
A Novel Role for Plasminogen Activator Inhibitor Type-2 as a Hypochlorite-Resistant Serine Protease Inhibitor and Holdase Chaperone

 ,
,
Abstract
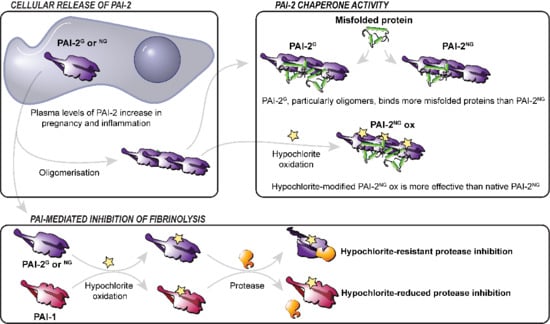
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Protein Determination
2.3. Hypochlorite (NaOCl) Treatments
2.4. Size Exclusion Chromatography (SEC)
2.5. 4,4′-Dianilino-1,1′-binaphthyl-5,5′-disulfonic Acid (Bis-ANS) Assay
2.6. Dityrosine Fluorescence and Circular Dichroism (CD) Measurements
2.7. SDS-PAGE
2.8. Native PAGE
2.9. uPA Activity Assay
2.10. Thioflavin-T (ThT) Assay
2.11. Western Blot
2.12. Aβ1–42 Cytotoxicity Assays
2.13. Immunohistochemistry and Immunofluorescence
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Lee, J.; Cochran, B.; Lobov, S.; Ranson, M. Forty years later and the role of plasminogen activator inhibitor type 2/SERPINB2 is still an enigma. Semin. Thromb. Hemost. 2011, 37, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Thorsen, S.; Philips, M.; Selmer, J.; Lecander, I.; Åstedt, B. Kinetics of inhibition of tissue-type and urokinase-type plasminogen activator by plasminogen-activator inhibitor type 1 and type 2. Eur. J. Biochem. 1988, 175, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Al-Ejeh, F.; Croucher, D.; Ranson, M. Kinetic analysis of plasminogen activator inhibitor type-2: Urokinase complex formation and subsequent internalisation by carcinoma cell lines. Exp. Cell Res. 2004, 297, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.A.; Croucher, D.R.; Ranson, M. Differential endocytosis of tissue plasminogen activator by serpins PAI-1 and PAI-2 on human peripheral blood monocytes. Thromb. Haemost. 2010, 104, 1133–1142. [Google Scholar] [CrossRef]
- Medcalf, R.L.; Stasinopoulos, S.J. The undecided serpin. The ins and outs of plasminogen activator inhibitor type 2. FEBS J. 2005, 272, 4858–4867. [Google Scholar] [CrossRef]
- Schroder, W.A.; Major, L.; Suhrbier, A. The role of SerpinB2 in immunity. Crit. Rev. Immunol. 2011, 31, 15–30. [Google Scholar] [CrossRef]
- Vaughan, D. PAI-1 and atherothrombosis. J. Thromb. Haemost. 2005, 3, 1879–1883. [Google Scholar] [CrossRef]
- Astedt, B.; Lindoff, C.; Lecander, I. Significance of the plasminogen activator inhibitor of placental type (PAI-2) in pregnancy. Semin. Thromb. Hemost. 1998, 24, 431–435. [Google Scholar] [CrossRef]
- Lee, J.A.; Yerbury, J.J.; Farrawell, N.; Shearer, R.F.; Constantinescu, P.; Hatters, D.M.; Schroder, W.A.; Suhrbier, A.; Wilson, M.R.; Saunders, D.N. SerpinB2 (PAI-2) modulates proteostasis via binding misfolded proteins and promotion of cytoprotective inclusion formation. Public Libr. Sci. One 2015, 10, e0130136. [Google Scholar] [CrossRef] [Green Version]
- Baker, M.S.; Green, S.P.; Goss, N.; Katrantzis, M.; Doe, W.F. Plasminogen activator inhibitor 2 (PAI-2) is not inactivated by exposure to oxidants which can be released from activated neutrophils. Biochem. Biophys. Res. Commun. 1990, 166, 993–1000. [Google Scholar] [CrossRef]
- Strandberg, L.; Lawrence, D.; Johansson, L.; Ny, T. The oxidative inactivation of plasminogen activator inhibitor type 1 results from a conformational change in the molecule and does not require the involvement of the P1’methionine. J. Biol. Chem. 1991, 266, 13852–13858. [Google Scholar] [CrossRef]
- Kruithof, E.; Tran-Thang, C.; Gudinchet, A.; Hauert, J.; Nicoloso, G.; Genton, C.; Welti, H.; Bachmann, F. Fibrinolysis in pregnancy: A study of plasminogen activator inhibitors. Blood 1987, 69, 460–466. [Google Scholar] [CrossRef] [Green Version]
- Wohlwend, A.; Belin, D.; Vassalli, J.D. Plasminogen activator-specific inhibitors produced by human monocytes/macrophages. J. Exp. Med. 1987, 165, 320–339. [Google Scholar] [CrossRef]
- Rohne, P.; Prochnow, H.; Wolf, S.; Renner, B.; Koch-Brandt, C. The Chaperone Activity of Clusterin is Dependent on Glycosylation and Redox Environment. Cell. Physiol. Biochem. 2014, 34, 1626–1639. [Google Scholar] [CrossRef]
- Fox, N.; Liu, B.; Scavelli, K.; Castillejos, A.; Kang, M.H.; Rhee, D.J. Glycosylation of SPARC is Critical for Binding to Extracellular Matrix Proteins and Protection from Matrix Metalloproteinase Degradation. Investig. Ophthalmol. Vis. Sci. 2019, 60, 5150. [Google Scholar]
- Martínek, V.; Sklenář, J.; Dračínský, M.; Šulc, M.; Hofbauerová, K.; Bezouška, K.; Frei, E.; Stiborová, M. Glycosylation Protects Proteins against Free Radicals Generated from Toxic Xenobiotics. Toxicol. Sci. 2010, 117, 359–374. [Google Scholar] [CrossRef] [Green Version]
- Uchida, E.; Morimoto, K.; Kawasaki, N.; Ahmed, Y.; Said, A.; Hayakawa, T. Effect of active oxygen radicals on protein and carbohydrate moieties of recombinant human erythropoietin. Free Radic. Res. 1997, 27, 311–323. [Google Scholar] [CrossRef]
- Hammer, A.; Desoye, G.; Dohr, G.; Sattler, W.; Malle, E. Myeloperoxidase-dependent generation of hypochlorite-modified proteins in human placental tissues during normal pregnancy. Lab. Investig. 2001, 81, 543–554. [Google Scholar] [CrossRef] [Green Version]
- Estelles, A.; Gilabert, J.; Aznar, J.; Loskutoff, D.J.; Schleef, R.R. Changes in the plasma levels of type 1 and type 2 plasminogen activator inhibitors in normal pregnancy and in patients with severe preeclampsia. Blood 1989, 74, 1332–1338. [Google Scholar] [CrossRef] [Green Version]
- Reith, A.; Booth, N.; Moore, N.; Cruickshank, D.; Bennett, B. Plasminogen activator inhibitors (PAI-1 and PAI-2) in normal pregnancies, pre-eclampsia and hydatidiform mole. Br. J. Obstet. Gynaecol. Int. J. Obstet. Gynaecol. 1993, 100, 370–374. [Google Scholar] [CrossRef]
- Grancha, S.; Estellés, A.; Gilabert, J.; Chirivella, M.; España, F.; Aznar, J. Decreased expression of PAI-2 mRNA and protein in pregnancies complicated with intrauterine fetal growth retardation. Thromb. Haemost. 1996, 76, 761–767. [Google Scholar] [CrossRef]
- Buhimschi, I.A.; Nayeri, U.A.; Zhao, G.; Shook, L.L.; Pensalfini, A.; Funai, E.F.; Bernstein, I.M.; Glabe, C.G.; Buhimschi, C.S. Protein misfolding, congophilia, oligomerization, and defective amyloid processing in preeclampsia. Sci. Transl. Med. 2014, 6, 245ra292. [Google Scholar] [CrossRef]
- Heinecke, J.W.; Li, W.; Daehnke, H.r.; Goldstein, J.A. Dityrosine, a specific marker of oxidation, is synthesized by the myeloperoxidase-hydrogen peroxide system of human neutrophils and macrophages. J. Biol. Chem. 1993, 268, 4069–4077. [Google Scholar] [CrossRef]
- Cochran, B.J.; Gunawardhana, L.P.; Vine, K.L.; Lee, J.A.; Lobov, S.; Ranson, M. The CD-loop of PAI-2 (SERPINB2) is redundant in the targeting, inhibition and clearance of cell surface uPA activity. BioMed Cent. Biotechnol. 2009, 9, 1. [Google Scholar] [CrossRef] [Green Version]
- Zusterzeel, P.L.; Mulder, T.P.; Peters, W.H.; Wiseman, S.A.; Steegers, E.A. Plasma protein carbonyls in nonpregnant, healthy pregnant and preeclamptic women. Free Radic. Res. 2000, 33, 471–476. [Google Scholar] [CrossRef]
- Zusterzeel, P.L.; Rutten, H.; Roelofs, H.M.; Peters, W.H.; Steegers, E.A. Protein carbonyls in decidua and placenta of pre-eclamptic women as markers for oxidative stress. Placenta 2001, 22, 213–219. [Google Scholar] [CrossRef]
- Hazen, S.L.; Heinecke, J.W. 3-Chlorotyrosine, a specific marker of myeloperoxidase-catalyzed oxidation, is markedly elevated in low density lipoprotein isolated from human atherosclerotic intima. J. Clin. Investig. 1997, 99, 2075. [Google Scholar] [CrossRef] [Green Version]
- Vlasova, I.; Vakhrusheva, T.; Sokolov, A.; Kostevich, V.; Ragimov, A. Peroxidase-induced degradation of single-walled carbon nanotubes: Hypochlorite is a major oxidant capable of in vivo degradation of carbon nanotubes. J. Phys. Conf. Ser. 2011, 291, 012056. [Google Scholar] [CrossRef] [Green Version]
- Weiss, S.J. Tissue destruction by neutrophils. New Engl. J. Med. 1989, 320, 365–376. [Google Scholar] [PubMed]
- Boncela, J.; Przygodzka, P.; Wyroba, E.; Papiewska-Pajak, I.; Cierniewski, C.S. Secretion of SerpinB2 from endothelial cells activated with inflammatory stimuli. Exp. Cell Res. 2013, 319, 1213–1219. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, H.; Booth, N.A. Secretion of plasminogen activator inhibitor 2 by human peripheral blood monocytes occurs via an endoplasmic reticulum–golgi-independent pathway. Exp. Cell Res. 1998, 242, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Sen, P.; Helmke, A.; Liao, C.M.; Sörensen-Zender, I.; Rong, S.; Bräsen, J.-H.; Melk, A.; Haller, H.; von Vietinghoff, S.; Schmitt, R. SerpinB2 Regulates Immune Response in Kidney Injury and Aging. J. Am. Soc. Nephrol. 2020, 31, 983–995. [Google Scholar] [CrossRef] [PubMed]
- Schroder, W.A.; Anraku, I.; Le, T.T.; Hirata, T.D.; Nakaya, H.I.; Major, L.; Ellis, J.J.; Suhrbier, A. SerpinB2 Deficiency Results in a Stratum Corneum Defect and Increased Sensitivity to Topically Applied Inflammatory Agents. Am. J. Pathol. 2016, 186, 1511–1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westrick, R.J.; Røjkjaer, L.P.; Yang, A.Y.; Roh, M.H.; Siebert, A.E.; Ginsburg, D. Deficiency of plasminogen activator inhibitor-2 results in accelerated tumor growth. J. Thromb. Haemost. 2020, 18, 2968–2975. [Google Scholar] [CrossRef]
- Gandley, R.E.; Rohland, J.; Zhou, Y.; Shibata, E.; Harger, G.F.; Rajakumar, A.; Kagan, V.E.; Markovic, N.; Hubel, C.A. Increased myeloperoxidase in the placenta and circulation of women with preeclampsia. Hypertension 2008, 52, 387–393. [Google Scholar] [CrossRef] [Green Version]
- Tanjung, M.T.; Siddik, H.D.; Hariman, H.; Koh, S.C. Coagulation and fibrinolysis in preeclampsia and neonates. Clin. Appl. Thromb. Hemost. Off. J. Int. Acad. Clin. Appl. Thromb. Hemost. 2005, 11, 467–473. [Google Scholar] [CrossRef]
- Fan, J.; Zhang, Y.-Q.; Li, P.; Hou, M.; Tan, L.; Wang, X.; Zhu, Y.-S. Interaction of plasminogen activator inhibitor-2 and proteasome subunit, beta type 1. Acta Biochim. Biophys. Sin. 2004, 36, 42–46. [Google Scholar] [CrossRef]
- Wilson, M.R.; Yerbury, J.J.; Poon, S. Potential roles of abundant extracellular chaperones in the control of amyloid formation and toxicity. Mol. Biosyst. 2008, 4, 42–52. [Google Scholar] [CrossRef] [Green Version]
- Stewart, E.M.; Aquilina, J.A.; Easterbrook-Smith, S.B.; Murphy-Durland, D.; Jacobsen, C.; Moestrup, S.; Wilson, M.R. Effects of glycosylation on the structure and function of the extracellular chaperone clusterin. Biochemistry 2007, 46, 1412–1422. [Google Scholar] [CrossRef]
- Haslbeck, M.; Weinkauf, S.; Buchner, J. Small heat shock proteins: Simplicity meets complexity. J. Biol. Chem. 2019, 294, 2121–2132. [Google Scholar] [CrossRef] [Green Version]
- Wyatt, A.R.; Yerbury, J.J.; Wilson, M.R. Structural characterization of clusterin-chaperone client protein complexes. J. Biol. Chem. 2009, 284, 21920–21927. [Google Scholar] [CrossRef] [Green Version]
- Wyatt, A.R.; Kumita, J.R.; Mifsud, R.W.; Gooden, C.A.; Wilson, M.R.; Dobson, C.M. Hypochlorite-induced structural modifications enhance the chaperone activity of human alpha2-macroglobulin. Proc. Natl. Acad. Sci. USA 2014, 111, E2081–E2090. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, U.; Nilson, A.N.; Kayed, R. The role of amyloid-β oligomers in toxicity, propagation, and immunotherapy. EBioMedicine 2016, 6, 42–49. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, H.; Ikeda, K.; Kondo, H.; Kato, M.; McGeer, P.L. Microglia express the type 2 plasminogen activator inhibitor in the brain of control subjects and patients with Alzheimer’s disease. Neurosci. Lett. 1993, 164, 233–235. [Google Scholar] [CrossRef]
- Chennakesava, C.S.; Di Santo, S.; Ziemiecki, A.; Schneider, H.; Andres, A.C. Differential expression of the receptor tyrosine kinase EphB4 and its ligand Ephrin-B2 during human placental development. Placenta 2006, 27, 959–967. [Google Scholar] [CrossRef]
- Lyden, T.W.; Robinson, J.M.; Tridandapani, S.; Teillaud, J.L.; Garber, S.A.; Osborne, J.M.; Frey, J.; Budde, P.; Anderson, C.L. The Fc receptor for IgG expressed in the villus endothelium of human placenta is Fc gamma RIIb2. J. Immunol. 2001, 166, 3882–3889. [Google Scholar] [CrossRef] [Green Version]
- McIntire, R.H.; Sifers, T.; Platt, J.S.; Ganacias, K.G.; Langat, D.K.; Hunt, J.S. Novel HLA-G-binding leukocyte immunoglobulin-like receptor (LILR) expression patterns in human placentas and umbilical cords. Placenta 2008, 29, 631–638. [Google Scholar] [CrossRef] [Green Version]
- Yui, J.; Hemmings, D.; Garcia-Lloret, M.; Guilbert, L.J. Expression of the human p55 and p75 tumor necrosis factor receptors in primary villous trophoblasts and their role in cytotoxic signal transduction. Biol. Reprod. 1996, 55, 400–409. [Google Scholar] [CrossRef] [Green Version]
- Koizumi, K.; Wang, G.; Park, L. Endothelial Dysfunction and Amyloid-β-Induced Neurovascular Alterations. Cell. Mol. Neurobiol. 2016, 36, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Smits, H.A.; van Beelen, A.J.; de Vos, N.M.; Rijsmus, A.; van der Bruggen, T.; Verhoef, J.; van Muiswinkel, F.L.; Nottet, H.S. Activation of human macrophages by amyloid-beta is attenuated by astrocytes. J. Immunol. 2001, 166, 6869–6876. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, D.A.; Loskutoff, D.J. Inactivation of plasminogen activator inhibitor by oxidants. Biochemistry 1986, 25, 6351–6355. [Google Scholar] [CrossRef]
- Wilczynska, M.; Lobov, S.; Ohlsson, P.I.; Ny, T. A redox-sensitive loop regulates plasminogen activator inhibitor type 2 (PAI-2) polymerization. EMBO J. 2003, 22, 1753–1761. [Google Scholar] [CrossRef] [Green Version]
- Foy, C.A.; Grant, P.J. PCR-RFLP Detection od PAI-2 Gene Variants: Prevelence in Ethnic Groups and Disease Relationship in patients Undergoing corony Angiography. Thromb. Haemost. 1997, 77, 0955–0958. [Google Scholar] [CrossRef]
- Li, X.; Luo, J.-Y.; Zhang, L.; Yang, Y.-N.; Xie, X.; Liu, F.; Chen, B.-D.; Ma, Y.-T. Variant of PAI-2 gene is associated with coronary artery disease and recurrent coronary event risk in Chinese Han population. Lipids Health Dis. 2015, 14, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malle, E.; Woenckhaus, C.; Waeg, G.; Esterbauer, H.; Grone, E.F.; Grone, H.J. Immunological evidence for hypochlorite-modified proteins in human kidney. Am. J. Pathol. 1997, 150, 603–615. [Google Scholar]
- Song, C.; Burgess, S.; Eicher, J.D.; O’Donnell, C.J.; Johnson, A.D. Causal Effect of Plasminogen Activator Inhibitor Type 1 on Coronary Heart Disease. J. Am. Heart Assoc. 2017, 6, e004918. [Google Scholar] [CrossRef]
- Cao, C.; Smith, Q. Crevicular fluid myeloperoxidase at healthy, gingivitis and periodontitis sites. J. Clin. Periodontol. 1989, 16, 17–20. [Google Scholar] [CrossRef]
- Dagar, M.; Deepa, D.K.; Molly, M.; Sharma, A.; Khattak, B.P. Effect of nonsurgical periodontal therapy on salivary myeloperoxidase levels: A biochemical study. J. Indian Soc. Periodontol. 2015, 19, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Neilands, J.; Bikker, F.J.; Kinnby, B. PAI-2/SerpinB2 inhibits proteolytic activity in a P. gingivalis-dominated multispecies bacterial consortium. Arch. Oral. Biol. 2016, 70, 1–8. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, J.M.P.; Harish, S.; Pai, V.R.; Shriyan, C. Increased Oxidatively Modified Forms of Albumin in Association with Decreased Total Antioxidant Activity in Different Types of Hypertensive Disorders of Pregnancy. Indian J. Clin. Biochem. 2017, 32, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.-T.; Zhong, M.; Tian, J.-W.; Hou, F.-F. Higher plasma AOPP is associated with increased proteinuria excretion and decreased glomerular filtration rate in pre-eclamptic women. Pregnancy Hypertens. Int. J. Women’s Cardiovasc. Health 2013, 3, 16–20. [Google Scholar] [CrossRef]
- Goemans, C.V.; Collet, J.F. Stress-induced chaperones: A first line of defense against the powerful oxidant hypochlorous acid. F1000Research 2019, 8, 1678. [Google Scholar] [CrossRef]
- Mañucat-Tan, N.; Zeineddine Abdallah, R.; Kaur, H.; Saviane, D.; Wilson, M.R.; Wyatt, A.R. Hypochlorite-induced aggregation of fibrinogen underlies a novel antioxidant role in blood plasma. Redox Biol. 2021, 40, 101847. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cater, J.H.; Mañucat-Tan, N.B.; Georgiou, D.K.; Zhao, G.; Buhimschi, I.A.; Wyatt, A.R.; Ranson, M. A Novel Role for Plasminogen Activator Inhibitor Type-2 as a Hypochlorite-Resistant Serine Protease Inhibitor and Holdase Chaperone. Cells 2022, 11, 1152. https://doi.org/10.3390/cells11071152
Cater JH, Mañucat-Tan NB, Georgiou DK, Zhao G, Buhimschi IA, Wyatt AR, Ranson M. A Novel Role for Plasminogen Activator Inhibitor Type-2 as a Hypochlorite-Resistant Serine Protease Inhibitor and Holdase Chaperone. Cells. 2022; 11(7):1152. https://doi.org/10.3390/cells11071152
Chicago/Turabian StyleCater, Jordan H., Noralyn B. Mañucat-Tan, Demi K. Georgiou, Guomao Zhao, Irina A. Buhimschi, Amy R. Wyatt, and Marie Ranson. 2022. "A Novel Role for Plasminogen Activator Inhibitor Type-2 as a Hypochlorite-Resistant Serine Protease Inhibitor and Holdase Chaperone" Cells 11, no. 7: 1152. https://doi.org/10.3390/cells11071152