
Clinical Grade Human Pluripotent Stem Cell-Derived Engineered Skin Substitutes Promote Keratinocytes Wound Closure In Vitro

 , , , , and
, , , , and 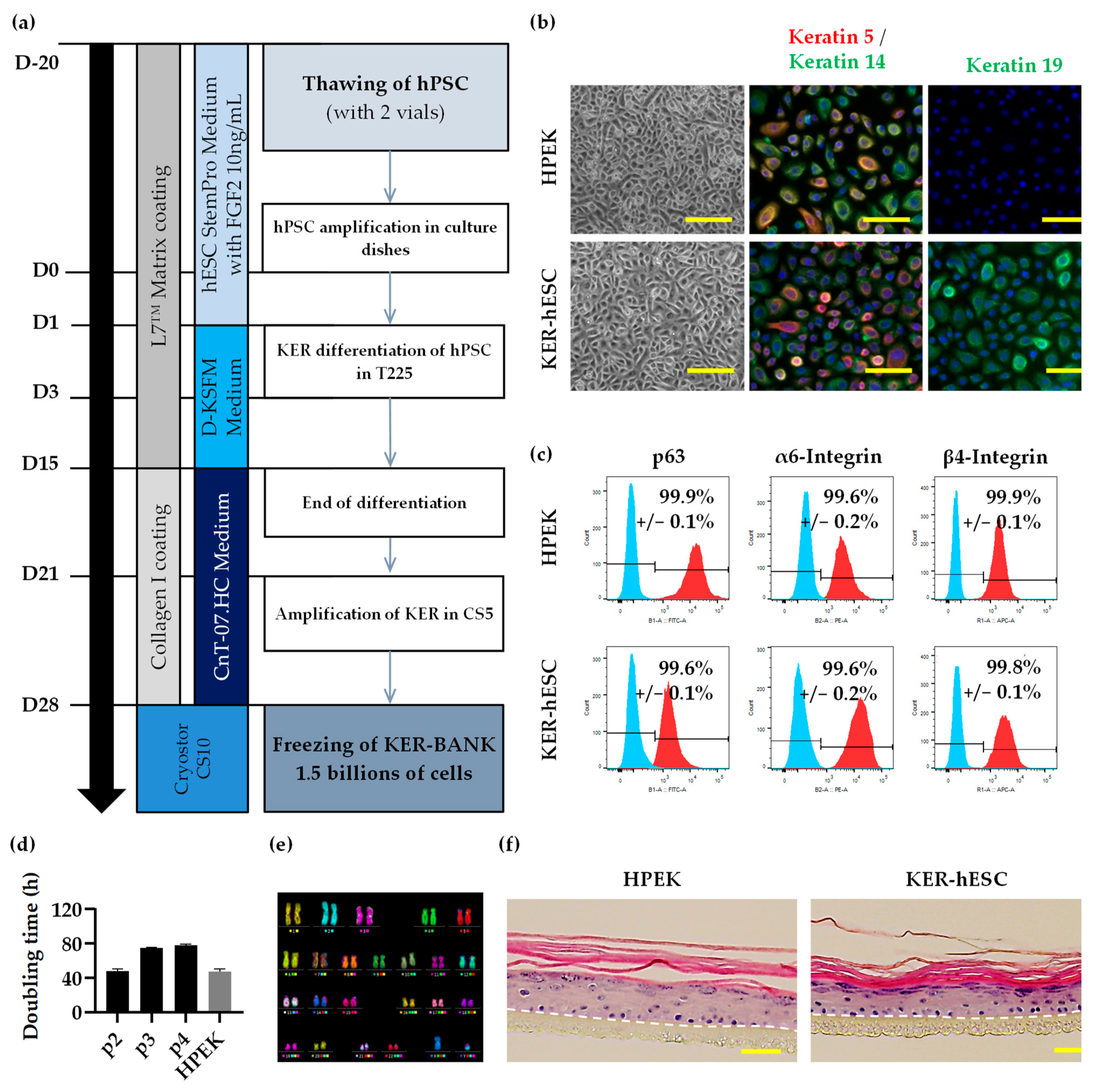{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. hPSC Culture
2.2. Karyotype Analysis
2.3. Immunofluorescence Analysis
2.4. Flow Cytometry Analysis
2.5. RT-qPCR
2.6. Keratinocyte Differentiation and Culture
2.7. Fibroblast and Myofibroblast Differentiation and Culture
2.8. 3D Cell Culture and Analysis
2.9. Scratch Wound Assay and Viability Analysis
2.10. Mycoplasma Detection
2.11. Statistical Analysis
3. Results
3.1. Characterization of Compatible Clinical Grade hESC
3.2. Development of Keratinocyte Differentiation Protocol Compatible with Clinical Grade Regulatory Standards
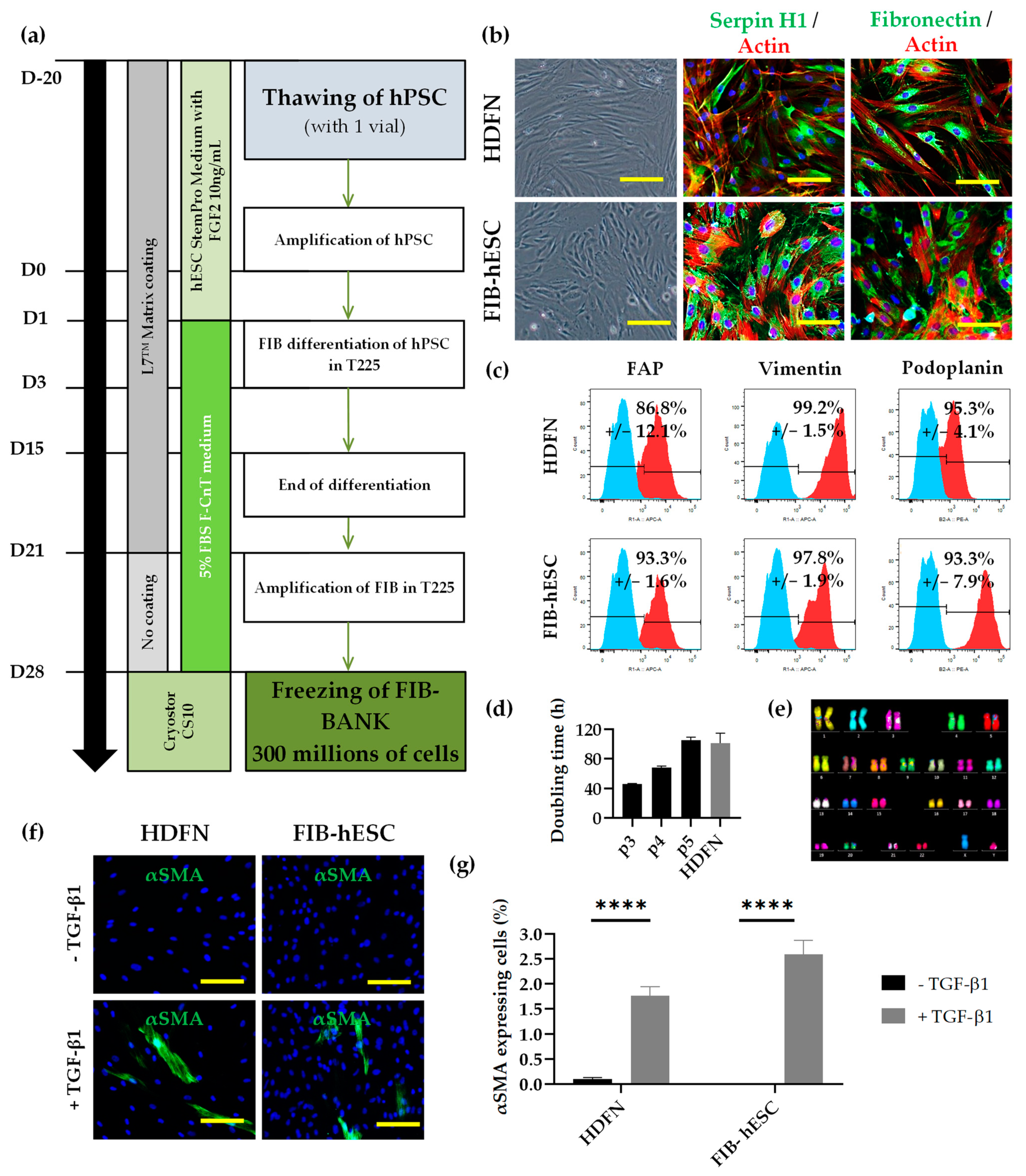
3.3. Development of Fibroblast Differentiation Protocol Compatible with Clinical Grade Regulatory Standards
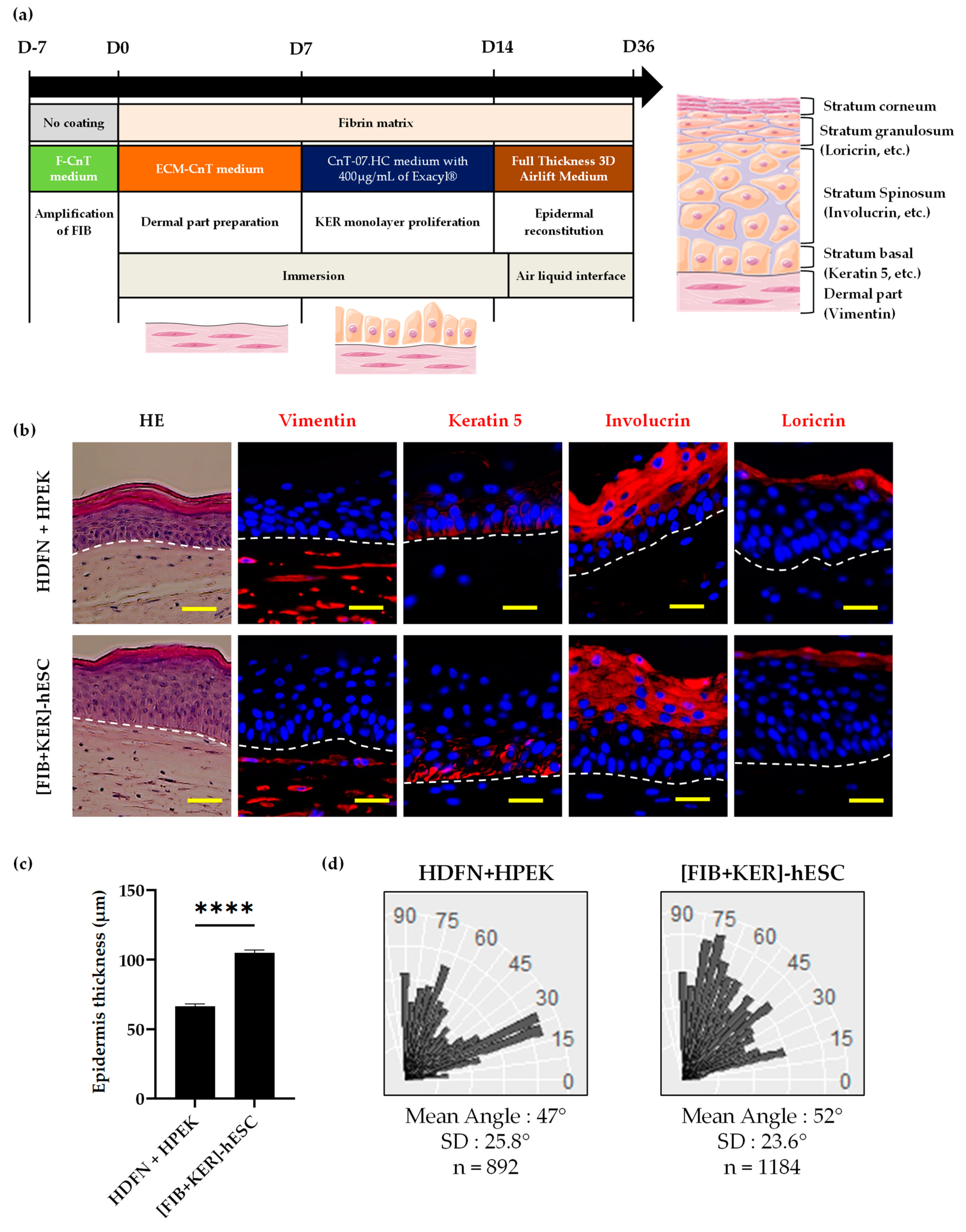
3.4. Characterization of Dermo-Epidermal Tissue Using GMPc Plasma-Based Fibrin Matrix
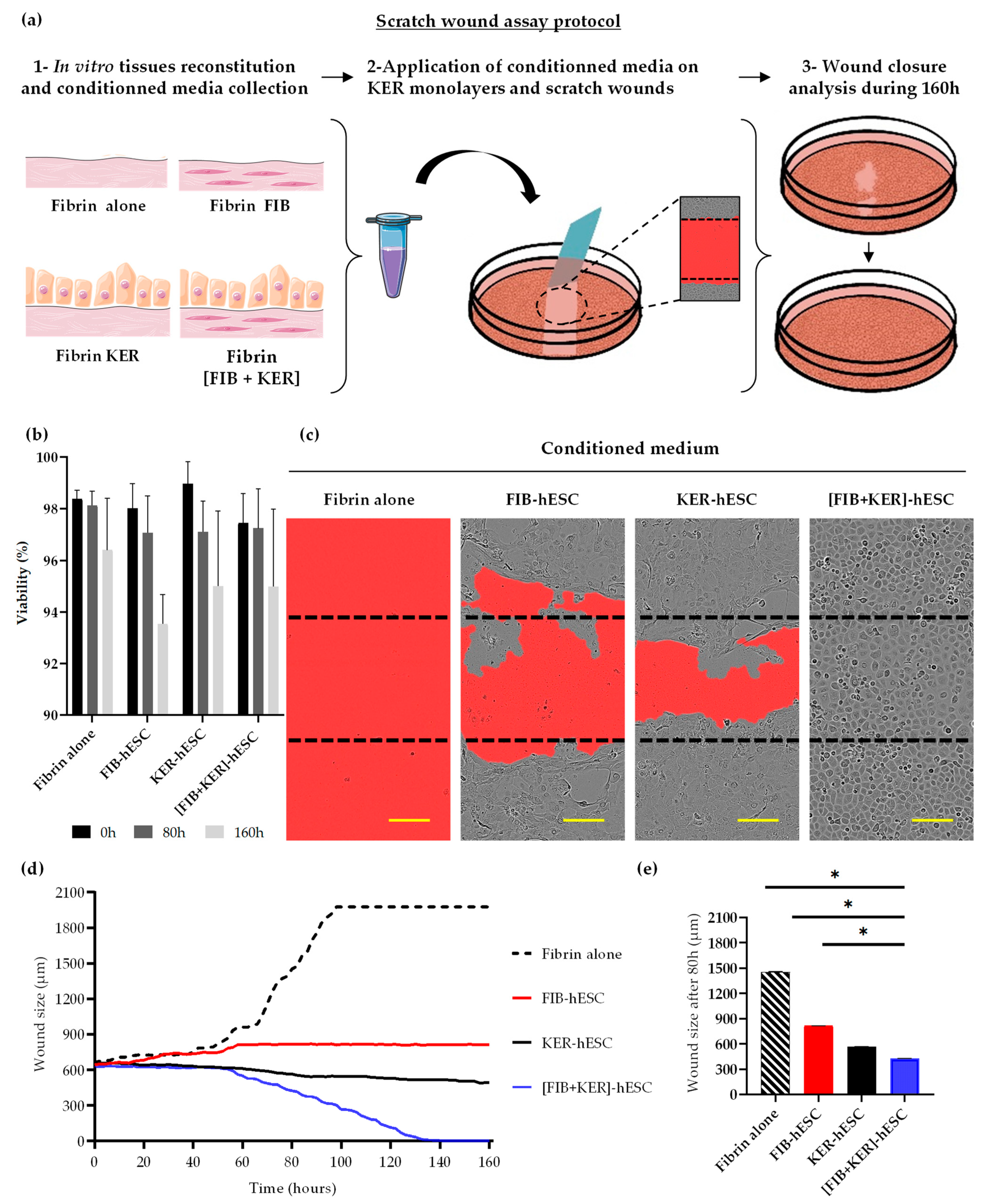
3.5. Conditioned Media from GMPc Human Embryonic Stem Cell-Derived Engineered Skin Substitutes Improve Keratinocytes Wound Closure In Vitro
3.6. Evaluation of GMPc Protocols Using Human-Induced Pluripotent Stem Cells
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sharma, P.; Kumar, A.; Dey, A.D.; Behl, T.; Chadha, S. Stem Cells and Growth Factors-Based Delivery Approaches for Chronic Wound Repair and Regeneration: A Promise to heal from within. Life Sci. 2021, 268, 118932. [Google Scholar] [CrossRef]
- Boniakowski, A.E.; Kimball, A.S.; Jacobs, B.N.; Kunkel, S.L.; Gallagher, K.A. Macrophage-Mediated Inflammation in Normal and Diabetic Wound Healing. J. Immunol. 2017, 199, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Nourian Dehkordi, A.; Mirahmadi Babaheydari, F.; Chehelgerdi, M.; Raeisi Dehkordi, S. Skin Tissue Engineering: Wound Healing Based on Stem-Cell-Based Therapeutic Strategies. Stem Cell Res. Ther. 2019, 10, 111. [Google Scholar] [CrossRef] [Green Version]
- Monfort, J.-B.; Senet, P. Leg Ulcers in Sickle-Cell Disease: Treatment Update. Adv. Wound Care 2020, 9, 348–356. [Google Scholar] [CrossRef]
- Olsson, M.; Järbrink, K.; Divakar, U.; Bajpai, R.; Upton, Z.; Schmidtchen, A.; Car, J. The Humanistic and Economic Burden of Chronic Wounds: A Systematic Review. Wound Repair Regen. 2019, 27, 114–125. [Google Scholar] [CrossRef] [Green Version]
- Han, G.; Ceilley, R. Chronic Wound Healing: A Review of Current Management and Treatments. Adv. Ther. 2017, 34, 599–610. [Google Scholar] [CrossRef] [Green Version]
- Dai, C.; Shih, S.; Khachemoune, A. Skin Substitutes for Acute and Chronic Wound Healing: An Updated Review. J. Dermatol. Treat. 2020, 31, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Duscher, D.; Barrera, J.; Wong, V.W.; Maan, Z.N.; Whittam, A.J.; Januszyk, M.; Gurtner, G.C. Stem Cells in Wound Healing: The Future of Regenerative Medicine? A Mini-Review. Gerontology 2016, 62, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Ojeh, N.; Pastar, I.; Tomic-Canic, M.; Stojadinovic, O. Stem Cells in Skin Regeneration, Wound Healing, and Their Clinical Applications. Int. J. Mol. Sci. 2015, 16, 25476–25501. [Google Scholar] [CrossRef]
- Ohnuki, M.; Takahashi, K. Present and Future Challenges of Induced Pluripotent Stem Cells. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20140367. [Google Scholar] [CrossRef]
- Guenou, H.; Nissan, X.; Larcher, F.; Feteira, J.; Lemaitre, G.; Saidani, M.; Del Rio, M.; Barrault, C.C.; Bernard, F.-X.; Peschanski, M.; et al. Human Embryonic Stem-Cell Derivatives for Full Reconstruction of the Pluristratified Epidermis: A Preclinical Study. Lancet 2009, 374, 1745–1753. [Google Scholar] [CrossRef]
- Itoh, M.; Kiuru, M.; Cairo, M.S.; Christiano, A.M. Generation of Keratinocytes from Normal and Recessive Dystrophic Epidermolysis Bullosa-Induced Pluripotent Stem Cells. Proc. Natl. Acad. Sci. USA 2011, 108, 8797–8802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sah, S.K.; Kanaujiya, J.K.; Chen, I.-P.; Reichenberger, E.J. Generation of Keratinocytes from Human Induced Pluripotent Stem Cells Under Defined Culture Conditions. Cell. Reprogramming 2021, 23, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Shamis, Y.; Hewitt, K.J.; Carlson, M.W.; Margvelashvilli, M.; Dong, S.; Kuo, C.K.; Daheron, L.; Egles, C.; Garlick, J.A. Fibroblasts Derived from Human Embryonic Stem Cells Direct Development and Repair of 3D Human Skin Equivalents. Stem Cell Res. Ther. 2011, 2, 10. [Google Scholar] [CrossRef] [Green Version]
- Itoh, M.; Umegaki-Arao, N.; Guo, Z.; Liu, L.; Higgins, C.A.; Christiano, A.M. Generation of 3D Skin Equivalents Fully Reconstituted from Human Induced Pluripotent Stem Cells (IPSCs). PLoS ONE 2013, 8, e77673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sachamitr, P.; Leishman, A.J.; Davies, T.J.; Fairchild, P.J. Directed Differentiation of Human Induced Pluripotent Stem Cells into Dendritic Cells Displaying Tolerogenic Properties and Resembling the CD141 + Subset. Front. Immunol. 2017, 8, 1935. [Google Scholar] [CrossRef] [PubMed]
- Kobold, S.; Guhr, A.; Mah, N.; Bultjer, N.; Seltmann, S.; Seiler Wulczyn, A.E.M.; Stacey, G.; Jie, H.; Liu, W.; Löser, P.; et al. A Manually Curated Database on Clinical Studies Involving Cell Products Derived from Human Pluripotent Stem Cells. Stem Cell Rep. 2020, 15, 546–555. [Google Scholar] [CrossRef]
- De Sousa, P.A.; Downie, J.M.; Tye, B.J.; Bruce, K.; Dand, P.; Dhanjal, S.; Serhal, P.; Harper, J.; Turner, M.; Bateman, M. Development and Production of Good Manufacturing Practice Grade Human Embryonic Stem Cell Lines as Source Material for Clinical Application. Stem Cell Res. 2016, 17, 379–390. [Google Scholar] [CrossRef] [Green Version]
- Cavallero, S.; Neves Granito, R.; Stockholm, D.; Azzolin, P.; Martin, M.T.; Fortunel, N.O. Exposure of Human Skin Organoids to Low Genotoxic Stress Can Promote Epithelial-to-Mesenchymal Transition in Regenerating Keratinocyte Precursor Cells. Cells 2020, 9, 1912. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Park, N.; Rim, Y.A.; Nam, Y.; Jung, H.; Lee, K.; Ju, J.H. Establishment of a Complex Skin Structure via Layered Co-Culture of Keratinocytes and Fibroblasts Derived from Induced Pluripotent Stem Cells. Stem Cell Res. Ther. 2018, 9, 217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soares, E.; Xu, Q.; Li, Q.; Qu, J.; Zheng, Y.; Raeven, H.H.M.; Brandao, K.O.; Petit, I.; van den Akker, W.M.R.; van Heeringen, S.J.; et al. Single-Cell RNA-Seq Identifies a Reversible Mesodermal Activation in Abnormally Specified Epithelia of P63 EEC Syndrome. Proc. Natl. Acad. Sci. USA 2019, 116, 17361–17370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Tian, L.; Shen, M.; Tu, C.; Wu, H.; Gu, M.; Paik, D.T.; Wu, J.C. Generation of Quiescent Cardiac Fibroblasts From Human Induced Pluripotent Stem Cells for In Vitro Modeling of Cardiac Fibrosis. Circ. Res. 2019, 125, 552–566. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, G.; Ranno, R.; Stracuzzi, G.; Bondanza, S.; Guerra, L.; Zambruno, G.; Micali, G.; De Luca, M. The Control of Epidermal Stem Cells (Holoclones) in the Treatment of Massive Full-Thickness Burns with Autologous Keratinocytes Cultured on Fibrin. Transplantation 1999, 68, 868–879. [Google Scholar] [CrossRef] [PubMed]
- Ronfard, V.; Rives, J.M.; Neveux, Y.; Carsin, H.; Barrandon, Y. Long-Term Regeneration of Human Epidermis on Third Degree Burns Transplanted with Autologous Cultured Epithelium Grown on a Fibrin Matrix. Transplantation 2000, 70, 1588–1598. [Google Scholar] [CrossRef] [PubMed]
- Janson, D.G.; Saintigny, G.; van Adrichem, A.; Mahé, C.; El Ghalbzouri, A. Different Gene Expression Patterns in Human Papillary and Reticular Fibroblasts. J. Investig. Dermatol. 2012, 132, 2565–2572. [Google Scholar] [CrossRef] [Green Version]
- Sriram, G.; Bigliardi, P.L.; Bigliardi-Qi, M. Fibroblast Heterogeneity and Its Implications for Engineering Organotypic Skin Models in Vitro. Eur. J. Cell Biol. 2015, 94, 483–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Corte, P.; Verween, G.; Verbeken, G.; Rose, T.; Jennes, S.; De Coninck, A.; Roseeuw, D.; Vanderkelen, A.; Kets, E.; Haddow, D.; et al. Feeder Layer- and Animal Product-Free Culture of Neonatal Foreskin Keratinocytes: Improved Performance, Usability, Quality and Safety. Cell Tissue Bank. 2012, 13, 175–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kljenak, A.; Tominac Trcin, M.; Bujić, M.; Dolenec, T.; Jevak, M.; Mršić, G.; Zmiš, G.; Barčot, Z.; Muljačić, A.; Popović, M. Fibrin Gel as a Scaffold for Skin Substitute–Production and Clinical Experience. Acta Clin. Croat. 2016, 55, 279–289. [Google Scholar] [CrossRef] [Green Version]
- Llames, S.G.; Del Rio, M.; Larcher, F.; García, E.; García, M.; Escamez, M.J.; Jorcano, J.L.; Holguín, P.; Meana, A. Human Plasma as a Dermal Scaffold for the Generation of a Completely Autologous Bioengineered Skin. Transplantation 2004, 77, 350–355. [Google Scholar] [CrossRef]
- Sun, T.; Jackson, S.; Haycock, J.W.; MacNeil, S. Culture of Skin Cells in 3D Rather than 2D Improves Their Ability to Survive Exposure to Cytotoxic Agents. J. Biotechnol. 2006, 122, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Boehnke, K.; Mirancea, N.; Pavesio, A.; Fusenig, N.E.; Boukamp, P.; Stark, H.-J. Effects of Fibroblasts and Microenvironment on Epidermal Regeneration and Tissue Function in Long-Term Skin Equivalents. Eur. J. Cell Biol. 2007, 86, 731–746. [Google Scholar] [CrossRef] [PubMed]
- Kubo, K.; Kuroyanagi, Y. A Study of Cytokines Released from Fibroblasts in Cultured Dermal Substitute. Artif. Organs 2005, 29, 845–849. [Google Scholar] [CrossRef] [PubMed]
- Allen-Hoffmann, B.L.; Schlosser, S.J.; Ivarie, C.A.; Sattler, C.A.; Meisner, L.F.; O’Connor, S.L. Normal Growth and Differentiation in a Spontaneously Immortalized Near-Diploid Human Keratinocyte Cell Line, NIKS. J. Investig. Dermatol. 2000, 114, 444–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamanaka, S. Pluripotent Stem Cell-Based Cell Therapy-Promise and Challenges. Cell Stem Cell 2020, 27, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Lemaître, G.; Nissan, X.; Baldeschi, C.; Peschanski, M. Concise Review: Epidermal Grafting: The Case for Pluripotent Stem Cells. Stem Cells 2011, 29, 895–899. [Google Scholar] [CrossRef]
- Sullivan, S.; Fairchild, P.J.; Marsh, S.G.E.; Müller, C.R.; Turner, M.L.; Song, J.; Turner, D. Haplobanking Induced Pluripotent Stem Cells for Clinical Use. Stem Cell Res. 2020, 49, 102035. [Google Scholar] [CrossRef]
- Bogomiakova, M.E.; Sekretova, E.K.; Eremeev, A.V.; Shuvalova, L.D.; Bobrovsky, P.A.; Zerkalenkova, E.A.; Lebedeva, O.S.; Lagarkova, M.A. Derivation of Induced Pluripotent Stem Cells Line (RCPCMi007-A-1) with Inactivation of the Beta-2-Microglobulin Gene by CRISPR/Cas9 Genome Editing. Stem Cell Res. 2021, 55, 102451. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Domingues, S.; Darle, A.; Masson, Y.; Saidani, M.; Lagoutte, E.; Bejanariu, A.; Coutier, J.; Ayata, R.E.; Bouschbacher, M.; Peschanski, M.; et al. Clinical Grade Human Pluripotent Stem Cell-Derived Engineered Skin Substitutes Promote Keratinocytes Wound Closure In Vitro. Cells 2022, 11, 1151. https://doi.org/10.3390/cells11071151
Domingues S, Darle A, Masson Y, Saidani M, Lagoutte E, Bejanariu A, Coutier J, Ayata RE, Bouschbacher M, Peschanski M, et al. Clinical Grade Human Pluripotent Stem Cell-Derived Engineered Skin Substitutes Promote Keratinocytes Wound Closure In Vitro. Cells. 2022; 11(7):1151. https://doi.org/10.3390/cells11071151
Chicago/Turabian StyleDomingues, Sophie, Annabelle Darle, Yolande Masson, Manoubia Saidani, Emilie Lagoutte, Ana Bejanariu, Julien Coutier, Raif Eren Ayata, Marielle Bouschbacher, Marc Peschanski, and et al. 2022. "Clinical Grade Human Pluripotent Stem Cell-Derived Engineered Skin Substitutes Promote Keratinocytes Wound Closure In Vitro" Cells 11, no. 7: 1151. https://doi.org/10.3390/cells11071151