nácher, M.; Pitaval, C.; Radovanovic, I.; Fukui, Y.; et al. Neutrophils scan for activated platelets to initiate inflammation. Science 2014, 346, 1234–1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, N.; Yoshikawa, T.; Nakamura, Y.; Sakamoto, K.; Takenaka, S.; Boku, Y.; Kassai, K.; Kondo, M. Interactions of neutrophils and endothelial cells under low flow conditions in vitro. Shock 1997, 8, 125–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidtke, D.; Diamond, S.L. Direct Observation of Membrane Tethers Formed during Neutrophil Attachment to Platelets or P-Selectin under Physiological Flow. J. Cell Biol. 2000, 149, 719–730. [Google Scholar] [CrossRef]
- Simon, D.I.; Chen, Z.; Xu, H.; Li, C.Q.; Dong, J.F.; McIntire, L.V.; Ballantyne, C.M.; Zhang, L.; Furman, M.I.; Berndt, M.C.; et al. Platelet glycoprotein ibalpha is a counterreceptor for the leukocyte integrin Mac-1 (CD11b/CD18). J. Exp. Med. 2000, 192, 193–204. [Google Scholar] [CrossRef] [Green Version]
- Santoso, S.; Sachs, U.J.H.; Kroll, H.; Linder, M.; Ruf, A.; Preissner, K.T.; Chavakis, T. The Junctional Adhesion Molecule 3 (JAM-3) on Human Platelets is a Counterreceptor for the Leukocyte Integrin Mac-1. J. Exp. Med. 2002, 196, 679–691. [Google Scholar] [CrossRef]
- Weber, C.; Springer, A.T. Neutrophil accumulation on activated, surface-adherent platelets in flow is mediated by interaction of Mac-1 with fibrinogen bound to alphaIIbbeta3 and stimulated by platelet-activating factor. J. Clin. Investig. 1997, 100, 2085–2093. [Google Scholar] [CrossRef]
- Wetterholm, E.; Linders, J.; Merza, M.; Regner, S.; Thorlacius, H. Platelet-derived CXCL4 regulates neutrophil infiltration and tissue damage in severe acute pancreatitis. Transl. Res. 2016, 176, 105–118. [Google Scholar] [CrossRef]
- Assinger, A.; Laky, M.; Badrnya, S.; Esfandeyari, A.; Volf, I. Periodontopathogens induce expression of CD40L on human platelets via TLR2 and TLR4. Thromb. Res. 2012, 130, e73–e78. [Google Scholar] [CrossRef]
- Gaertner, F.; Ahmad, Z.; Rosenberger, G.; Fan, S.; Nicolai, L.; Busch, B.; Yavuz, G.; Luckner, M.; Ishikawa-Ankerhold, H.; Hennel, R.; et al. Migrating Platelets Are Mechano-scavengers that Collect and Bundle Bacteria. Cell 2017, 171, 1368–1382.e23. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Metzler, K.D.; Goosmann, C.; Lubojemska, A.; Zychlinsky, A.; Papayannopoulos, V. A Myeloperoxidase-Containing Complex Regulates Neutrophil Elastase Release and Actin Dynamics during NETosis. Cell Rep. 2014, 8, 883–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papayannopoulos, V. Neutrophils Stepping Through (to the Other Side). Immunity 2018, 49, 992–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Brühl, M.L.; Stark, K.; Steinhart, A.; Chandraratne, S.; Konrad, I.; Lorenz, M.; Khandoga, A.; Tirniceriu, A.; Coletti, R.; Köllnberger, M.; et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J. Exp. Med. 2012, 209, 819–835. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hottz, E.D.; Lopes, J.F.; Freitas, C.; Valls-De-Souza, R.; Oliveira, M.F.; Bozza, M.; Da Poian, A.; Weyrich, A.; Zimmerman, G.A.; Bozza, F.A.; et al. Platelets mediate increased endothelium permeability in dengue through NLRP3-inflammasome activation. Blood 2013, 122, 3405–3414. [Google Scholar] [CrossRef] [Green Version]
- Feng, G.; Yang, X.; Li, Y.; Wang, X.; Tan, S.; Chen, F. LPS enhances platelets aggregation via TLR4, which is related to mitochondria damage caused by intracellular ROS, but not extracellular ROS. Cell. Immunol. 2018, 328, 86–92. [Google Scholar] [CrossRef]
- Brown, G.T.; Narayanan, P.; Li, W.; Silverstein, R.L.; McIntyre, T.M. Lipopolysaccharide Stimulates Platelets through an IL-1β Autocrine Loop. J. Immunol. 2013, 191, 5196–5203. [Google Scholar] [CrossRef] [Green Version]
- Borges-Rodriguez, M.; Shields, C.A.; Travis, O.K.; Tramel, R.W.; Baik, C.H.; Giachelli, C.A.; Tardo, G.A.; Williams, J.M.; Cornelius, D.C. Platelet Inhibition Prevents NLRP3 Inflammasome Activation and Sepsis-Induced Kidney Injury. Int. J. Mol. Sci. 2021, 22, 10330. [Google Scholar] [CrossRef]
- Ouyang, Y.; Wang, Y.; Liu, B.; Ma, X.; Ding, R. Effects of antiplatelet therapy on the mortality rate of patients with sepsis: A meta-analysis. J. Crit. Care 2018, 50, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Du, F.; Jiang, P.; He, S.; Song, D.; Xu, F. Antiplatelet Therapy for Critically Ill Patients: A Pairwise and Bayesian Network Meta-Analysis. Shock 2018, 49, 616–624. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Li, H.; Gu, X.; Wang, Z.; Liu, S.; Chen, L. Effect of Antiplatelet Therapy on Acute Respiratory Distress Syndrome and Mortality in Critically Ill Patients: A Meta-Analysis. PLoS ONE 2016, 11, e0154754. [Google Scholar] [CrossRef] [Green Version]
- Trauer, J.; Muhi, S.; McBryde, E.S.; Al Harbi, S.A.; Arabi, Y.M.; Boyle, A.J.; Cartin-Ceba, R.; Chen, W.; Chen, Y.; Falcone, M.; et al. Quantifying the Effects of Prior Acetyl-Salicylic Acid on Sepsis-Related Deaths: An Individual Patient Data Meta-Analysis Using Propensity Matching. Crit. Care Med. 2017, 45, 1871–1879. [Google Scholar] [CrossRef] [PubMed]
- Li, H.L.; Feng, Q.; Tsoi, M.F.; Fei, Y.; Cheung, B.M.Y. Risk of infections in patients treated with ticagrelor vs. clopidogrel: A systematic review and meta-analysis. Eur. Hear. J. Cardiovasc. Pharmacother. 2020, 7, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Eisen, D.P.; Leder, K.; Woods, R.L.; Lockery, J.E.; McGuinness, S.L.; Wolfe, R.; Pilcher, D.; Moore, E.M.; Shastry, A.; Nelson, M.R.; et al. Effect of aspirin on deaths associated with sepsis in healthy older people (ANTISEPSIS): A randomised, double-blind, placebo-controlled primary prevention trial. Lancet Respir. Med. 2021, 9, 186–195. [Google Scholar] [CrossRef]
- McNeil, J.J.; Nelson, M.R.; Woods, R.L.; Lockery, J.E.; Wolfe, R.; Reid, C.M.; Kirpach, B.; Shah, R.C.; Ives, D.G.; Storey, E.; et al. Effect of Aspirin on All-Cause Mortality in the Healthy Elderly. N. Engl. J. Med. 2018, 379, 1519–1528. [Google Scholar] [CrossRef]
- Akinosoglou, K.; Alexopoulos, D. Use of antiplatelet agents in sepsis: A glimpse into the future. Thromb. Res. 2014, 133, 131–138. [Google Scholar] [CrossRef]
- Liverani, E.; Rico, M.C.; Tsygankov, A.Y.; Kilpatrick, L.E.; Kunapuli, S.P. P2Y12 Receptor Modulates Sepsis-Induced Inflammation. Arter. Thromb. Vasc. Biol. 2016, 36, 961–971. [Google Scholar] [CrossRef] [Green Version]
- Omarjee, L.; Meilhac, O.; Perrot, F.; Janin, A.; Mahe, G. Can Ticagrelor be used to prevent sepsis-induced coagulopathy in COVID-19? Clin. Immunol. 2020, 216, 108468. [Google Scholar] [CrossRef]
- Schmitt, C.; Abt, M.; Ciorciaro, C.; Kling, D.; Jamois, C.; Schick, E.; Solier, C.; Benghozi, R.; Gaudreault, J. First-in-man Study with Inclacumab, a Human Monoclonal Antibody Against P-selectin. J. Cardiovasc. Pharmacol. 2015, 65, 611–619. [Google Scholar] [CrossRef] [Green Version]
- Greinacher, A.; Thiele, T.; Warkentin, T.E.; Weisser, K.; Kyrle, P.A.; Eichinger, S. Thrombotic Thrombocytopenia after ChAdOx1 nCov-19 Vaccination. N. Engl. J. Med. 2021, 384, 2092–2101. [Google Scholar] [CrossRef]
- Scully, M.; Singh, D.; Lown, R.; Poles, A.; Solomon, T.; Levi, M.; Goldblatt, D.; Kotoucek, P.; Thomas, W.; Lester, W. Pathologic Antibodies to Platelet Factor 4 after ChAdOx1 nCoV-19 Vaccination. N. Engl. J. Med. 2021, 384, 2202–2211. [Google Scholar] [CrossRef]
- Schultz, N.H.; Sørvoll, I.H.; Michelsen, A.E.; Munthe, L.A.; Lund-Johansen, F.; Ahlen, M.T.; Wiedmann, M.; Aamodt, A.-H.; Skattør, T.H.; Tjønnfjord, G.E.; et al. Thrombosis and Thrombocytopenia after ChAdOx1 nCoV-19 Vaccination. N. Engl. J. Med. 2021, 384, 2124–2130. [Google Scholar] [CrossRef]
- Sessa, F.; Salerno, M.; Esposito, M.; Di Nunno, N.; Zamboni, P.; Pomara, C. Autopsy Findings and Causality Relationship between Death and COVID-19 Vaccination: A Systematic Review. J. Clin. Med. 2021, 10, 5876. [Google Scholar] [CrossRef]
- Schneider, J.; Sottmann, L.; Greinacher, A.; Hagen, M.; Kasper, H.-U.; Kuhnen, C.; Schlepper, S.; Schmidt, S.; Schulz, R.; Thiele, T.; et al. Postmortem investigation of fatalities following vaccination with COVID-19 vaccines. Int. J. Legal. Med. 2021, 135, 2335–2345. [Google Scholar] [CrossRef]
- Burgelman, M.; Vandendriessche, C.; Vandenbroucke, R.E. Extracellular Vesicles: A Double-Edged Sword in Sepsis. Pharmaceuticals 2021, 14, 829. [Google Scholar] [CrossRef]
- Van der Pol, E.; Böing, A.N.; Gool, E.L.; Nieuwland, R. Recent developments in the nomenclature, presence, isolation, detection and clinical impact of extracellular vesicles. J. Thromb. Haemost. 2016, 14, 48–56. [Google Scholar] [CrossRef] [Green Version]
- Berckmans, R.J.; Lacroix, R.; Hau, C.M.; Sturk, A.; Nieuwland, R. Extracellular vesicles and coagulation in blood from healthy humans revisited. J. Extracell. Vesicles 2019, 8, 1688936. [Google Scholar] [CrossRef]
- Flaumenhaft, R.; Dilks, J.R.; Richardson, J.; Alden, E.; Patel-Hett, S.R.; Battinelli, E.; Klement, G.L.; Sola-Visner, M.; Italiano, J.E., Jr. Megakaryocyte-derived microparticles: Direct visualization and distinction from platelet-derived microparticles. Blood 2009, 113, 1112–1121. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Feng, Y.; Jeyaram, A.; Jay, S.M.; Zou, L.; Chao, W. Circulating Plasma Extracellular Vesicles from Septic Mice Induce Inflammation via MicroRNA- and TLR7-Dependent Mechanisms. J. Immunol. 2018, 201, 3392–3400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zafrani, L.; Gerotziafas, G.; Byrnes, C.; Hu, X.; Perez, J.; Lévi, C.; Placier, S.; Letavernier, E.; Leelahavanichkul, A.; Haymann, J.-P.; et al. Calpastatin Controls Polymicrobial Sepsis by Limiting Procoagulant Microparticle Release. Am. J. Respir. Crit. Care Med. 2012, 185, 744–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mooberry, M.J.; Bradford, R.; Hobl, E.L.; Lin, F.C.; Jilma, B.; Key, N.S. Procoagulant microparticles promote coagulation in a factor XI-dependent manner in human endotoxemia. J. Thromb. Haemost. 2016, 14, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Meng, H.; Ma, R.; He, Z.; Wu, X.; Cao, M.; Yao, Z.; Zhao, L.; Li, T.; Deng, R.; et al. Circulating Microparticles, Blood Cells, and Endothelium Induce Procoagulant Activity in Sepsis Through Phosphatidylserine Exposure. Shock 2016, 45, 299–307. [Google Scholar] [CrossRef]
- Popova, T.G.; Teunis, A.; Espina, V.; Liotta, L.A.; Popov, S.G. Chemokine-Releasing Microparticles Improve Bacterial Clearance and Survival of Anthrax Spore-Challenged Mice. PLoS ONE 2016, 11, e0163163. [Google Scholar] [CrossRef] [Green Version]
- Köffel, R.; Wolfmeier, H.; Larpin, Y.; Besançon, H.; Schoenauer, R.; Babiychuk, V.S.; Drücker, P.; Pabst, T.; Mitchell, T.J.; Babiychuk, E.B.; et al. Host-Derived Microvesicles Carrying Bacterial Pore-Forming Toxins Deliver Signals to Macrophages: A Novel Mechanism of Shaping Immune Responses. Front. Immunol. 2018, 9, 1688. [Google Scholar] [CrossRef] [Green Version]
- Record, M.; Carayon, K.; Poirot, M.; Silvente-Poirot, S. Exosomes as new vesicular lipid transporters involved in cell–cell communication and various pathophysiologies. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2014, 1841, 108–120. [Google Scholar] [CrossRef]
- Duchez, A.-C.; Boudreau, L.H.; Naika, G.S.; Bollinger, J.; Belleannée, C.; Cloutier, N.; Laffont, B.; Mendoza-Villarroel, R.E.; Levesque, T.; Rollet-Labelle, E.; et al. Platelet microparticles are internalized in neutrophils via the concerted activity of 12-lipoxygenase and secreted phospholipase A2-IIA. Proc. Natl. Acad. Sci. USA 2015, 112, E3564–E3573. [Google Scholar] [CrossRef] [Green Version]
- Maugeri, N.; Capobianco, A.; Rovere-Querini, P.; Ramirez, G.A.; Tombetti, E.; Della Valle, P.; Monno, A.; D’Alberti, V.; Gasparri, A.M.; Franchini, S.; et al. Platelet microparticles sustain autophagy-associated activation of neutrophils in systemic sclerosis. Sci. Transl. Med. 2018, 10, eaao3089. [Google Scholar] [CrossRef] [Green Version]
- Lindemann, S.; Tolley, N.D.; Dixon, D.A.; McIntyre, T.M.; Prescott, S.M.; Zimmerman, G.A.; Weyrich, A.S. Activated platelets mediate inflammatory signaling by regulated interleukin 1beta synthesis. J. Cell Biol. 2001, 154, 485–490. [Google Scholar] [CrossRef] [Green Version]
- Bei, J.J.; Liu, C.; Peng, S.; Liu, C.H.; Zhao, W.B.; Qu, X.L.; Chen, Q.; Zhou, Z.; Yu, Z.; Peter, K.; et al. Staphylococcal SSL5-induced platelet microparticles provoke proinflammatory responses via the CD40/TRAF6/NFκB signalling pathway in monocytes. Thromb. Haemost. 2016, 115, 632–645. [Google Scholar] [CrossRef]
- Qu, M.; Zou, X.; Fang, F.; Wang, S.; Xu, L.; Zeng, Q.; Fan, Z.; Chen, L.; Yue, W.; Xie, X.; et al. Platelet-derived microparticles enhance megakaryocyte differentiation and platelet generation via miR-1915-3p. Nat. Commun. 2020, 11, 4964. [Google Scholar] [CrossRef]
- French, S.L.; Butov, K.R.; Allaeys, I.; Canas, J.; Morad, G.; Davenport, P.; Laroche, A.; Trubina, N.M.; Italiano, J.E., Jr.; Moses, M.A.; et al. Platelet-derived extracellular vesicles infiltrate and modify the bone marrow during inflammation. Blood Adv. 2020, 4, 3011–3023. [Google Scholar] [CrossRef]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Szilágyi, B.; Fejes, Z.; Rusznyák, Á.; Fenyvesi, F.; Pócsi, M.; Halmi, S.; Griger, Z.; Kunapuli, S.P.; Kappelmayer, J.; Nagy, B.J. Platelet Microparticles Enriched in miR-223 Reduce ICAM-1-Dependent Vascular Inflammation in Septic Conditions. Front. Physiol. 2021, 12, 658524. [Google Scholar] [CrossRef]
- Johnnidis, J.B.; Harris, M.H.; Wheeler, R.T.; Stehling-Sun, S.; Lam, M.H.; Kirak, O.; Brummelkamp, T.R.; Fleming, M.D.; Camargo, F.D. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature 2008, 451, 1125–1129. [Google Scholar] [CrossRef]
- Kirito, K.; Kaushansky, K. Transcriptional regulation of megakaryopoiesis: Thrombopoietin signaling and nuclear factors. Curr. Opin. Hematol. 2006, 13, 151–156. [Google Scholar] [CrossRef]
- Vainchenker, W.; Constantinescu, S.N. JAK/STAT signaling in hematological malignancies. Oncogene 2012, 32, 2601–2613. [Google Scholar] [CrossRef] [Green Version]
- Park, H.J.; Li, J.; Hannah, R.; Biddie, S.; Leal-Cervantes, A.I.; Kirschner, K.; Cruz, D.F.S.; Sexl, V.; Göttgens, B.; Green, A.R. Cytokine-induced megakaryocytic differentiation is regulated by genome-wide loss of a uSTAT transcriptional program. EMBO J. 2015, 35, 580–594. [Google Scholar] [CrossRef]
- Cunin, P.; Bouslama, R.; Machlus, K.; Martínez-Bonet, M.; Lee, P.Y.; Wactor, A.; Nelson-Maney, N.; Morris, A.; Guo, L.; Weyrich, A.; et al. Megakaryocyte emperipolesis mediates membrane transfer from intracytoplasmic neutrophils to platelets. eLife 2019, 8, e44031. [Google Scholar] [CrossRef]
- Valet, C.; Magnen, M.; Qiu, L.; Cleary, S.J.; Wang, K.M.; Ranucci, S.; Grockowiak, E.; Boudra, R.; Conrad, C.; Seo, Y.; et al. Sepsis promotes splenic production of a protective platelet pool with high CD40 ligand expression. J. Clin. Investig. 2022, 132, e153920. [Google Scholar] [CrossRef]
- Battina, H.L.; Alentado, V.J.; Srour, E.F.; Moliterno, A.R.; Kacena, M.A. Interaction of the inflammatory response and megakaryocytes in COVID-19 infection. Exp. Hematol. 2021, 104, 32–39. [Google Scholar] [CrossRef]
- Coomes, E.A.; Haghbayan, H. Interleukin-6 in COVID-19: A systematic review and meta-analysis. Rev. Med. Virol. 2020, 30, 1–9. [Google Scholar] [CrossRef]
- Navarro, S.; Debili, N.; Le Couedic, J.P.; Klein, B.; Breton-Gorius, J.; Doly, J.; Vainchenker, W. Interleukin-6 and its receptor are expressed by human megakaryocytes: In vitro effects on proliferation and endoreplication. Blood 1991, 77, 461–471. [Google Scholar] [CrossRef] [Green Version]
- Kaser, A.; Brandacher, G.; Steurer, W.; Kaser, S.; Offner, F.A.; Zoller, H.; Theurl, I.; Widder, W.; Molnar, C.; Ludwiczek, O.; et al. Interleukin-6 stimulates thrombopoiesis through thrombopoietin: Role in inflammatory thrombocytosis. Blood 2001, 98, 2720–2725. [Google Scholar] [CrossRef] [Green Version]
- Dan, K.; Gomi, S.; Inokuchi, K.; Ogata, K.; Yamada, T.; Ohki, I.; Hasegawa, S.; Nomura, T. Effects of interleukin-1 and tumor necrosis factor on megakaryocytopoiesis: Mechanism of reactive thrombocytosis. Acta Haematol. 1995, 93, 67–72. [Google Scholar] [CrossRef]
- Haas, S.; Hansson, J.; Klimmeck, D.; Loeffler, D.; Velten, L.; Uckelmann, H.; Wurzer, S.; Prendergast, M.; Schnell, A.; Hexel, K.; et al. Inflammation-Induced Emergency Megakaryopoiesis Driven by Hematopoietic Stem Cell-like Megakaryocyte Progenitors. Cell Stem Cell 2015, 17, 422–434. [Google Scholar] [CrossRef] [Green Version]
- Lefrancais, E.; Ortiz-Muñoz, G.; Caudrillier, A.; Mallavia, B.; Liu, F.; Sayah, D.M.; Thornton, E.E.; Headley, M.; David, T.; Coughlin, T.D.S.R.; et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature 2017, 544, 105–109. [Google Scholar] [CrossRef]
- Khatib-Massalha, E.; Méndez-Ferrer, S. Megakaryocyte Diversity in Ontogeny, Functions and Cell-Cell Interactions. Front. Oncol. 2022, 12, 840044. [Google Scholar] [CrossRef]
- Liu, C.; Wu, D.; Xia, M.; Li, M.; Sun, Z.; Shen, B.; Liu, Y.; Jiang, E.; Wang, H.; Su, P.; et al. Characterization of Cellular Heterogeneity and an Immune Subpopulation of Human Megakaryocytes. Adv. Sci. 2021, 8, 2100921. [Google Scholar] [CrossRef]
- Wang, J.; Xie, J.; Han, X.; Wang, D.; Chen, M.; Jiang, L.; Shi, G.; Zhao, M. Megakaryocyte derived immune-stimulating cells regulate host-defense immunity against bacterial pathogens. Cell Biol. 2021. Available online: http://biorxiv.org/lookup/doi/10.1101/2021.06.09.447810 (accessed on 29 April 2022).
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D., Jr.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pariser, D.N.; Hilt, Z.T.; Ture, S.K.; Blick-Nitko, S.K.; Looney, M.R.; Cleary, S.J.; Roman-Pagan, E.; Ii, J.S.; Georas, S.N.; Veazey, J.M.; et al. Lung megakaryocytes are immune modulatory cells. J. Clin. Investig. 2021, 131, 137377. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, S.; Matsubara, T.; Yone, K.; Hirano, Y.; Okumura, K.; Yabuta, K. Kawasaki disease differs from anaphylactoid purpura and measles with regard to tumour necrosis factor-α and interleukin 6 in serum. Eur. J. Pediatr. 1992, 151, 44–47. [Google Scholar] [CrossRef]
- Zufferey, A.; Speck, E.R.; Machlus, K.R.; Aslam, R.; Guo, L.; McVey, M.J.; Kim, M.; Kapur, R.; Boilard, E.; Italiano, J.E.; et al. Mature murine megakaryocytes present antigen-MHC class I molecules to T cells and transfer them to platelets. Blood Adv. 2017, 1, 1773–1785. [Google Scholar] [CrossRef]
- Lin, J.; Weiss, A. The tyrosine phosphatase CD148 is excluded from the immunologic synapse and down-regulates prolonged T cell signaling. J. Cell Biol. 2003, 162, 673–682. [Google Scholar] [CrossRef] [Green Version]
- Senis, Y.A.; Tomlinson, M.G.; Ellison, S.; Mazharian, A.; Lim, J.; Zhao, Y.; Kornerup, K.N.; Auger, J.M.; Thomas, S.G.; Dhanjal, T.; et al. The tyrosine phosphatase CD148 is an essential positive regulator of platelet activation and thrombosis. Blood 2009, 113, 4942–4954. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Babushok, D.V. Secondary myelodysplastic syndrome and leukemia in acquired aplastic anemia and paroxysmal nocturnal hemoglobinuria. Blood 2020, 136, 36–49. [Google Scholar] [CrossRef]
- Vainchenker, W.; Raslova, H. The megakaryocyte: A cell with 3 faces as a mythic god? Blood 2021, 138, 1199–1200. [Google Scholar] [CrossRef]
- Boilard, E.; Flamand, L. The role of the megakaryocyte in immunity has gone viral. Blood 2019, 133, 2001–2002. [Google Scholar] [CrossRef]
- Tacchini-Cottier, F.; Vesin, C.; Redard, M.; Buurman, W.; Piguet, P.F. Role of TNFR1 and TNFR2 in TNF-induced platelet consumption in mice. J. Immunol. 1998, 160, 6182–6186. [Google Scholar] [PubMed]
- Feng, E.; Balint, E.; Poznanski, S.; Ashkar, A.; Loeb, M. Aging and Interferons: Impacts on Inflammation and Viral Disease Outcomes. Cells 2021, 10, 708. [Google Scholar] [CrossRef] [PubMed]
- D’Atri, L.P.; Etulain, J.; Rivadeneyra, L.; Lapponi, M.J.; Centurion, M.; Cheng, K.; Yin, H.; Schattner, M. Expression and functionality of Toll-like receptor 3 in the megakaryocytic lineage. J. Thromb. Haemost. 2015, 13, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Pozner, R.G.; Ure, A.E.; Jaquenod de Giusti, C.; D’Atri, L.P.; Italiano, J.E.; Torres, O.; Romanowski, V.; Schattner, M.; Gómez, R.M. Junín virus infection of human hematopoietic progenitors impairs in vitro proplatelet formation and platelet release via a bystander effect involving type I IFN signaling. PLoS Pathog. 2010, 6, e1000847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negrotto, S.; De Giusti, C.J.; Lapponi, M.J.; Etulain, J.; Rivadeneyra, L.; Pozner, R.G.; Gomez, R.M.; Schattner, M. Expression and functionality of type I interferon receptor in the megakaryocytic lineage. J. Thromb. Haemost. 2011, 9, 2477–2485. [Google Scholar] [CrossRef]
- Ning, S.; Pagano, J.S.; Barber, G.N. IRF7: Activation, regulation, modification and function. Genes Immun. 2011, 12, 399–414. [Google Scholar] [CrossRef] [Green Version]
- Kochs, G.; Haller, O. Interferon-induced human MxA GTPase blocks nuclear import of Thogoto virus nucleocapsids. Proc. Natl. Acad. Sci. USA 1999, 96, 2082–2086. [Google Scholar] [CrossRef] [Green Version]
- Brass, A.L.; Huang, I.-C.; Benita, Y.; John, S.P.; Krishnan, M.N.; Feeley, E.; Ryan, B.J.; Weyer, J.L.; Van Der Weyden, L.; Fikrig, E.; et al. The IFITM Proteins Mediate Cellular Resistance to Influenza A H1N1 Virus, West Nile Virus, and Dengue Virus. Cell 2009, 139, 1243–1254. [Google Scholar] [CrossRef] [Green Version]
- Lood, C.; Tydén, H.; Gullstrand, B.; Jönsen, A.; Källberg, E.; Mörgelin, M.; Kahn, R.; Gunnarsson, I.; Leanderson, T.; Ivars, F.; et al. Platelet-Derived S100A8/A9 and Cardiovascular Disease in Systemic Lupus Erythematosus. Arthritis Rheumatol. 2016, 68, 1970–1980. [Google Scholar] [CrossRef] [Green Version]
- Amini-Bavil-Olyaee, S.; Choi, Y.J.; Lee, J.H.; Shi, M.; Huang, I.C.; Farzan, M.; Jung, J.U. The antiviral effector IFITM3 disrupts intracellular cholesterol homeostasis to block viral entry. Cell Host Microbe. 2013, 13, 452–464. [Google Scholar] [CrossRef] [Green Version]
- Manne, B.K.; Denorme, F.; Middleton, E.A.; Portier, I.; Rowley, J.W.; Stubben, C.; Petrey, A.C.; Tolley, N.D.; Guo, L.; Cody, M.; et al. Platelet gene expression and function in patients with COVID-19. Blood 2020, 136, 1317–1329. [Google Scholar] [CrossRef] [PubMed]
- Prelli Bozzo, C.; Nchioua, R.; Volcic, M.; Koepke, L.; Krüger, J.; Schütz, D.; Heller, S.; Stürzel, C.M.; Kmiec, D.; Conzelmann, C.; et al. IFITM proteins promote SARS-CoV-2 infection and are targets for virus inhibition in vitro. Nat. Commun. 2021, 12, 4584. [Google Scholar] [CrossRef] [PubMed]
- Assinger, A. Platelets and infection—An emerging role of platelets in viral infection. Front. Immunol. 2014, 5, 649. [Google Scholar] [CrossRef] [Green Version]
- Campbell, R.A.; Schwertz, H.; Hottz, E.D.; Rowley, J.W.; Manne, B.K.; Washington, A.V.; Hunter-Mellado, R.; Tolley, N.D.; Christensen, M.; Eustes, A.S.; et al. Human megakaryocytes possess intrinsic antiviral immunity through regulated induction of IFITM3. Blood 2019, 133, 2013–2026. [Google Scholar] [CrossRef] [PubMed]
- Everitt, A.R.; Clare, S.; Pertel, T.; John, S.P.; Wash, R.S.; Smith, S.E.; Chin, C.R.; Feeley, E.M.; Sims, J.S.; Adams, D.J.; et al. IFITM3 restricts the morbidity and mortality associated with influenza. Nature 2012, 484, 519–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dann, R.; Hadi, T.; Montenont, E.; Boytard, L.; Alebrahim, D.; Feinstein, J.; Allen, N.; Simon, R.; Barone, K.; Uryu, K.; et al. Platelet-Derived MRP-14 Induces Monocyte Activation in Patients With Symptomatic Peripheral Artery Disease. J. Am. Coll. Cardiol. 2018, 71, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Ometto, F.; Friso, L.; Astorri, D.; Botsios, C.; Raffeiner, B.; Punzi, L.; Doria, A. Calprotectin in rheumatic diseases. Exp. Biol. Med. 2016, 242, 859–873. [Google Scholar] [CrossRef]
- Barrett, T.J.; Cornwell, M.; Myndzar, K.; Rolling, C.C.; Xia, Y.; Drenkova, K.; Biebuyck, A.; Fields, A.T.; Tawil, M.; Luttrell-Williams, E.; et al. Platelets amplify endotheliopathy in COVID-19. Sci. Adv. 2021, 7, eabh2434. [Google Scholar] [CrossRef]
- Björk, P.; Björk, A.; Vogl, T.; Stenström, M.; Liberg, D.; Olsson, A.; Roth, J.; Ivars, F.; Leanderson, T. Identification of human S100A9 as a novel target for treatment of autoimmune disease via binding to quinoline-3-carboxamides. PLoS Biol. 2009, 7, e97. [Google Scholar] [CrossRef] [Green Version]
- Van Zoelen, M.A.D.; Vogl, T.; Foell, D.; Van Veen, S.Q.; van Till, J.W.O.; Florquin, S.; Tanck, M.W.; Wittebole, X.; Laterre, P.; Boermeester, M.A.; et al. Expression and role of myeloid-related protein-14 in clinical and experimental sepsis. Am. J. Respir. Crit. Care Med. 2009, 180, 1098–1106. [Google Scholar] [CrossRef] [Green Version]
- Brennan, M.P.; Loughman, A.; Devocelle, M.; Arasu, S.; Chubb, A.J.; Foster, T.J.; Cox, D. Elucidating the role of Staphylococcus epidermidis serine-aspartate repeat protein G in platelet activation. J. Thromb. Haemost. 2009, 7, 1364–1372. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia, C.; Compagnon, B.; Poëtte, M.; Gratacap, M.-P.; Lapébie, F.-X.; Voisin, S.; Minville, V.; Payrastre, B.; Vardon-Bounes, F.; Ribes, A. Platelet Versus Megakaryocyte: Who Is the Real Bandleader of Thromboinflammation in Sepsis? Cells 2022, 11, 1507. https://doi.org/10.3390/cells11091507
Garcia C, Compagnon B, Poëtte M, Gratacap M-P, Lapébie F-X, Voisin S, Minville V, Payrastre B, Vardon-Bounes F, Ribes A. Platelet Versus Megakaryocyte: Who Is the Real Bandleader of Thromboinflammation in Sepsis? Cells. 2022; 11(9):1507. https://doi.org/10.3390/cells11091507
Chicago/Turabian StyleGarcia, Cédric, Baptiste Compagnon, Michaël Poëtte, Marie-Pierre Gratacap, François-Xavier Lapébie, Sophie Voisin, Vincent Minville, Bernard Payrastre, Fanny Vardon-Bounes, and Agnès Ribes. 2022. "Platelet Versus Megakaryocyte: Who Is the Real Bandleader of Thromboinflammation in Sepsis?" Cells 11, no. 9: 1507. https://doi.org/10.3390/cells11091507