
Adaptive Remodeling of the Neuromuscular Junction with Aging

Abstract
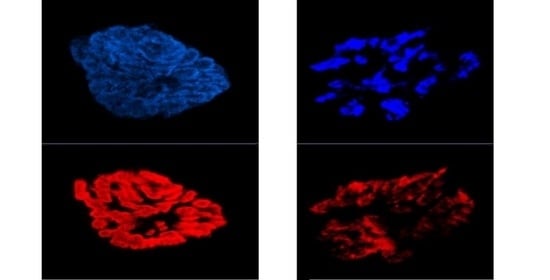
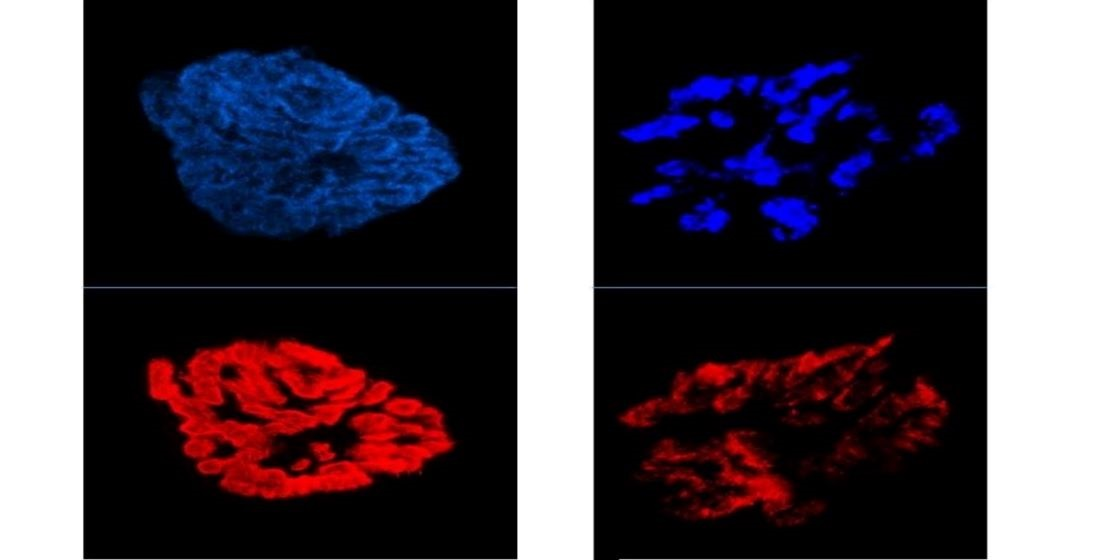
:
1. Introduction
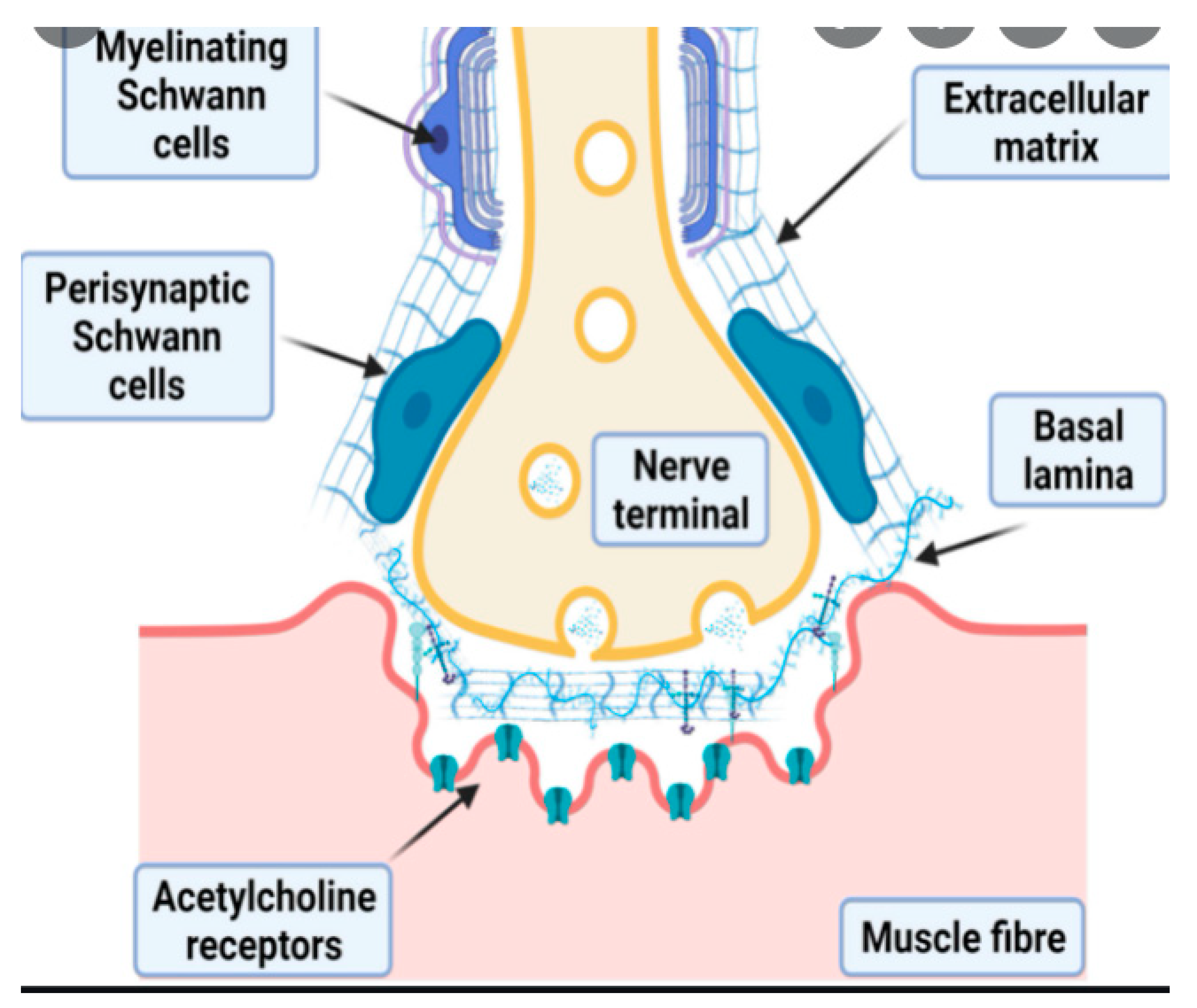
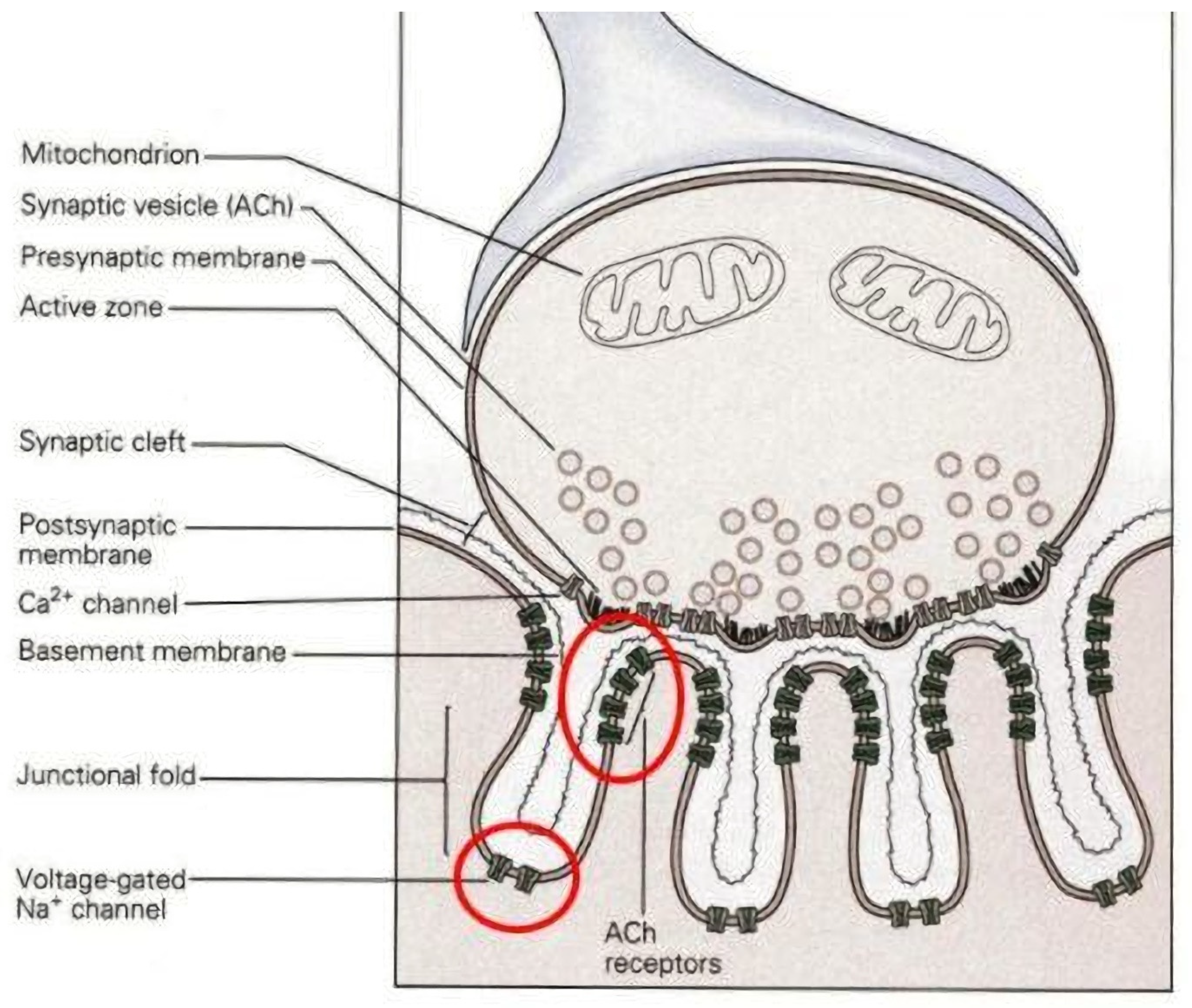
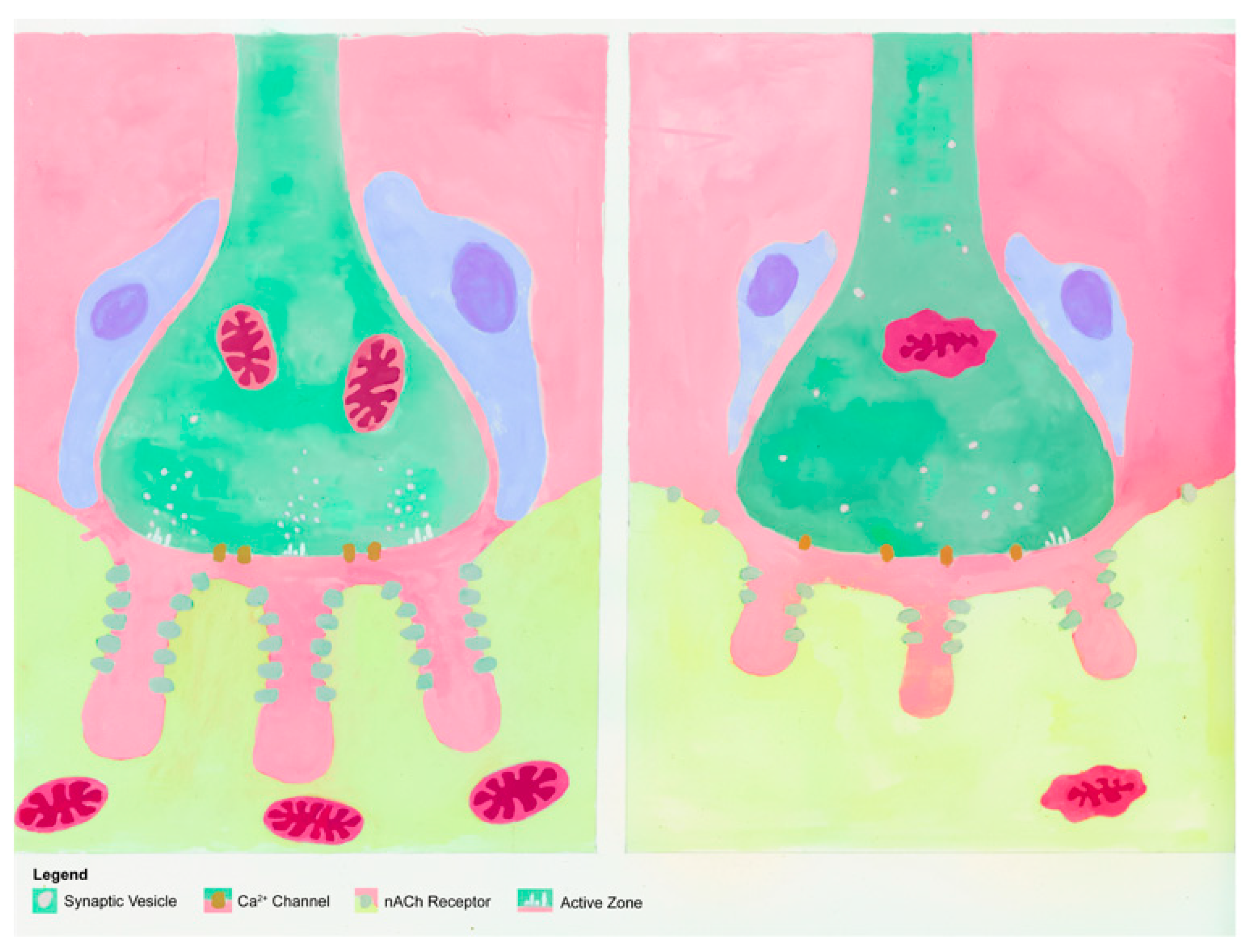
2. Design of NMJs and Motor Units
3. Effects of Aging on the NMJ
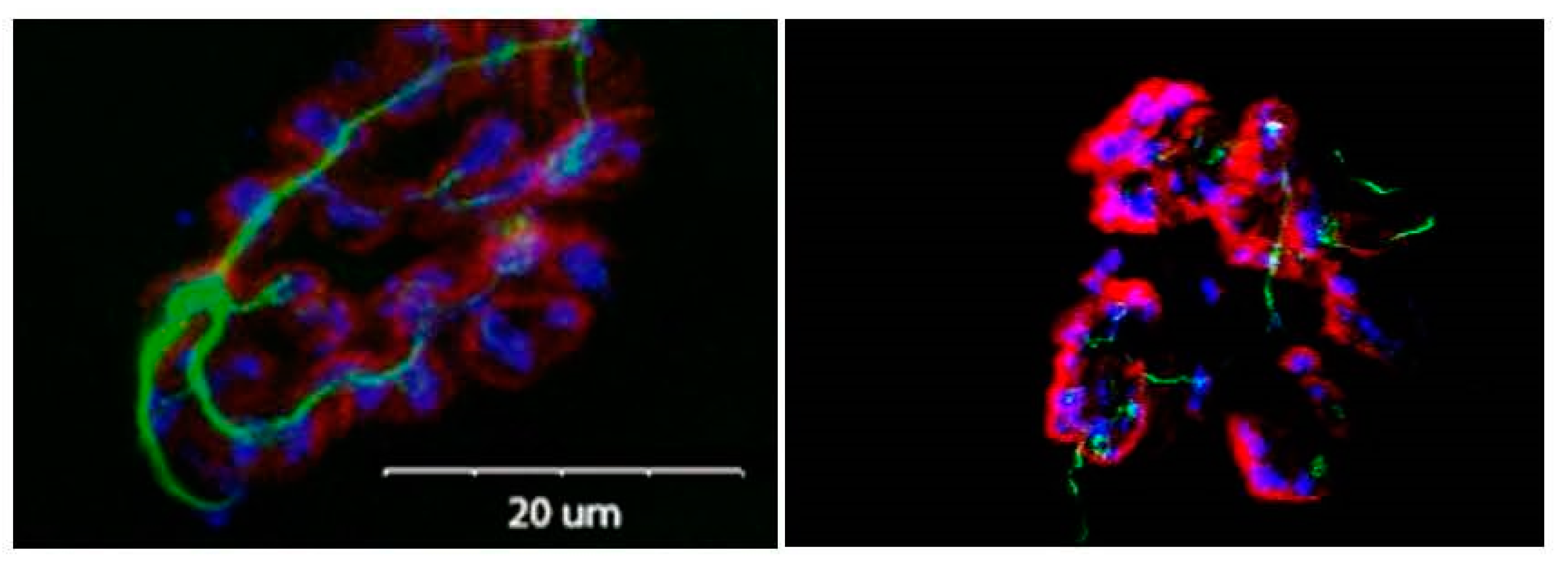
3.1. Morphological Adaptations
3.2. Physiological Adaptations
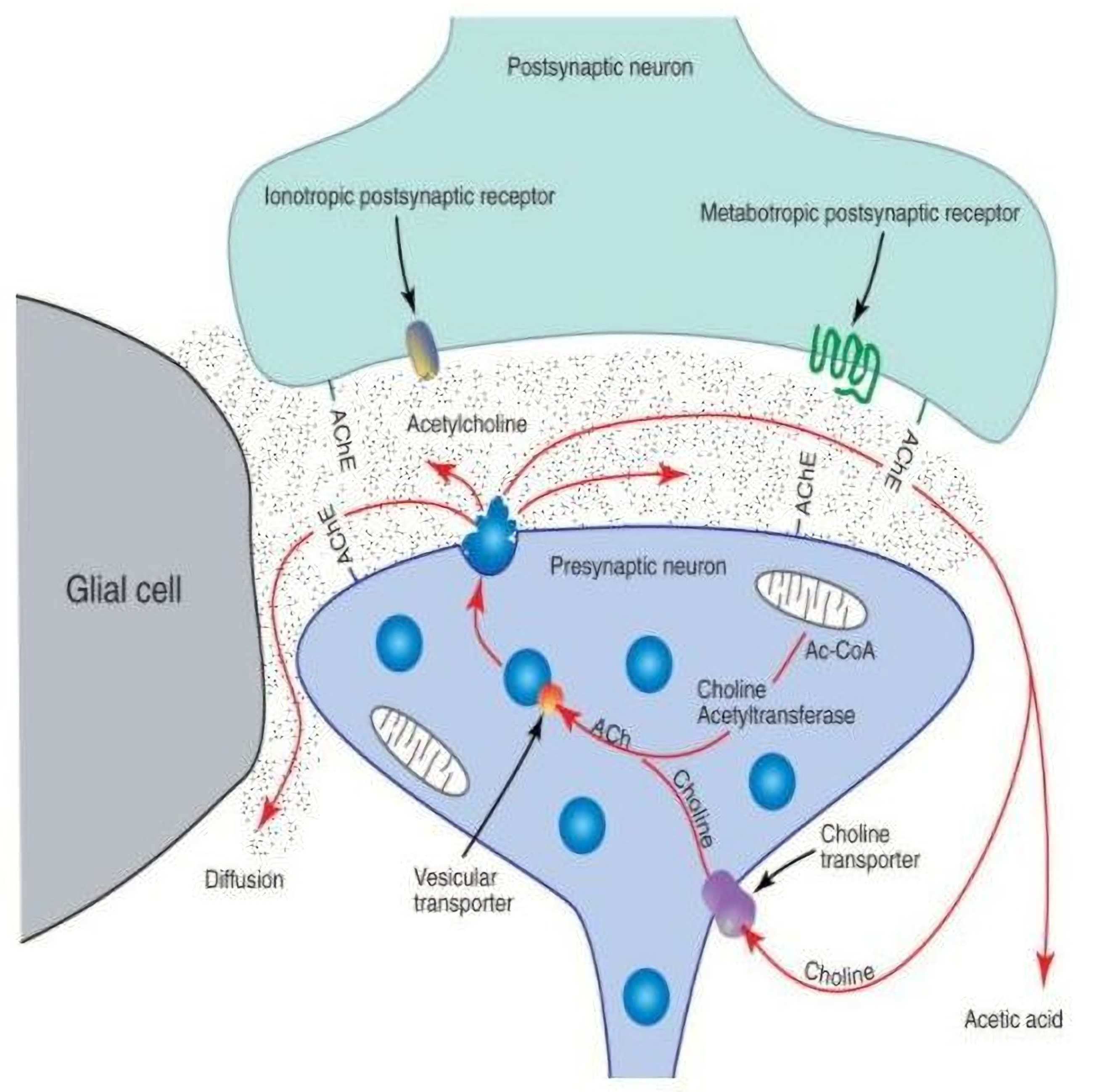
3.2.1. Presynaptic
3.2.2. Postsynaptic
3.3. Countering Aging-Related NMJ Adaptations
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kinsella, K.; Velkoff, V.A. Health and disability. Aging Clin. Exp. Res. 2002, 14, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Kaiser Family Foundation. Aging in the United States. Popul. Bull. 2019, 70. [Google Scholar]
- Ibebunjo, C.; Chick, J.M.; Kendall, T.; Eash, J.K.; Li, C.; Zhang, Y.; Vickers, C.; Wu, Z.; Clarke, B.A.; Shi, J.; et al. Genomic and proteomic profiling reveals reduced mitochondrial function and disruption of the neuromuscular junction driving rat sarcopenia. Mol. Cell. Biol. 2013, 33, 194–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudolf, R.; Khan, M.M.; Labeit, S.; Deschenes, M.R. Degeneration of neuromuscular junction in age and dystrophy. Front. Aging Neurosci. 2014, 6, 99. [Google Scholar] [CrossRef] [Green Version]
- Punga, A.R.; Ruegg, M.A. Signaling and aging at the neuromuscular synapse: Lessons learnt from neuromuscular diseases. Curr. Opin. Pharmacol. 2012, 12, 340–346. [Google Scholar] [CrossRef]
- Rosenberg, I.H. Sarcopenia: Origins and clinical relevance. J. Nutr. 1997, 127, 990S–991S. [Google Scholar] [CrossRef] [Green Version]
- Lexell, J.; Taylor, C.C.; Sjostrom, M. What is the cause of the ageing atrophy? Total number, size and proportion of different fiber types studied in whole vastus lateralis muscle from 15- to 83-year-old men. J. Neurol. Sci. 1988, 84, 275–294. [Google Scholar] [CrossRef]
- Vandervoort, A.A. Aging of the human neuromuscular system. Muscle Nerve 2002, 25, 17–25. [Google Scholar] [CrossRef]
- Gonzalez-Freire, M.; de Cabo, R.; Studenski, S.A.; Ferrucci, L. The Neuromuscular Junction: Aging at the Crossroad between Nerves and Muscle. Front. Aging Neurosci. 2014, 6, 208. [Google Scholar] [CrossRef] [Green Version]
- Pannérec, A.; Springer, M.; Migliavacca, E.; Ireland, A.; Piasecki, M.; Karaz, S.; Jacot, G.; Métairon, S.; Danenberg, E.; Raymond, F.; et al. A robust neuromuscular system protects rat and human skeletal muscle from sarcopenia. Aging 2016, 8, 712–729. [Google Scholar] [CrossRef] [Green Version]
- Monti, E.; Reggiani, C.; Franchi, M.V.; Toniolo, L.; Sandri, M.; Armani, A.; Zampieri, S.; Giacomello, E.; Sarto, F.; Sirago, G.; et al. Neuromuscular junction instability and altered intracellular calcium handling as early determinants of force loss during unloading in humans. J. Physiol. 2021, 599, 3037–3061. [Google Scholar] [CrossRef] [PubMed]
- Eisen, A. The Dying Forward Hypothesis of ALS: Tracing Its History. Brain Sci. 2021, 11, 300. [Google Scholar] [CrossRef] [PubMed]
- Cappello, V.; Francolini, M. Neuromuscular Junction Dismantling in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2017, 18, 2092. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.R.; Shah, S.B.; Lovering, R.M. The Neuromuscular Junction: Roles in Aging and Neuromuscular Disease. Int. J. Mol. Sci. 2021, 22, 8058. [Google Scholar] [CrossRef]
- Clark, J.A.; Southam, K.A.; Blizzard, C.A.; King, A.E.; Dickson, T.C. Axonal degeneration, distal collateral branching and neuromuscular junction architecture alterations occur prior to symptom onset in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. J. Chem. Neuroanat. 2016, 76, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Fischer, L.R.; Culver, D.G.; Tennant, P.; Davis, A.A.; Wang, M.; Castellano-Sanchez, A.; Khan, J.; Polak, M.A.; Glass, J.D. Amyotrophic lateral sclerosis is a distal axonopathy: Evidence in mice and man. Exp. Neurol. 2004, 185, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Khurana, S.; Vats, A.; Sahu, B.; Ganguly, N.K.; Chakraborti, P.; Gourie-Devi, M.; Taneja, V. Neuromuscular Junction Dysfunction in Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2022, 59, 1502–1527. [Google Scholar] [CrossRef]
- Lipka, A.F.; Boldingh, M.I.; van Zwet, E.W.; Schreurs, M.W.; Kuks, J.B.; Tallaksen, C.M.; Titulaer, M.J.; Verschuuren, J.J. Long-term follow-up, quality of life, and survival of patients with Lambert-Eaton myasthenic syndrome. Neurology 2020, 94, e511–e520. [Google Scholar] [CrossRef] [Green Version]
- Kesner, V.G.; Oh, S.J.; Dimachkie, M.M.; Barohn, R.J. Lambert-Eaton Myasthenic Syndrome. Neurol. Clin. 2018, 36, 379–394. [Google Scholar] [CrossRef]
- Koneczny, I.; Herbst, R. Myasthenia Gravis: Pathogenic Effects of Autoantibodies on Neuromuscular Architecture. Cells 2019, 8, 671. [Google Scholar] [CrossRef] [Green Version]
- Farrugia, M.E.; Goodfellow, J.A. A Practical Approach to Managing Patients With Myasthenia Gravis-Opinions and a Review of the Literature. Front. Neurol. 2020, 11, 604. [Google Scholar] [CrossRef]
- Wang, S.; Breskovska, I.; Gandhy, S.; Punga, A.R.; Guptill, J.T.; Kaminski, H.J. Advances in autoimmune myasthenia gravis management. Expert Rev. Neurother. 2018, 18, 573–588. [Google Scholar] [CrossRef] [PubMed]
- Slater, C.R. The Structure of Human Neuromuscular Junctions: Some Unanswered Molecular Questions. Int. J. Mol. Sci. 2017, 18, 2183. [Google Scholar] [CrossRef]
- Slater, C.R. Structural factors influencing the efficacy of neuromuscular transmission. Ann. N. Y. Acad. Sci. 2008, 1132, 1–12. [Google Scholar] [CrossRef]
- Deschenes, M.R. Motor unit and neuromuscular junction remodeling with aging. Curr. Aging Sci. 2011, 4, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Arbour, D.; Vande Velde, C.; Robitaille, R. New perspectives on amyotrophic lateral sclerosis: The role of glial cells at the neuromuscular junction. J. Physiol. 2017, 593, 647–661. [Google Scholar] [CrossRef]
- Barik, A.; Li, L.; Sathyamurthy, A.; Xiong, W.C.; Mei, L. Schwann Cells in Neuromuscular Junction Formation and Maintenance. J. Neurosci. 2016, 36, 9770–9781. [Google Scholar] [CrossRef] [Green Version]
- Duregotti, E.; Negro, S.; Scorzeto, M.; Zornetta, I.; Dickinson, B.C.; Chang, C.J.; Montecucco, C.; Rigoni, M. Mitochondrial alarmins released by degenerating motor axon terminals activate perisynaptic Schwann cells. Proc. Natl. Acad. Sci. USA 2015, 112, E497–E505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, R.J.; Vukovic, J.; Dunlop, S.; Grounds, M.D.; Shavlakadze, T. Striking denervation of neuromuscular junctions without lumbar motoneuron loss in geriatric mouse muscle. PLoS ONE 2011, 6, e28090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lichtman, J.W.; Sanes, J.R. Watching the neuromuscular junction. J. Neurocytol. 2003, 32, 767–775. [Google Scholar] [CrossRef]
- Sanes, J.R.; Lichtman, J.W. Development of the vertebrate neuromuscular junction. Annu. Rev. Neurosci. 1999, 22, 389–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henneman, E. Relation between size of neurons and their susceptibility to discharge. Science 1957, 126, 1345–1347. [Google Scholar] [CrossRef] [PubMed]
- Burke, R.E. Motor unit types of cat triceps surae muscle. J. Physiol. 1967, 193, 141–160. [Google Scholar] [CrossRef] [PubMed]
- Delgado, R.; Maureira, C.; Oliva, C.; Kidokoro, Y.; Labarca, P. Size of vesicle pools, rates of mobilization, and recycling at neuromuscular synapses of a Drosophila mutant, shibire. Neuron 2000, 28, 941–953. [Google Scholar] [CrossRef] [Green Version]
- Maggio, S.; Ceccaroli, P.; Polidori, E.; Cioccoloni, A.; Stocchi, V.; Guescini, M. Signal Exchange through Extracellular Vesicles in Neuromuscular Junction Establishment and Maintenance: From Physiology to Pathology. Int. J. Mol. Sci. 2019, 20, 2804. [Google Scholar] [CrossRef] [Green Version]
- Blunk, A.D.; Akbergenova, Y.; Cho, R.W.; Lee, J.; Walldorf, U.; Xu, K.; Zhong, G.; Zhuang, X.; Littleton, J.T. Postsynaptic actin regulates active zone spacing and glutamate receptor apposition at the Drosophila neuromuscular junction. Mol. Cell. Neurosci. 2014, 61, 241–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homan, A.E.; Meriney, S.D. Active zone structure-function relationships at the neuromuscular junction. Synapse 2018, 72, e22057. [Google Scholar] [CrossRef]
- Laghaei, R.; Ma, J.; Tarr, T.B.; Homan, A.E.; Kelly, L.S.; Tilvawala, M.S.; Vuocolo, B.S.; Rajasekaran, H.P.; Meriney, S.D.; Dittrich, M. Transmitter release site organization can predict synaptic function at the neuromuscular junction. J. Neurophysiol. 2018, 119, 1340–1355. [Google Scholar] [CrossRef]
- Blotnick-Rubin, E.; Anglister, L. Fine Localization of Acetylcholinesterase in the Synaptic Cleft of the Vertebrate Neuromuscular Junction. Front. Mol. Neurosci. 2018, 11, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soreq, H.; Seidman, S. Acetylcholinesterase--new roles for an old actor. Nat. Rev. Neurosci. 2001, 2, 294–302. [Google Scholar] [CrossRef]
- Balice-Gordon, R.J. Age-related changes in neuromuscular innervation. Muscle Nerve Suppl. 1997, 5, 83–87. [Google Scholar] [CrossRef]
- Cardasis, C.A.; Padykula, H.A. Ultrastructural evidence indicating reorganization at the neuromuscular junction in the normal rat soleus muscle. Anat. Rec. 1981, 200, 41–59. [Google Scholar] [CrossRef] [PubMed]
- Robbins, N.; Fahim, M.A. Progression of age changes in mature mouse motor nerve terminals and its relation to locomotor activity. J. Neurocytol. 1985, 14, 1019–1036. [Google Scholar] [CrossRef] [PubMed]
- Deschenes, M.R.; Tufts, H.L.; Oh, J.; Li, S.; Noronha, A.L.; Adan, M.A. Effects of exercise training on neuromuscular junctions and their active zones in young and aged muscles. Neurobiol. Aging 2020, 95, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Deschenes, M.R.; Kressin, K.A.; Garratt, R.N.; Leathrum, C.M.; Shaffrey, E.C. Effects of exercise training on neuromuscular junction morphology and pre- to post-synaptic coupling in young and aged rats. Neuroscience 2016, 316, 167–177. [Google Scholar] [CrossRef] [Green Version]
- Prakash, Y.S.; Sieck, G.C. Age-related remodeling of neuromuscular junctions on type-identified diaphragm fibers. Muscle Nerve 1998, 21, 887–895. [Google Scholar] [CrossRef]
- Khosa, S.; Trikamji, B.; Khosa, G.S.; Khanli, H.M.; Mishra, S.K. An Overview of Neuromuscular Junction Aging Findings in Human and Animal Studies. Curr. Aging Sci. 2019, 12, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Willadt, S.; Nash, M.; Slater, C. Age-related changes in the structure and function of mammalian neuromuscular junctions. Ann. N. Y. Acad. Sci. 2018, 1412, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Andonian, M.H.; Fahim, M.A. Nerve terminal morphology in C57BL/6NNia mice at different ages. J. Gerontol. 1989, 44, B43–B51. [Google Scholar] [CrossRef]
- Fahim, M.A. Endurance exercise modulates neuromuscular junction of C57BL/6NNia aging mice. J. Appl. Physiol. 1997, 83, 59–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fish, L.A.; Fallon, J.R. Multiple MuSK signaling pathways and the aging neuromuscular junction. Neurosci. Lett. 2020, 731, 135014. [Google Scholar] [CrossRef] [PubMed]
- Deschenes, M.R.; Maresh, C.M.; Crivello, J.F.; Armstrong, L.E.; Kraemer, W.J.; Covault, J. The effects of exercise training of different intensities on neuromuscular junction morphology. J. Neurocytol. 1993, 22, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Deschenes, M.R.; Sherman, E.G.; Roby, M.A.; Glass, E.K.; Harris, M.B. Effect of resistance training on neuromuscular junctions of young and aged muscles featuring different recruitment patterns. J. Neurosci. Res. 2015, 93, 504–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taetzsch, T.; Valdez, G. NMJ maintenance and repair in aging. Curr. Opin. Physiol. 2018, 4, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Nishimune, H.; Badawi, Y.; Mori, S.; Shigemoto, K. Dual-color STED microscopy reveals a sandwich structure of Bassoon and Piccolo in active zones of adult and aged mice. Sci. Rep. 2016, 6, 27935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Mizushige, T.; Nishimune, H. Active zone density is conserved during synaptic growth but impaired in aged mice. J. Comp. Neurol. 2012, 520, 434–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milde, S.; Adalbert, R.; Elaman, M.H.; Coleman, M.P. Axonal transport declines with age in two distinct phases separated by a period of relative stability. Neurobiol. Aging 2015, 36, 971–981. [Google Scholar] [CrossRef] [Green Version]
- Minoshima, S.; Cross, D. In vivo imaging of axonal transport using MRI: Aging and Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2008, 35 (Suppl. 1), S89–S92. [Google Scholar] [CrossRef]
- Deschenes, M.R.; Hurst, T.E.; Ramser, A.E.; Sherman, E.G. Presynaptic to postsynaptic relationships of the neuromuscular junction are held constant across age and muscle fiber type. Dev. Neurobiol. 2013, 73, 744–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Billings, S.E.; Nishimune, H. Calcium channels link the muscle-derived synapse organizer laminin beta2 to Bassoon and CAST/Erc2 to organize presynaptic active zones. J. Neurosci. 2011, 31, 512–525. [Google Scholar] [CrossRef] [Green Version]
- Fahim, M.A.; Robbins, N. Ultrastructural studies of young and old mouse neuromuscular junctions. J. Neurocytol. 1982, 11, 641–656. [Google Scholar] [CrossRef] [PubMed]
- Arnold, A.-S.; Gill, J.; Christe, M.; Ruiz, R.; McGuirk, S.; St-Pierre, J.; Tabares, L.; Handschin, C. Morphological and functional remodelling of the neuromuscular junction by skeletal muscle PGC-1alpha. Nat. Commun. 2014, 5, 3569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deschenes, M.R.; Adan, M.A.; Kapral, M.C.; Kressin, K.A.; Leathrum, C.M.; Seo, A.; Li, S.; Schaffrey, E.C. Neuromuscular adaptability of male and female rats to muscle unloading. J. Neurosci. Res. 2018, 96, 284–296. [Google Scholar] [CrossRef]
- Rosenheimer, J.L.; Smith, D.O. Differential changes in the end-plate architecture of functionally diverse muscles during aging. J. Neurophysiol. 1985, 53, 1567–1581. [Google Scholar] [CrossRef] [PubMed]
- Hunter, S.K.; Pereira, H.M.; Keenan, K.G. The aging neuromuscular system and motor performance. J. Appl. Physiol. 2016, 121, 982–995. [Google Scholar] [CrossRef]
- Wilson, M.H.; Deschenes, M.R. The neuromuscular junction: Anatomical features and adaptations to various forms of increased, or decreased neuromuscular activity. Int. J. Neurosci. 2005, 115, 803–828. [Google Scholar] [CrossRef]
- Willadt, S.; Nash, M.; Slater, C.R. Age-related fragmentation of the motor endplate is not associated with impaired neuromuscular transmission in the mouse diaphragm. Sci. Rep. 2016, 20, 24849. [Google Scholar] [CrossRef]
- Slater, C.R. ‘Fragmentation’ of NMJs: A sign of degeneration or regeneration? A long journey with many junctions. Neuroscience 2020, 439, 28–40. [Google Scholar] [CrossRef]
- Bao, Z.; Cui, C.; Chow, S.K.; Qin, L.; Wong, R.M.Y.; Cheung, W.H. AChRs Degeneration at NMJ in Aging-Associated Sarcopenia-A Systematic Review. Front. Aging Neurosci. 2020, 12, 597811. [Google Scholar] [CrossRef]
- Jang, Y.C.; Van Remmen, H. Age-associated alterations of the neuromuscular junction. Exp. Gerontol. 2011, 46, 193–198. [Google Scholar] [CrossRef] [Green Version]
- Vaughan, S.K.; Sutherland, N.M.; Valdez, G. Attenuating Cholinergic Transmission Increases the Number of Satellite Cells and Preserves Muscle Mass in Old Age. Front. Aging Neurosci. 2019, 11, 262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anagnostou, M.E.; Hepple, R.T. Mitochondrial Mechanisms of Neuromuscular Junction Degeneration with Aging. Cells 2020, 9, 197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canto-Santos, J.; Grau-Junyent, J.M.; Garrabou, G. The Impact of Mitochondrial Deficiencies in Neuromuscular Diseases. Antioxidants 2020, 9, 964. [Google Scholar] [CrossRef] [PubMed]
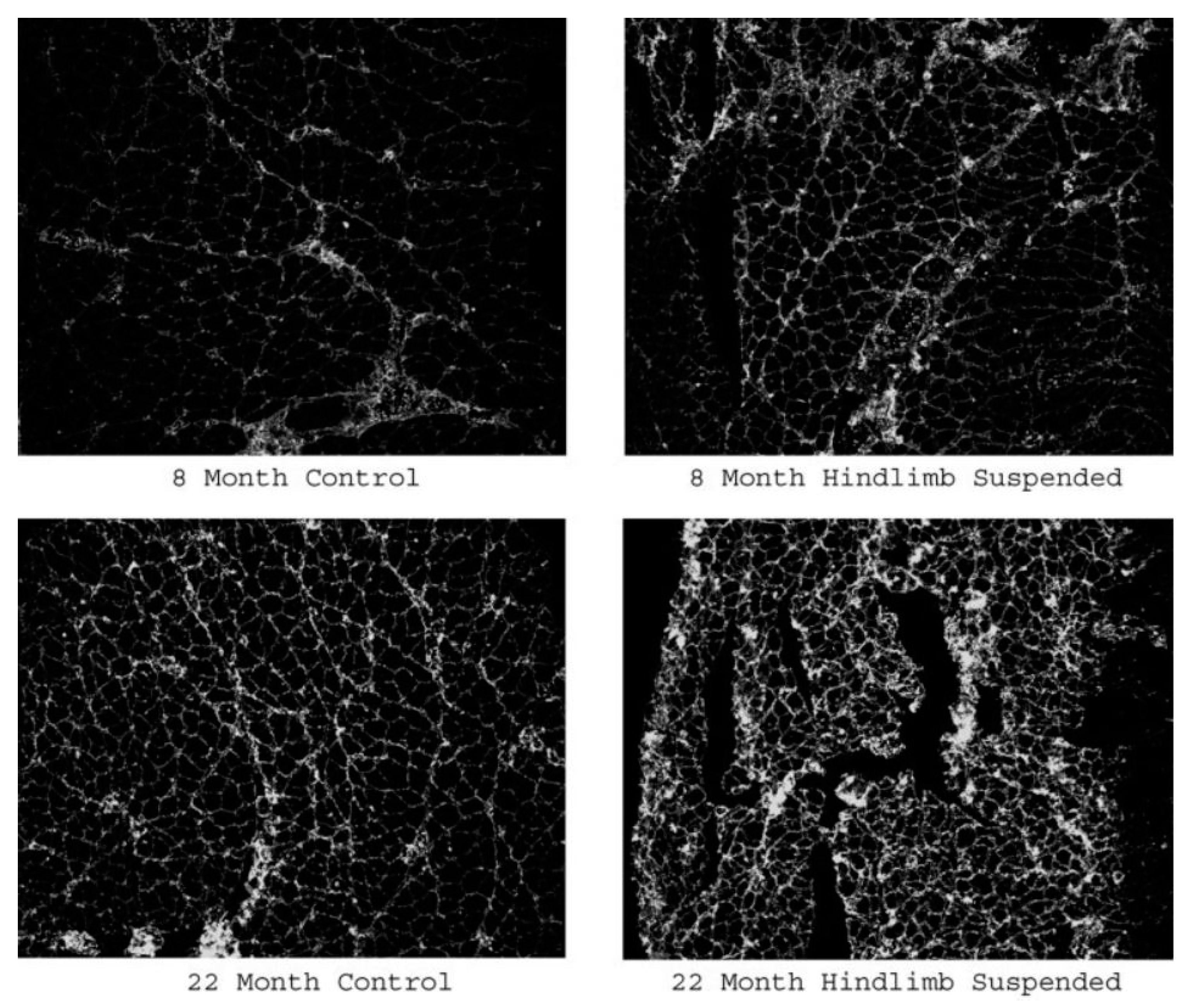
- Baehr, L.M.; West, D.W.D.; Marcotte, G.; Marshall, A.G.; De Sousa, L.G.; Baar, K.; Bodine, S.C. Age-related deficits in skeletal muscle recovery following disuse are associated with neuromuscular junction instability and ER stress, not impaired protein synthesis. Aging 2016, 8, 127–146. [Google Scholar] [CrossRef] [Green Version]
- Genin, E.C.; Hounoum, B.M.; Bannwarth, S.; Fragaki, K.; Lacas-Gervais, S.; Mauri-Crouzet, A.; Lespinasse, F.; Neveu, J.; Ropert, B.; Augé, G.; et al. Mitochondrial defect in muscle precedes neuromuscular junction degeneration and motor neuron death in CHCHD10(S59L/+) mouse. Acta Neuropathol. 2019, 138, 123–145. [Google Scholar] [CrossRef]
- Covault, J.; Merlie, J.P.; Goridis, C.; Sanes, J.R. Molecular forms of N-CAM and its RNA in developing and denervated skeletal muscle. J. Cell Biol. 1986, 102, 731–739. [Google Scholar] [CrossRef] [Green Version]
- Covault, J.; Sanes, J.R. Neural cell adhesion molecule (N-CAM) accumulates in denervated and paralyzed skeletal muscles. Proc. Natl. Acad. Sci. USA 1985, 82, 4544–4548. [Google Scholar] [CrossRef] [Green Version]
- Covault, J.; Sanes, J.R. Distribution of N-CAM in synaptic and extrasynaptic portions of developing and adult skeletal muscle. J. Cell Biol. 1986, 102, 716–730. [Google Scholar] [CrossRef] [Green Version]
- Elkerdany, M.K.; Fahim, M.A. Age changes in neuromuscular junctions of masseter muscle. Anat. Rec. 1993, 237, 291–295. [Google Scholar] [CrossRef]
- Lee, S.B.; Oh, J.H.; Park, J.H.; Choi, J.H.; Wee, J.H. Differences in youngest-old, middle-old, and odest-old patients who visit the emergency department. Clin. Exp. Emerg. Med. 2018, 5, 249–255. [Google Scholar] [CrossRef]
- Büttner, R.; Schulz, A.; Reuter, M.; Akula, A.K.; Mindos, T.; Carlstedt, A.; Riecken, L.B.; Baader, S.L.; Bauer, R.; Morrison, H. Inflammaging impairs peripheral nerve maintenance and regeneration. Aging Cell 2018, 17, e12833. [Google Scholar] [CrossRef] [PubMed]
- Fuertes-Alvarez, S.; Izeta, A. Terminal schwann cell aging: Implications for age-associated neuromuscualr dysfunction. Aging Dis. 2021, 12, 494. [Google Scholar] [CrossRef] [PubMed]
- Alshuaib, W.B.; Fahim, M.A. Effect of exercise on physiological age-related change at mouse neuromuscular junctions. Neurobiol. Aging 1990, 11, 555–561. [Google Scholar] [CrossRef]
- Mahoney, R.E.; Rawson, J.M.; Eaton, B.A. An age-dependent change in the set point of synaptic homeostasis. J. Neurosci. 2014, 34, 2111–2119. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.A.; Reich, C.D.; Dissanayake, K.N.; Kristmundsdottir, F.; Findlater, G.S.; Ribchester, R.R.; Simmen, M.W.; Gillingwater, T.H. NMJ-morph reveals principal components of synaptic morphology influencing structure-function relationships at the neuromuscular junction. Open Biol. 2016, 6, 160240. [Google Scholar] [CrossRef]
- Tintignac, L.A.; Brenner, H.R.; Ruegg, M.A. Mechanisms Regulating Neuromuscular Junction Development and Function and Causes of Muscle Wasting. Physiol. Rev. 2015, 95, 809–852. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.O. Non-uniform changes in nerve-terminal calcium homeostasis during aging. Neurobiol. Aging 1987, 8, 366–368. [Google Scholar] [CrossRef]
- Smith, D.O.; Rosenheimer, J.L. Decreased sprouting and degeneration of nerve terminals of active muscles in aged rats. J. Neurophysiol. 1982, 48, 100–109. [Google Scholar] [CrossRef]
- Smith, D.O. Acetylcholine storage, release and leakage at the neuromuscular junction of mature adult and aged rats. J. Physiol. 1984, 347, 161–176. [Google Scholar] [CrossRef]
- Giovannini, F.; Sher, E.; Webster, R.; Boot, J.; Lang, B. Calcium channel subtypes contributing to acetylcholine release from normal, 4-aminopyridine-treated and myasthenic syndrome auto-antibodies-affected neuromuscular junctions. Br. J. Pharmacol. 2002, 136, 1135–1145. [Google Scholar] [CrossRef] [Green Version]
- Sudhof, T.C.; Rizo, J. Synaptic vesicle exocytosis. Cold Spring Harb. Perspect. Biol. 2011, 3, a005637. [Google Scholar] [CrossRef]
- Liu, Y.; Sugiura, Y.; Sudhof, T.C.; Lin, W. Ablation of all synaptobrevin vSNARES blocks evoked but not spontaneous neurotransmitter release at neuromuscular synapses. J. Neurosci. 2019, 31, 6049–6066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, T.P.; Dai, Z.S. The neuromuscular junction revisited: Ca2+ channels and transmitter release in cholinergic neurones in Xenopus nerve and muscle cell culture. J. Exp. Biol. 1990, 153, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Deschenes, M.R.; Tufts, H.L.; Noronha, A.L.; Li, S. Both aging and exercise training alter the rate of recovery of neuromuscular performance of male soleus muscles. Biogerontology 2019, 20, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Pratt, J.; De Vito, G.; Narici, M.; Segurado, R.; Dolan, J.; Conroy, J.; Boreham, C. Grip strength performance from 9431 participants of the GenoFit study: Normative data and associated factors. Geroscience 2021, 43, 2533–2546. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J. Ca2+ mediated coupling between neuromuscular junction and mitochondria in skeletal muscle. Neurosci. Lett. 2021, 754, 135899. [Google Scholar] [CrossRef] [PubMed]
- Spendiff, S.; Vuda, M.; Gouspillou, G.; Aare, S.; Perez, A.; Morais, J.A.; Jagoe, R.T.; Filion, M.-E.; Glicksman, R.; Kapchinsky, S.; et al. Denervation drives mitochondrial dysfunction in skeletal muscle of octogenarians. J. Physiol. 2016, 594, 7361–7379. [Google Scholar] [CrossRef] [PubMed]
- Austin, S.; St-Pierre, J. PGC1alpha and mitochondrial metabolism—emerging concepts and relevance in ageing and neurodegenerative disorders. J. Cell Sci. 2012, 125, 4963–4971. [Google Scholar] [CrossRef] [Green Version]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jager, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef] [Green Version]
- Fulop, T.; Larbi, A.; Pawelec, G.; Khalil, A.; Cohen, A.A.; Hirokawa, K.; Witkowski, J.M.; Franceschi, C. Immunology of Aging: The Birth of Inflammaging. Clin. Rev. Allergy Immunol. 2021, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Zhao, R.; Wan, Q.-Y.; Wu, Y.; Zhou, Y.; Wang, Y.; Cui, Y.; Shen, X.; Wu, X. Sarcopenia and adverse health-related outcomes: An umbrella review of meta-analyses of observational studies. Cancer Med. 2020, 9, 7964–7978. [Google Scholar] [CrossRef]
- Patton, B.L.; Miner, J.H.; Chiu, A.Y.; Sanes, J.R. Distribution and function of laminins in the neuromuscular system of developing, adult, and mutant mice. J. Cell Biol. 1997, 139, 1507–1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuel, M.A.; Valdez, G.; Tapia, J.C.; Lichtman, J.W.; Sanes, J.R. Agrin and synaptic laminin are required to maintain adult neuromuscular junctions. PLoS ONE 2012, 7, e46663. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.M.; Chand, K.K.; Hammond, L.A.; Lavidis, N.A.; Noakes, P.G. Functional decline at the aging neuromuscular junction is associated with altered laminin-alpha4 expression. Aging 2017, 9, 880–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, K.; Boppart, M.D. Influence of exercise and aging on extracellular matrix composition in the skeletal muscle stem cell niche. J. Appl. Physiol. 2016, 121, 1053–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, R.S.; Nishimune, H. The role of laminins in the organization and function of neuromuscular junctions. Matrix Biol. 2017, 57–58, 86–105. [Google Scholar] [CrossRef] [Green Version]
- Sugita, S.; Fleming, L.L.; Wood, C.; Vaughan, S.; Gomes, M.P.S.M.; Camargo, W.; Naves, L.A.; Prado, V.; Prado, M.A.M.; Guatimosim, C.; et al. VAChT overexpression increases acetylcholine at the synaptic cleft and accelerates aging of neuromuscular junctions. Skelet Muscle 2016, 6, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, M.C.; Lin, W.; Yang, J.; Dominguez, B.; Padgett, D.; Sugiura, Y.; Aryal, P.; Gould, T.W.; Oppenheim, R.W.; Hester, M.E.; et al. Acetylcholine negatively regulates development of the neuromuscular junction through distinct cellular mechanisms. Proc. Natl. Acad. Sci. USA 2010, 107, 10702–10707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, K.; Shen, C.; Li, L.; Wu, H.; Xing, G.; Dong, Z.; Jing, H.; Chen, W.; Zhang, H.; Tan, Z.; et al. Sarcoglycan alpha mitigates neuromuscular junction decline in aged mice by stabilizing LRP4. J. Neurosci. 2018, 38, 8860–8873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, M.E.; Kramarcy, N.; Krall, S.P.; Rossi, S.G.; Rotundo, R.L.; Sealock, R.; Froehner, S.C. Absence of alpha-syntrophin leads to structurally aberrant neuromuscular synapses deficient in utrophin. J. Cell Biol. 2000, 150, 1385–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courtney, J.; Steinbach, J.H. Age changes in neuromuscular junction morphology and acetylcholine receptor distribution on rat skeletal muscle fibres. J. Physiol. 1981, 320, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.R.; Robbins, N.; Fang, Z.P. Plasticity of presynaptic and postsynaptic elements of neuromuscular junctions repeatedly observed in living adult mice. J. Neurocytol. 1991, 20, 165–182. [Google Scholar] [CrossRef] [PubMed]
- Wigston, D.J. Repeated in vivo visualization of neuromuscular junctions in adult mouse lateral gastrocnemius. J. Neurosci. 1990, 10, 1753–1761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banker, B.Q.; Kelly, S.S.; Robbins, N. Neuromuscular transmission and correlative morphology in young and old mice. J. Physiol. 1983, 339, 355–377. [Google Scholar] [CrossRef] [PubMed]
- Burke, S.K.; Fenton, A.I.; Konokhova, Y.; Hepple, R.T. Variation in muscle and neuromuscular junction morphology between atrophy-resistant and atrophy-prone muscles supports failed re-innervation in aging muscle atrophy. Exp. Gerontol. 2021, 156, 111613. [Google Scholar] [CrossRef] [PubMed]
- Moreira-Pais, A.; Ferreira, R.; Oliveira, P.A.; Duarte, J.A. A neuromuscular perspective of sarcopenia pathogenesis: Deciphering the signaling pathways involved. Geroscience 2022, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Jacob, J.M.; Robbins, N. Differential effects of age on neuromuscular transmission in partially denervated mouse muscle. J. Neurosci. 1990, 10, 1522–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robbins, N. Compensatory plasticity of aging at the neuromuscular junction. Exp. Gerontol. 1992, 27, 75–81. [Google Scholar] [CrossRef]
- Veronese, N.; Demurtas, J.; Soysal, P.; Smith, L.; Torbahn, G.; Schoene, D.; Schwingshackl, L.; Sieber, C.; Bauer, J.; Cesari, M.; et al. Sarcopenia and health-related outcomes: An umbrella review of observational studies. Eur. Geriatr. Med. 2019, 10, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.C.; Liu, Y.; Hayworth, C.R.; Bhattacharya, A.; Lustgarten, M.S.; Muller, F.L.; Chaudhuri, A.; Qi, W.; Li, Y.; Huang, J.-Y.; et al. Dietary restriction attenuates age-associated muscle atrophy by lowering oxidative stress in mice even in complete absence of CuZnSOD. Aging Cell 2012, 11, 770–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockinger, J.; Maxwell, N.; Shapiro, D.; deCabo, R.; Valdez, G. Caloric Restriction Mimetics Slow Aging of Neuromuscular Synapses and Muscle Fibers. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 73, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Valdez, G.; Tapia, J.C.; Kang, H.; Clemenson, G.D., Jr.; Gage, F.H.; Lichtman, J.W.; Sanes, J.R. Attenuation of age-related changes in mouse neuromuscular synapses by caloric restriction and exercise. Proc. Natl. Acad. Sci. USA 2010, 107, 14863–14868. [Google Scholar] [CrossRef] [Green Version]
- Nishimune, H.; Stanford, J.A.; Mori, Y. Role of exercise in maintaining the integrity of the neuromuscular junction. Muscle Nerve 2014, 49, 315–324. [Google Scholar] [CrossRef] [Green Version]
- Neto, W.K.; Ciena, A.P.; Anaruma, C.A.; de Souza, R.R.; Gama, E.F. Effects of exercise on neuromuscualr junction components across age: Systematic review of animal experimental studies. BMC Res. Notes 2015, 8, 713. [Google Scholar] [CrossRef] [Green Version]
- Deschenes, M.R.; Roby, M.A.; Eason, M.K.; Harris, M.B. Remodeling of the neuromuscular junction precedes sarcopenia related alterations in myofibers. Exp. Gerontol. 2010, 45, 389–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, A.; Morsch, M.; Murata, Y.; Ghazanfari, N.; Reddel, S.W.; Phillips, W.D. Sequence of age-associated changes to the mouse neuromuscular junction and the protective effects of voluntary exercise. PLoS ONE 2013, 8, e67970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deschenes, M.R.; Holdren, A.N.; McCoy, R.W. Adaptations to short-term muscle unloading in young and aged men. Med. Sci. Sports Exerc. 2008, 40, 856–863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hugon, M.; Massion, J.; Wiesendanger, M. Anticipatory postural changes induced by active unloading and comparison with passive unloading in man. Pflugers Arch. 1982, 393, 292–296. [Google Scholar] [CrossRef]
- Deschenes, M.R.; McCoy, R.W.; Mangis, K.A. Chronic Resistance Training Does Not Ameliorate Unloading-Induced Decrements in Neuromuscular Function. Am. J. Phys. Med. Rehabil. 2017, 96, 549–556. [Google Scholar] [CrossRef]
- Deschenes, M.R.; McCoy, R.W.; Mangis, K.A. Factors relating to gender specificity of unloading-induced declines in strength. Muscle Nerve 2012, 46, 210–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deschenes, M.R.; Wilson, M.H. Age-related differences in synaptic plasticity following muscle unloading. J. Neurobiol. 2003, 57, 246–256. [Google Scholar] [CrossRef] [PubMed]
- Deschenes, M.R.; Will, K.M.; Booth, F.W.; Gordon, S.E. Unlike myofibers, neuromuscular junctions remain stable during prolonged muscle unloading. J. Neurol. Sci. 2003, 210, 5–10. [Google Scholar] [CrossRef]
- Pratt, J.; De Vito, G.; Narici, M.; Boreham, C. Neuromuscular Junction Aging: A Role for Biomarkers and Exercise. J. Gerontol. A Biol. Sci. Med. Sci. 2021, 76, 576–585. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Presynaptic | Authors |
| Increased complexity of nerve terminal branching | Khosa et al., 2019; Andonian and Fahim, 1989; Fahim, 1997 |
| Increased nerve terminal branch number | Deschenes et al., 2020; Prakash and Sieck, 1998; Deschene et al., 2010 |
| Increased total nerve terminal branch length | Robbins and Fahim, 1985; Deschenes et al., 2020; Deschenes et al., 2016 |
| Increased planar area of nerve terminal branch length | Fahim, 1997; Prakash and Sieck, 1998 |
| Increased area of vesicle clusters | Deschenes et al., 2011; Deschenes et al., 2010 |
| Decreased total number of vesicles | Deschenes et al., 2015; Taetzch and Valdez, 2018 |
| Decreased number and concentration of active zones | Nishimune et al., 2016 |
| Postsynaptic | Authors |
| Abandoned incidence of abandoned endplate gutters | Rosenheimer and Smith, 1985; Bao et al., 2020 |
| Increased fragmentation of receptors | Willadt et al., 2016; Deschenes et al., 2015; Hunter, 2016 |
| Decreased length of endplate | Vaughn et al., 2019; Arnold et al., 2014 |
| Decreased total area of endplate | Prakash and Sieck, 1998; Fahim and Robbins, 1982 |
| Decreased perimeter length around endplate | Jang and Van Remmen, 2011; Elkerdany and Fahim, 1993 |
| Increased expression of NCAM | Deschenes and Wilson, 2003 |
| Presynaptic Alterations | Authors |
| Increased quantal content | Fahim, 1997; Alshuaib and Fahim, 1990; Mahoney et al., 2014 |
| Increased quantal size | Fahim, 1997; Jones et al., 2016 |
| Increased spontaneous release of ACh | Smith, 1984; Smith and Weiler, 1987 |
| Decreased calcium clearance from nerve terminal | Smith, 1987 |
| Postsynaptic | Authors |
| Increased endplate potential amplitude | Iyer, 2021; Smith, 1987 |
| Reduced safety factor of endplate potential | Giovannini et al., 2002; Liu et al., 2019 |
| Increased synaptic depression during train of stimuli | Feng and Dai, 1990 |
| Increased incidence of neurotransmission failure | Fahim, 1997; Smith and Weiler, 1987 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deschenes, M.R.; Flannery, R.; Hawbaker, A.; Patek, L.; Mifsud, M. Adaptive Remodeling of the Neuromuscular Junction with Aging. Cells 2022, 11, 1150. https://doi.org/10.3390/cells11071150
Deschenes MR, Flannery R, Hawbaker A, Patek L, Mifsud M. Adaptive Remodeling of the Neuromuscular Junction with Aging. Cells. 2022; 11(7):1150. https://doi.org/10.3390/cells11071150
Chicago/Turabian StyleDeschenes, Michael R., Rachel Flannery, Alexis Hawbaker, Leah Patek, and Mia Mifsud. 2022. "Adaptive Remodeling of the Neuromuscular Junction with Aging" Cells 11, no. 7: 1150. https://doi.org/10.3390/cells11071150