
Microtubules as Regulators of Neural Network Shape and Function: Focus on Excitability, Plasticity and Memory


Abstract
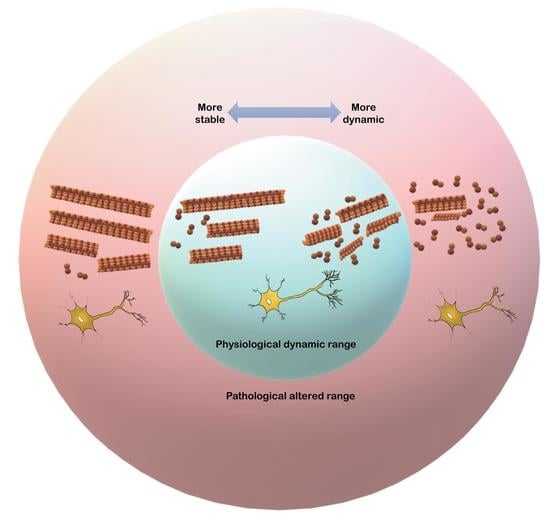
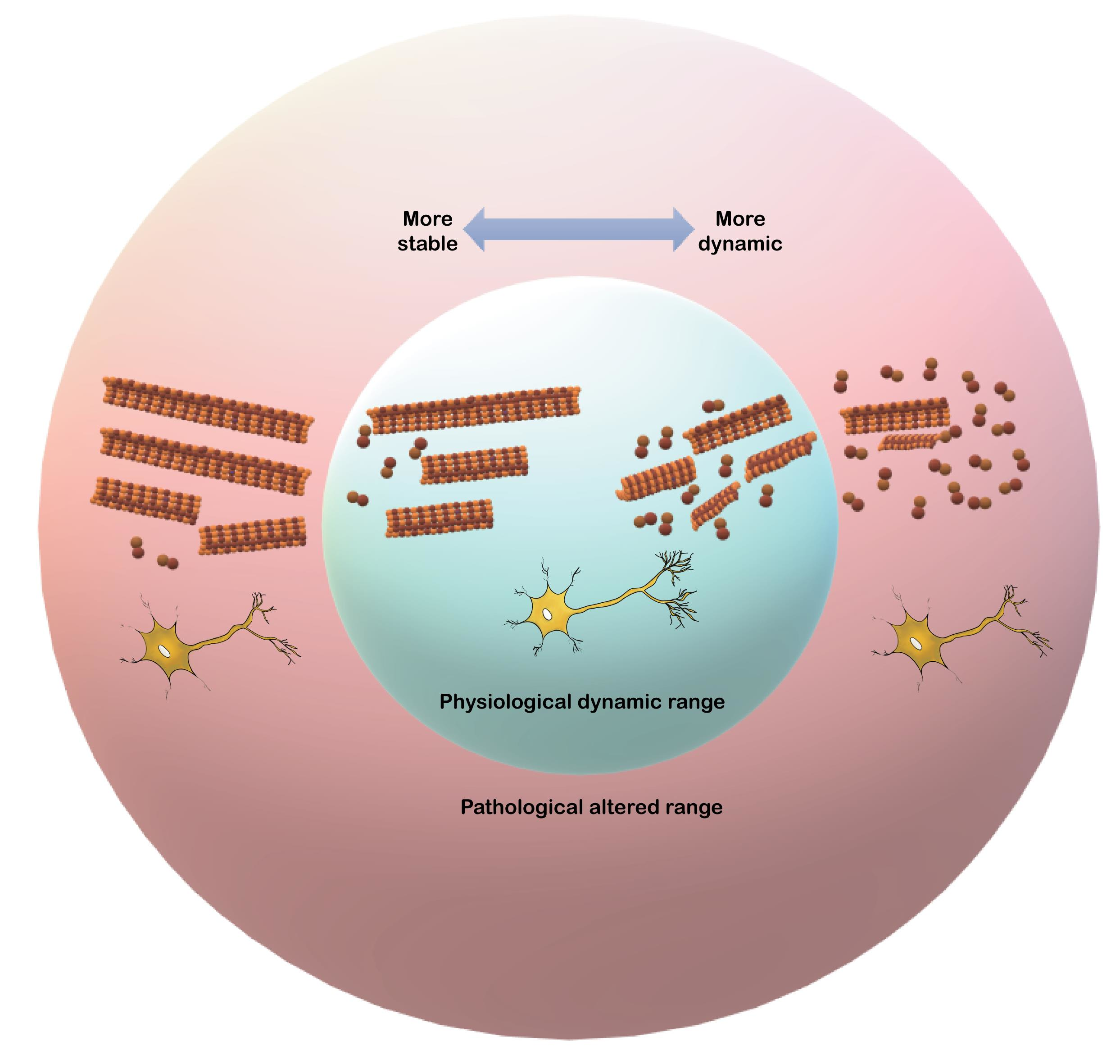
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Modulation of MT Stability and Its Impact on Brain Function
3. Pharmacological Modulators of MT Stability
4. Microtubular Reconfiguration during Brain Function
5. Microtubular Reconfiguration during Neuronal Activity
6. Changes in Excitability and Synaptic Transmission, and Their Morphological Correlates, Induced by Microtubular Reconfiguration
7. Changes in Long-Term Synaptic Plasticity and in Dendritic Spines Induced by Microtubular Modulation
8. Changes in Learning and Memory Induced by Microtubular Modulation
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kapitein, L.C.; Hoogenraad, C.C. Which Way to Go? Cytoskeletal Organization and Polarized Transport in Neurons. Mol. Cell. Neurosci. 2011, 46, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Witte, H.; Bradke, F. The Role of the Cytoskeleton during Neuronal Polarization. Curr. Opin. Neurobiol. 2008, 18, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Solís-Chagoyán, H.; Calixto, E.; Figueroa, A.; Montaño, L.M.; Berlanga, C.; Rodríguez-Verdugo, M.S.; Romo, F.; Jiménez, M.; Gurrola, C.Z.; Riquelme, A.; et al. Microtubule Organization and L-Type Voltage-Activated Calcium Current in Olfactory Neuronal Cells Obtained from Patients with Schizophrenia and Bipolar Disorder. Schizophr. Res. 2013, 143, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Mondragón-Rodríguez, S.; Gu, N.; Manseau, F.; Williams, S. Alzheimer’s Transgenic Model Is Characterized by Very Early Brain Network Alterations and β-CTF Fragment Accumulation: Reversal by β-Secretase Inhibition. Front. Cell. Neurosci. 2018, 12, 121. [Google Scholar] [CrossRef]
- Mondragón-Rodríguez, S.; Salgado-Burgos, H.; Peña-Ortega, F. Circuitry and Synaptic Dysfunction in Alzheimer’s Disease: A New Tau Hypothesis. Neural Plast. 2020, 2020, 2960343. [Google Scholar] [CrossRef]
- Dent, E.W.; Merriam, E.B.; Hu, X. The Dynamic Cytoskeleton: Backbone of Dendritic Spine Plasticity. Curr. Opin. Neurobiol. 2011, 21, 175–181. [Google Scholar] [CrossRef] [Green Version]
- Baas, P.W.; Rao, A.N.; Matamoros, A.J.; Leo, L. Stability Properties of Neuronal Microtubules. Cytoskeleton 2016, 73, 442–460. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Huertas, C.; Freixo, F.; Viais, R.; Lacasa, C.; Soriano, E.; Lüders, J. Non-Centrosomal Nucleation Mediated by Augmin Organizes Microtubules in Post-Mitotic Neurons and Controls Axonal Microtubule Polarity. Nat. Commun. 2016, 7, 12187. [Google Scholar] [CrossRef]
- Nogales, E.; Wang, H.-W. Structural Intermediates in Microtubule Assembly and Disassembly: How and Why? Curr. Opin. Cell Biol. 2006, 18, 179–184. [Google Scholar] [CrossRef]
- Weisenberg, R.; Taylor, E.W. Studies on ATPase Activity of Sea Urchin Eggs and the Isolated Mitotic Apparatus. Exp. Cell Res. 1968, 53, 372–384. [Google Scholar] [CrossRef]
- Bryan, J.; Wilson, L. Are Cytoplasmic Microtubules Heteropolymers? Proc. Natl. Acad. Sci. USA 1971, 68, 1762–1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, R.A.; O’Brien, E.T.; Pryer, N.K.; Soboeiro, M.F.; Voter, W.A.; Erickson, H.P.; Salmon, E.D. Dynamic Instability of Individual Microtubules Analyzed by Video Light Microscopy: Rate Constants and Transition Frequencies. J. Cell Biol. 1988, 107, 1437–1448. [Google Scholar] [CrossRef] [PubMed]
- Burns, R.G.; Farrell, K.W. Getting to the Heart of β-Tubulin. Trends Cell Biol. 1996, 6, 297–303. [Google Scholar] [CrossRef]
- Mejillano, M.R.; Barton, J.S.; Nath, J.P.; Himes, R.H. GTP Analogs Interact with the Tubulin Exchangeable Site during Assembly and upon Binding. Biochemistry 1990, 29, 1208–1216. [Google Scholar] [CrossRef]
- Weisenberg, R.C.; Deery, W.J.; Dickinson, P.J. Tubulin-Nucleotide Interactions during the Polymerization and Depolymerization of Microtubules. Biochemistry 1976, 15, 4248–4254. [Google Scholar] [CrossRef]
- David-Pfeuty, T.; Erickson, H.P.; Pantaloni, D. Guanosinetriphosphatase Activity of Tubulin Associated with Microtubule Assembly. Proc. Natl. Acad. Sci. USA 1977, 74, 5372–5376. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Centonze, V.; Ahmad, F.; Baas, P. Microtubule Nucleation and Release from the Neuronal Centrosome. J. Cell Biol. 1993, 122, 349–359. [Google Scholar] [CrossRef]
- Kapitein, L.C.; Hoogenraad, C.C. Building the Neuronal Microtubule Cytoskeleton. Neuron 2015, 87, 492–506. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, F.J.; Baas, P.W. Microtubules Released from the Neuronal Centrosome Are Transported into the Axon. J. Cell Sci. 1995, 108, 2761–2769. [Google Scholar] [CrossRef]
- Dent, E.W. Of Microtubules and Memory: Implications for Microtubule Dynamics in Dendrites and Spines. Mol. Biol. Cell 2017, 28, 1–8. [Google Scholar] [CrossRef]
- Heidemann, S.R.; Landers, J.M.; Hamborg, M.A. Polarity Orientation of Axonal Microtubules. J. Cell Biol. 1981, 91, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Vale, R.D. The Molecular Motor Toolbox for Intracellular Transport. Cell 2003, 112, 467–480. [Google Scholar] [CrossRef] [Green Version]
- Qu, X.; Kumar, A.; Blockus, H.; Waites, C.; Bartolini, F. Activity-Dependent Nucleation of Dynamic Microtubules at Presynaptic Boutons Controls Neurotransmission. Curr. Biol. 2019, 29, 4231–4240.e5. [Google Scholar] [CrossRef] [PubMed]
- Piriya Ananda Babu, L.; Wang, H.-Y.; Eguchi, K.; Guillaud, L.; Takahashi, T. Microtubule and Actin Differentially Regulate Synaptic Vesicle Cycling to Maintain High-Frequency Neurotransmission. J. Neurosci. 2020, 40, 131–142. [Google Scholar] [CrossRef]
- Baas, P.W.; Deitch, J.S.; Black, M.M.; Banker, G.A. Polarity Orientation of Microtubules in Hippocampal Neurons: Uniformity in the Axon and Nonuniformity in the Dendrite. Proc. Natl. Acad. Sci. USA 1988, 85, 8335–8339. [Google Scholar] [CrossRef] [Green Version]
- Baas, P.W.; Slaughter, T.; Brown, A.; Black, M.M. Microtubule Dynamics in Axons and Dendrites. J. Neurosci. Res. 1991, 30, 134–153. [Google Scholar] [CrossRef]
- Qiang, L.; Sun, X.; Austin, T.O.; Muralidharan, H.; Jean, D.C.; Liu, M.; Yu, W.; Baas, P.W. Tau Does Not Stabilize Axonal Microtubules but Rather Enables Them to Have Long Labile Domains. Curr. Biol. 2018, 28, 2181–2189.e4. [Google Scholar] [CrossRef] [Green Version]
- Poulain, F.E.; Sobel, A. The Microtubule Network and Neuronal Morphogenesis: Dynamic and Coordinated Orchestration through Multiple Players. Mol. Cell. Neurosci. 2010, 43, 15–32. [Google Scholar] [CrossRef]
- Kirschner, M. Beyond Self-Assembly: From Microtubules to Morphogenesis. Cell 1986, 45, 329–342. [Google Scholar] [CrossRef]
- Mitchison, T.; Kirschner, M. Dynamic Instability of Microtubule Growth. Nature 1984, 312, 237–242. [Google Scholar] [CrossRef]
- Holy, T.E.; Leibler, S. Dynamic Instability of Microtubules as an Efficient Way to Search in Space. Proc. Natl. Acad. Sci. USA 1994, 91, 5682–5685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roll-Mecak, A. The Tubulin Code in Microtubule Dynamics and Information Encoding. Dev. Cell 2020, 54, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Waites, C.; Qu, X.; Bartolini, F. The Synaptic Life of Microtubules. Curr. Opin. Neurobiol. 2021, 69, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Marchisella, F.; Coffey, E.T.; Hollos, P. Microtubule and Microtubule Associated Protein Anomalies in Psychiatric Disease. Cytoskeleton 2016, 73, 596–611. [Google Scholar] [CrossRef] [Green Version]
- Verhey, K.J.; Gaertig, J. The Tubulin Code. Cell Cycle 2007, 6, 2152–2160. [Google Scholar] [CrossRef]
- Yu, I.; Garnham, C.P.; Roll-Mecak, A. Writing and Reading the Tubulin Code. J. Biol. Chem. 2015, 290, 17163–17172. [Google Scholar] [CrossRef] [Green Version]
- Janke, C.; Bulinski, J.C. Post-Translational Regulation of the Microtubule Cytoskeleton: Mechanisms and Functions. Nat. Rev. Mol. Cell Biol. 2011, 12, 773–786. [Google Scholar] [CrossRef]
- Fukushima, N.; Furuta, D.; Hidaka, Y.; Moriyama, R.; Tsujiuchi, T. Post-Translational Modifications of Tubulin in the Nervous System. J. Neurochem. 2009, 109, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Dunn, S.; Morrison, E.E.; Liverpool, T.B.; Molina-París, C.; Cross, R.A.; Alonso, M.C.; Peckham, M. Differential Trafficking of Kif5c on Tyrosinated and Detyrosinated Microtubules in Live Cells. J. Cell Sci. 2008, 121, 1085–1095. [Google Scholar] [CrossRef] [Green Version]
- Konishi, Y.; Setou, M. Tubulin Tyrosination Navigates the Kinesin-1 Motor Domain to Axons. Nat. Neurosci. 2009, 12, 559–567. [Google Scholar] [CrossRef]
- Hammond, J.W.; Huang, C.-F.; Kaech, S.; Jacobson, C.; Banker, G.; Verhey, K.J. Posttranslational Modifications of Tubulin and the Polarized Transport of Kinesin-1 in Neurons. Mol. Biol. Cell 2010, 21, 572–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sudo, H.; Baas, P.W. Acetylation of Microtubules Influences Their Sensitivity to Severing by Katanin in Neurons and Fibroblasts. J. Neurosci. 2010, 30, 7215–7226. [Google Scholar] [CrossRef] [PubMed]
- Wade, R.H. On and Around Microtubules: An Overview. Mol. Biotechnol. 2009, 43, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Slaughter, T.; Black, M.M. STOP (Stable-Tubule-Only-Polypeptide) Is Preferentially Associated with the Stable Domain of Axonal Microtubules. J. Neurocytol. 2003, 32, 399–413. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, C.; Dı́az-Nido, J.; Avila, J. Phosphorylation of Microtubule-Associated Protein 2 and Its Relevance for the Regulation of the Neuronal Cytoskeleton Function. Prog. Neurobiol. 2000, 61, 133–168. [Google Scholar] [CrossRef]
- Dehmelt, L.; Halpain, S. The MAP2/Tau Family of Microtubule-Associated Proteins. Genome Biol. 2004, 6, 204. [Google Scholar] [CrossRef] [Green Version]
- Schwenk, B.M.; Lang, C.M.; Hogl, S.; Tahirovic, S.; Orozco, D.; Rentzsch, K.; Lichtenthaler, S.F.; Hoogenraad, C.C.; Capell, A.; Haass, C.; et al. The FTLD Risk Factor TMEM106B and MAP6 Control Dendritic Trafficking of Lysosomes. EMBO J. 2013, 33, 450–467. [Google Scholar] [CrossRef]
- Penazzi, L.; Bakota, L.; Brandt, R. Microtubule Dynamics in Neuronal Development, Plasticity, and Neurodegeneration. Int. Rev. Cell Mol. Biol. 2016, 321, 89–169. [Google Scholar]
- Binder, L.I.; Frankfurter, A.; Rebhun, L.I. The Distribution of Tau in the Mammalian Central Nervous System. J. Cell Biol. 1985, 101, 1371–1378. [Google Scholar] [CrossRef] [Green Version]
- Matus, A. Microtubule-Associated Proteins and the Determination of Neuronal Form. J. Physiol. 1990, 84, 134–137. [Google Scholar]
- Vale, R.D.; Schnapp, B.J.; Mitchison, T.; Steuer, E.; Reese, T.S.; Sheetz, M.P. Different axoplasmic proteins generate movement in opposite directions along microtubules in vitro. Cell 1985, 43, 623–632. [Google Scholar] [CrossRef]
- Aoki, C.; Siekevitz, P. Ontogenetic Changes in the Cyclic Adenosine 3′,5′-Monophosphate- Stimulatable Phosphorylation of Cat Visual Cortex Proteins, Particularly of Microtubule-Associated Protein 2 (MAP 2): Effects of Normal and Dark Rearing and of the Exposure to Light. J. Neurosci. 1985, 5, 2465–2483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaworski, J.; Kapitein, L.C.; Gouveia, S.M.; Dortland, B.R.; Wulf, P.S.; Grigoriev, I.; Camera, P.; Spangler, S.A.; di Stefano, P.; Demmers, J.; et al. Dynamic Microtubules Regulate Dendritic Spine Morphology and Synaptic Plasticity. Neuron 2009, 61, 85–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barten, D.M.; Fanara, P.; Andorfer, C.; Hoque, N.; Wong, P.Y.A.; Husted, K.H.; Cadelina, G.W.; DeCarr, L.B.; Yang, L.; Liu, V.; et al. Hyperdynamic Microtubules, Cognitive Deficits, and Pathology Are Improved in Tau Transgenic Mice with Low Doses of the Microtubule-Stabilizing Agent BMS-241027. J. Neurosci. 2012, 32, 7137–7145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fanara, P.; Husted, K.H.; Selle, K.; Wong, P.Y.A.; Banerjee, J.; Brandt, R.; Hellerstein, M.K. Changes in Microtubule Turnover Accompany Synaptic Plasticity and Memory Formation in Response to Contextual Fear Conditioning in Mice. Neuroscience 2010, 168, 167–178. [Google Scholar] [CrossRef]
- Uchida, S.; Martel, G.; Pavlowsky, A.; Takizawa, S.; Hevi, C.; Watanabe, Y.; Kandel, E.R.; Alarcon, J.M.; Shumyatsky, G.P. Learning-Induced and Stathmin-Dependent Changes in Microtubule Stability Are Critical for Memory and Disrupted in Ageing. Nat. Commun. 2014, 5, 4389. [Google Scholar] [CrossRef] [Green Version]
- Uchida, S.; Shumyatsky, G.P. Deceivingly Dynamic: Learning-Dependent Changes in Stathmin and Microtubules. Neurobiol. Learn. Mem. 2015, 124, 52–61. [Google Scholar] [CrossRef] [Green Version]
- Martel, G.; Uchida, S.; Hevi, C.; Chevere-Torres, I.; Fuentes, I.; Park, Y.J.; Hafeez, H.; Yamagata, H.; Watanabe, Y.; Shumyatsky, G.P. Genetic Demonstration of a Role for Stathmin in Adult Hippocampal Neurogenesis, Spinogenesis, and NMDA Receptor-Dependent Memory. J. Neurosci. 2016, 36, 1185–1202. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.E.; Slivicki, R.A.; Hohmann, A.G.; Crystal, J.D. The Chemotherapeutic Agent Paclitaxel Selectively Impairs Learning While Sparing Source Memory and Spatial Memory. Behav. Brain Res. 2017, 320, 48–57. [Google Scholar] [CrossRef] [Green Version]
- Matamoros, A.J.; Baas, P.W. Microtubules in Health and Degenerative Disease of the Nervous System. Brain Res. Bull. 2016, 126, 217–225. [Google Scholar] [CrossRef] [Green Version]
- Brandt, R.; Bakota, L. Microtubule Dynamics and the Neurodegenerative Triad of Alzheimer’s Disease: The Hidden Connection. J. Neurochem. 2017, 143, 409–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortal, S. Microtubule Dynamics in Cytoskeleton, Neurodegenerative and Psychiatric Disease. STEMedicine 2021, 2, e81. [Google Scholar] [CrossRef]
- Binet, S.; Meininger, V. Modifications of Microtubule Proteins in ALS Nerve Precede Detectable Histologic and Ultrastructural Changes. Neurology 1988, 38, 1596. [Google Scholar] [CrossRef] [PubMed]
- Fanara, P.; Banerjee, J.; Hueck, R.V.; Harper, M.R.; Awada, M.; Turner, H.; Husted, K.H.; Brandt, R.; Hellerstein, M.K. Stabilization of Hyperdynamic Microtubules Is Neuroprotective in Amyotrophic Lateral Sclerosis. J. Biol. Chem. 2007, 282, 23465–23472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Y.; Jiang, H.; Yang, F.; Nakaso, K.; Feng, J. Parkin Protects Dopaminergic Neurons against Microtubule-Depolymerizing Toxins by Attenuating Microtubule-Associated Protein Kinase Activation. J. Biol. Chem. 2009, 284, 4009–4017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trushina, E.; Heldebrant, M.P.; Perez-Terzic, C.M.; Bortolon, R.; Kovtun, I.V.; Badger, J.D.; Terzic, A.; Estevez, A.; Windebank, A.J.; Dyer, R.B.; et al. Microtubule Destabilization and Nuclear Entry Are Sequential Steps Leading to Toxicity in Huntington’s Disease. Proc. Natl. Acad. Sci. USA 2003, 100, 12171–12176. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, M.; Smith, M.J.; Goedert, M. Tau Proteins with FTDP-17 Mutations Have a Reduced Ability to Promote Microtubule Assembly. FEBS Lett. 1998, 437, 207–210. [Google Scholar] [CrossRef] [Green Version]
- Alonso, A.D.C.; Grundke-Iqbal, I.; Iqbal, K. Alzheimer’s Disease Hyperphosphorylated Tau Sequesters Normal Tau into Tangles of Filaments and Disassembles Microtubules. Nat. Med. 1996, 2, 783–787. [Google Scholar] [CrossRef]
- Kaufmann, W.E.; Macdonald, S.M.; Altamura, C.R. Dendritic Cytoskeletal Protein Expression in Mental Retardation: An Immunohistochemical Study of the Neocortex in Rett Syndrome. Cereb. Cortex 2000, 10, 992–1004. [Google Scholar] [CrossRef] [Green Version]
- Gozes, I. Microtubules (Tau) as an Emerging Therapeutic Target: NAP (Davunetide). Curr. Pharm. Des. 2011, 17, 3413–3417. [Google Scholar] [CrossRef]
- Shelton, M.A.; Newman, J.T.; Gu, H.; Sampson, A.R.; Fish, K.N.; MacDonald, M.L.; Moyer, C.E.; DiBitetto, J.V.; Dorph-Petersen, K.-A.; Penzes, P.; et al. Loss of Microtubule-Associated Protein 2 Immunoreactivity Linked to Dendritic Spine Loss in Schizophrenia. Biol. Psychiatry 2015, 78, 374–385. [Google Scholar] [CrossRef] [Green Version]
- Drago, A.; Crisafulli, C.; Sidoti, A.; Calabrò, M.; Serretti, A. The Microtubule-Associated Molecular Pathways May Be Genetically Disrupted in Patients with Bipolar Disorder. Insights from the Molecular Cascades. J. Affect. Dis. 2016, 190, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Cavaletti, G.; Marmiroli, P. Chemotherapy-Induced Peripheral Neurotoxicity. Nat. Rev. Neurol. 2010, 6, 657–666. [Google Scholar] [CrossRef]
- Mandilaras, V.; Wan-Chow-Wah, D.; Monette, J.; Gaba, F.; Monette, M.; Alfonso, L. The Impact of Cancer Therapy on Cognition in the Elderly. Front. Pharmacol. 2013, 4, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.-F.; Yang, L.; Li, S.; Wang, Q.; Yuan, X.-B.; Gao, X.; Bao, L.; Zhang, X. Fibroblast Growth Factor 13 Is a Microtubule-Stabilizing Protein Regulating Neuronal Polarization and Migration. Cell 2012, 149, 1549–1564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassimeris, L.; Spittle, C. Regulation of Microtubule-Associated Proteins. Int. Rev. Cytol. 2001, 210, 163–226. [Google Scholar] [PubMed]
- Fournet, V.; de Lavilléon, G.; Schweitzer, A.; Giros, B.; Andrieux, A.; Martres, M.-P. Both Chronic Treatments by Epothilone D and Fluoxetine Increase the Short-Term Memory and Differentially Alter the Mood Status of STOP/MAP6 KO Mice. J. Neurochem. 2012, 123, 982–996. [Google Scholar] [CrossRef] [Green Version]
- Neve, R.L.; Harris, P.; Kosik, K.S.; Kurnit, D.M.; Donlon, T.A. Identification of CDNA Clones for the Human Microtubule-Associated Protein Tau and Chromosomal Localization of the Genes for Tau and Microtubule-Associated Protein 2. Mol. Brain Res. 1986, 1, 271–280. [Google Scholar] [CrossRef]
- Guo, T.; Noble, W.; Hanger, D.P. Roles of Tau Protein in Health and Disease. Acta Neuropathol. 2017, 133, 665–704. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.; Neve, R.L.; Kosik, K.S. The Microtubule Binding Domain of Tau Protein. Neuron 1989, 2, 1615–1624. [Google Scholar] [CrossRef]
- Amos, L.A. Microtubule Structure and Its Stabilisation. Org. Biomol. Chem. 2004, 2, 2153. [Google Scholar] [CrossRef] [PubMed]
- Drechsel, D.N.; Hyman, A.A.; Cobb, M.H.; Kirschner, M.W. Modulation of the Dynamic Instability of Tubulin Assembly by the Microtubule-Associated Protein Tau. Mol. Biol. Cell 1992, 3, 1141–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papasozomenos, S.C.; Binder, L.I. Phosphorylation Determines Two Distinct Species of Tau in the Central Nervous System. Cell Motil. Cytoskelet. 1987, 8, 210–226. [Google Scholar] [CrossRef] [PubMed]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic Function of Tau Mediates Amyloid-β Toxicity in Alzheimer’s Disease Mouse Models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [Green Version]
- Martin, L.; Latypova, X.; Wilson, C.M.; Magnaudeix, A.; Perrin, M.-L.; Terro, F. Tau Protein Phosphatases in Alzheimer’s Disease: The Leading Role of PP2A. Ageing Res. Rev. 2013, 12, 39–49. [Google Scholar] [CrossRef]
- Martin, L.; Latypova, X.; Wilson, C.M.; Magnaudeix, A.; Perrin, M.-L.; Yardin, C.; Terro, F. Tau Protein Kinases: Involvement in Alzheimer’s Disease. Ageing Res. Rev. 2013, 12, 289–309. [Google Scholar] [CrossRef]
- Lindwall, G.; Cole, R.D. Phosphorylation Affects the Ability of Tau Protein to Promote Microtubule Assembly. J. Biol. Chem. 1984, 259, 5301–5305. [Google Scholar] [CrossRef]
- Alonso, A.C.; Zaidi, T.; Grundke-Iqbal, I.; Iqbal, K. Role of Abnormally Phosphorylated Tau in the Breakdown of Microtubules in Alzheimer Disease. Proc. Natl. Acad. Sci. USA 1994, 91, 5562–5566. [Google Scholar] [CrossRef] [Green Version]
- Merrick, S.E.; Trojanowski, J.Q.; Lee, V.M.-Y. Selective Destruction of Stable Microtubules and Axons by Inhibitors of Protein Serine/Threonine Phosphatases in Cultured Human Neurons (NT2N Cells). J. Neurosci. 1997, 17, 5726–5737. [Google Scholar] [CrossRef]
- Zhang, B.; Maiti, A.; Shively, S.; Lakhani, F.; McDonald-Jones, G.; Bruce, J.; Lee, E.B.; Xie, S.X.; Joyce, S.; Li, C.; et al. Microtubule-Binding Drugs Offset Tau Sequestration by Stabilizing Microtubules and Reversing Fast Axonal Transport Deficits in a Tauopathy Model. Proc. Natl. Acad. Sci. USA 2004, 102, 227–231. [Google Scholar] [CrossRef] [Green Version]
- Alonso, A.d.C.; Grundke-Iqbal, I.; Barra, H.S.; Iqbal, K. Abnormal Phosphorylation of Tau and the Mechanism of Alzheimer Neurofibrillary Degeneration: Sequestration of Microtubule-Associated Proteins 1 and 2 and the Disassembly of Microtubules by the Abnormal Tau. Proc. Natl. Acad. Sci. USA 1997, 94, 298–303. [Google Scholar] [CrossRef] [Green Version]
- Hoover, B.R.; Reed, M.N.; Su, J.; Penrod, R.D.; Kotilinek, L.A.; Grant, M.K.; Pitstick, R.; Carlson, G.A.; Lanier, L.M.; Yuan, L.L.; et al. Tau Mislocalization to Dendritic Spines Mediates Synaptic Dysfunction Independently of Neurodegeneration. Neuron 2010, 68, 1067–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tackenberg, C.; Brandt, R. Divergent Pathways Mediate Spine Alterations and Cell Death Induced by Amyloid-, Wild-Type Tau, and R406W Tau. J. Neurosci. 2009, 29, 14439–14450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golovyashkina, N.; Penazzi, L.; Ballatore, C.; Smith, A.B.; Bakota, L.; Brandt, R. Region-Specific Dendritic Simplification Induced by Aβ, Mediated by Tau via Dysregulation of Microtubule Dynamics: A Mechanistic Distinct Event from Other Neurodegenerative Processes. Mol. Neurodegener. 2015, 10, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arendt, T.; Bullmann, T. Neuronal Plasticity in Hibernation and the Proposed Role of the Microtubule-Associated Protein Tau as a “Master Switch” Regulating Synaptic Gain in Neuronal Networks. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 305, R478–R489. [Google Scholar] [CrossRef] [Green Version]
- Varidaki, A.; Hong, Y.; Coffey, E.T. Repositioning Microtubule Stabilizing Drugs for Brain Disorders. Front. Cell. Neurosci. 2018, 12, 226. [Google Scholar] [CrossRef] [PubMed]
- Goodin, S.; Kane, M.P.; Rubin, E.H. Epothilones: Mechanism of Action and Biologic Activity. J. Clin. Oncol. 2004, 22, 2015–2025. [Google Scholar] [CrossRef]
- Kolman, A. Epothilone D (Kosan/Roche). Curr. Opin. Investig. Drugs 2004, 5, 657–667. [Google Scholar]
- Michaelis, M.L. Ongoing In Vivo Studies with Cytoskeletal Drugs in Tau Transgenic Mice. Curr. Alzheimer Res. 2006, 3, 215–219. [Google Scholar] [CrossRef]
- Michaelis, M.L.; Ansar, S.; Chen, Y.; Reiff, E.R.; Seyb, K.I.; Himes, R.H.; Audus, K.L.; Georg, G.I. β-Amyloid-Induced Neurodegeneration and Protection by Structurally Diverse Microtubule-Stabilizing Agents. J. Pharmacol. Exp. Ther. 2005, 312, 659–668. [Google Scholar] [CrossRef]
- Shemesh, O.A.; Spira, M.E. Rescue of Neurons from Undergoing Hallmark Tau-Induced Alzheimer’s Disease Cell Pathologies by the Antimitotic Drug Paclitaxel. Neurobiol. Dis. 2011, 43, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Silva, D.F.F.; Esteves, A.R.; Arduino, D.M.; Oliveira, C.R.; Cardoso, S.M. Amyloid-β-Induced Mitochondrial Dysfunction Impairs the Autophagic Lysosomal Pathway in a Tubulin Dependent Pathway. J. Alzheimers Dis. 2011, 26, 565–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.J.; Swain, S.M. Peripheral Neuropathy Induced by Microtubule-Stabilizing Agents. J. Clin. Oncol. 2006, 24, 1633–1642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlson, K.; Ocean, A.J. Peripheral Neuropathy with Microtubule-Targeting Agents: Occurrence and Management Approach. Clin. Breast Cancer 2011, 11, 73–81. [Google Scholar] [CrossRef] [Green Version]
- Chiorazzi, A.; Nicolini, G.; Canta, A.; Oggioni, N.; Rigolio, R.; Cossa, G.; Lombardi, R.; Roglio, I.; Cervellini, I.; Lauria, G.; et al. Experimental Epothilone B Neurotoxicity: Results of in Vitro and in Vivo Studies. Neurobiol. Dis. 2009, 35, 270–277. [Google Scholar] [CrossRef]
- LaPointe, N.E.; Morfini, G.; Brady, S.T.; Feinstein, S.C.; Wilson, L.; Jordan, M.A. Effects of Eribulin, Vincristine, Paclitaxel and Ixabepilone on Fast Axonal Transport and Kinesin-1 Driven Microtubule Gliding: Implications for Chemotherapy-Induced Peripheral Neuropathy. NeuroToxicology 2013, 37, 231–239. [Google Scholar] [CrossRef] [Green Version]
- Ballatore, C.; Brunden, K.R.; Trojanowski, J.Q.; Lee, V.M.Y.; Smith, A.B. Non-Naturally Occurring Small Molecule Microtubule-Stabilizing Agents: A Potential Tactic for CNS-Directed Therapies. ACS Chem. Neurosci. 2017, 8, 5–7. [Google Scholar] [CrossRef] [Green Version]
- Nogales, E.; Grayer Wolf, S.; Khan, I.A.; Ludueña, R.F.; Downing, K.H. Structure of Tubulin at 6.5 Å and Location of the Taxol-Binding Site. Nature 1995, 375, 424–427. [Google Scholar]
- Nogales, E.; Wolf, S.G.; Downing, K.H. Erratum: Structure of the Aβ Tubulin Dimer by Electron Crystallography. Nature 1998, 393, 191. [Google Scholar] [CrossRef] [Green Version]
- Amos, L.A.; Löwe, J. How Taxol® Stabilises Microtubule Structure. Chem. Biol. 1999, 6, R65–R69. [Google Scholar] [CrossRef] [Green Version]
- Prota, A.E.; Bargsten, K.; Zurwerra, D.; Field, J.J.; Díaz, J.F.; Altmann, K.-H.; Steinmetz, M.O. Molecular Mechanism of Action of Microtubule-Stabilizing Anticancer Agents. Science 2013, 339, 587–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunden, K.R.; Zhang, B.; Carroll, J.; Yao, Y.; Potuzak, J.S.; Hogan, A.M.L.; Iba, M.; James, M.J.; Xie, S.X.; Ballatore, C.; et al. Epothilone D Improves Microtubule Density, Axonal Integrity, and Cognition in a Transgenic Mouse Model of Tauopathy. J. Neurosci. 2010, 30, 13861–13866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellal, F.; Hurtado, A.; Ruschel, J.; Flynn, K.C.; Laskowski, C.J.; Umlauf, M.; Kapitein, L.C.; Strikis, D.; Lemmon, V.; Bixby, J.; et al. Microtubule Stabilization Reduces Scarring and Causes Axon Regeneration After Spinal Cord Injury. Science 2011, 331, 928–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sengottuvel, V.; Leibinger, M.; Pfreimer, M.; Andreadaki, A.; Fischer, D. Taxol Facilitates Axon Regeneration in the Mature CNS. J. Neurosci. 2011, 31, 2688–2699. [Google Scholar] [CrossRef]
- Baas, P.W.; Ahmad, F.J. Beyond Taxol: Microtubule-Based Treatment of Disease and Injury of the Nervous System. Brain 2013, 136, 2937–2951. [Google Scholar] [CrossRef] [Green Version]
- Flatters, S.J.L.; Bennett, G.J. Studies of Peripheral Sensory Nerves in Paclitaxel-Induced Painful Peripheral Neuropathy: Evidence for Mitochondrial Dysfunction. Pain 2006, 122, 245–257. [Google Scholar] [CrossRef] [Green Version]
- Markman, M.; Mekhail, T.M. Paclitaxel in Cancer Therapy. Exp. Opin. Pharmacother. 2002, 3, 755–766. [Google Scholar] [CrossRef]
- Alloatti, G.; Penna, C.; Gallo, M.P.; Levi, R.C.; Bombardelli, E.; Appendino, G. Differential Effects of Paclitaxel and Derivatives on Guinea Pig Isolated Heart and Papillary Muscle. J. Pharmacol. Exp. Ther. 1998, 284, 561. [Google Scholar]
- Gallo, J.M.; Li, S.; Guo, P.; Reed, K.; Ma, J. The Effect of P-Glycoprotein on Paclitaxel Brain and Brain Tumor Distribution in Mice. Cancer Res. 2003, 63, 5114. [Google Scholar]
- Kemper, E.M.; van Zandbergen, A.E.; Cleypool, C.; Mos, H.A.; Boogerd, W.; Beijnen, J.H.; van Tellingen, O. Increased Penetration of Paclitaxel into the Brain by Inhibition of P-Glycoprotein. Clin. Cancer Res. 2003, 9, 2849. [Google Scholar]
- Furukawa, K.; Mattson, M.P. Taxol Stabilizes [Ca2+]i and Protects Hippocampal Neurons against Excitotoxicity. Brain Res. 1995, 689, 141–146. [Google Scholar] [CrossRef]
- Wolf, S.; Barton, D.; Kottschade, L.; Grothey, A.; Loprinzi, C. Chemotherapy-Induced Peripheral Neuropathy: Prevention and Treatment Strategies. Eur. J. Cancer 2008, 44, 1507–1515. [Google Scholar] [CrossRef]
- Postma, T.J.; Hoekman, K.; van Riel, J.M.G.H.; Heimans, J.J.; Vermorken, J.B. Peripheral Neuropathy Due to Biweekly Paclitaxel, Epirubicin and Cisplatin in Patients with Advanced Ovarian Cancer. J. Neurooncol. 1999, 45, 241–246. [Google Scholar] [CrossRef]
- Ahles, T.A.; Saykin, A.J.; Furstenberg, C.T.; Cole, B.; Mott, L.A.; Skalla, K.; Whedon, M.B.; Bivens, S.; Mitchell, T.; Greenberg, E.R.; et al. Neuropsychologic Impact of Standard-Dose Systemic Chemotherapy in Long-Term Survivors of Breast Cancer and Lymphoma. J. Clin. Oncol. 2002, 20, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Wefel, J.S.; Schagen, S.B. Chemotherapy-Related Cognitive Dysfunction. Curr. Neurol. Neurosci. Rep. 2012, 12, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Ferris, C.F.; Nodine, S.; Pottala, T.; Cai, X.; Knox, T.M.; Fofana, F.H.; Kim, S.; Kulkarni, P.; Crystal, J.D.; Hohmann, A.G. Alterations in Brain Neurocircuitry Following Treatment with the Chemotherapeutic Agent Paclitaxel in Rats. Neurobiol. Pain 2019, 6, 100034. [Google Scholar] [CrossRef]
- Otová, B.; Václavíková, R.; Danielová, V.; Holubová, J.; Ehrlichová, M.; Horský, S.; Souček, P.; Šimek, P.; Gut, I. Effects of Paclitaxel, Docetaxel and Their Combinations on Subcutaneous Lymphomas in Inbred Sprague–Dawley/Cub Rats. Eur. J. Pharm. Sci. 2006, 29, 442–450. [Google Scholar] [CrossRef]
- Persohn, E.; Canta, A.; Schoepfer, S.; Traebert, M.; Mueller, L.; Gilardini, A.; Galbiati, S.; Nicolini, G.; Scuteri, A.; Lanzani, F.; et al. Morphological and Morphometric Analysis of Paclitaxel and Docetaxel-Induced Peripheral Neuropathy in Rats. Eur. J. Cancer 2005, 41, 1460–1466. [Google Scholar] [CrossRef]
- Park, S.R.; Kim, H.K.; Kim, C.G.; Choi, I.J.; Lee, J.S.; Lee, J.H.; Ryu, K.W.; Kim, Y.-W.; Bae, J.-M.; Kim, N.K. Phase I/II Study of S-1 Combined with Weekly Docetaxel in Patients with Metastatic Gastric Carcinoma. Br. J. Cancer 2008, 98, 1305–1311. [Google Scholar] [CrossRef]
- Ziske, C.G.; Schöttker, B.; Gorschlüter, M.; Mey, U.; Kleinschmidt, R.; Schlegel, U.; Sauerbruch, T.; Schmidt-Wolf, I.G.H. Acute Transient Encephalopathy after Paclitaxel Infusion: Report of Three Cases. Ann. Oncol. 2002, 13, 629–631. [Google Scholar] [CrossRef]
- Mercado-Gómez, O.; Ferrera, P.; Arias, C. Histopathologic Changes Induced by the Microtubule-Stabilizing Agent Taxol in the Rat Hippocampus In Vivo. J. Neurosci. Res. 2004, 78, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Bollag, D.M.; McQueney, P.A.; Zhu, J.; Hensens, O.; Koupal, L.; Liesch, J.; Goetz, M.; Lazarides, E.; Woods, C.M. Epothilones, a New Class of Microtubule-Stabilizing Agents with a Taxol-like Mechanism of Action. Cancer Res. 1995, 55, 2325–2333. [Google Scholar] [PubMed]
- Giannakakou, P.; Gussio, R.; Nogales, E.; Downing, K.H.; Zaharevitz, D.; Bollbuck, B.; Poy, G.; Sackett, D.; Nicolaou, K.C.; Fojo, T. A Common Pharmacophore for Epothilone and Taxanes: Molecular Basis for Drug Resistance Conferred by Tubulin Mutations in Human Cancer Cells. Proc. Natl. Acad. Sci. USA 2000, 97, 2904–2909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nettles, J.H.; Li, H.; Cornett, B.; Krahn, J.M.; Snyder, J.P.; Downing, K.H. The Binding Mode of Epothilone A on α,ß-Tubulin by Electron Crystallography. Science 2004, 305, 866–869. [Google Scholar] [CrossRef]
- Andrieux, A.; Salin, P.; Schweitzer, A.; Bégou, M.; Pachoud, B.; Brun, P.; Gory-Fauré, S.; Kujala, P.; Suaud-Chagny, M.-F.; Höfle, G.; et al. Microtubule Stabilizer Ameliorates Synaptic Function and Behavior in a Mouse Model for Schizophrenia. Biol. Psychiatry 2006, 60, 1224–1230. [Google Scholar] [CrossRef] [Green Version]
- Brizuela, M.; Blizzard, C.A.; Chuckowree, J.A.; Dawkins, E.; Gasperini, R.J.; Young, K.M.; Dickson, T.C. The Microtubule-Stabilizing Drug Epothilone D Increases Axonal Sprouting Following Transection Injury in Vitro. Mol. Cell. Neurosci. 2015, 66, 129–140. [Google Scholar] [CrossRef]
- Sandner, B.; Puttagunta, R.; Motsch, M.; Bradke, F.; Ruschel, J.; Blesch, A.; Weidner, N. Systemic Epothilone D Improves Hindlimb Function after Spinal Cord Contusion Injury in Rats. Exp. Neurol. 2018, 306, 250–259. [Google Scholar] [CrossRef]
- Cartelli, D.; Casagrande, F.; Busceti, C.L.; Bucci, D.; Molinaro, G.; Traficante, A.; Passarella, D.; Giavini, E.; Pezzoli, G.; Battaglia, G.; et al. Microtubule Alterations Occur Early in Experimental Parkinsonism and The Microtubule Stabilizer Epothilone D Is Neuroprotective. Sci. Rep. 2013, 3, 1837. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Carroll, J.; Trojanowski, J.Q.; Yao, Y.; Iba, M.; Potuzak, J.S.; Hogan, A.M.L.; Xie, S.X.; Ballatore, C.; Smith, A.B.; et al. The Microtubule-Stabilizing Agent, Epothilone D, Reduces Axonal Dysfunction, Neurotoxicity, Cognitive Deficits, and Alzheimer-like Pathology in an Interventional Study with Aged Tau Transgenic Mice. J. Neurosci. 2012, 32, 3601–3611. [Google Scholar] [CrossRef]
- Chuckowree, J.A.; Zhu, Z.; Brizuela, M.; Lee, K.M.; Blizzard, C.A.; Dickson, T.C. The Microtubule-Modulating Drug Epothilone D Alters Dendritic Spine Morphology in a Mouse Model of Mild Traumatic Brain Injury. Front. Cell. Neurosci. 2018, 12, 223. [Google Scholar] [CrossRef] [Green Version]
- Ruschel, J.; Hellal, F.; Flynn, K.C.; Dupraz, S.; Elliott, D.A.; Tedeschi, A.; Bates, M.; Sliwinski, C.; Brook, G.; Dobrindt, K.; et al. Systemic Administration of Epothilone B Promotes Axon Regeneration after Spinal Cord Injury. Science 2015, 348, 347–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, E.-H.; Sim, A.; Im, S.-K.; Hur, E.-M. Effects of Microtubule Stabilization by Epothilone B Depend on the Type and Age of Neurons. Neural Plast. 2016, 2016, 5056418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Zhang, X.; Ge, H.; Liu, W.; Sun, E.; Ma, Y.; Zhao, H.; Li, R.; Chen, W.; Yuan, J.; et al. Epothilone B Benefits Nigrostriatal Pathway Recovery by Promoting Microtubule Stabilization After Intracerebral Hemorrhage. J. Am. Heart Assoc. 2018, 7, e007626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, L.; Gao, W.; Chen, S.; Song, Y.; Song, C.; Zhou, Z.; Zhao, H.; Zhou, K.; Wang, W.; Zhu, K.; et al. Epothilone B Impairs Functional Recovery after Spinal Cord Injury by Increasing Secretion of Macrophage Colony-Stimulating Factor. Cell Death Dis. 2017, 8, e3162. [Google Scholar] [CrossRef]
- Clark, J.A.; Blizzard, C.A.; Breslin, M.C.; Yeaman, E.J.; Lee, K.M.; Chuckowree, J.A.; Dickson, T.C. Epothilone D Accelerates Disease Progression in the SOD1 G93A Mouse Model of Amyotrophic Lateral Sclerosis. Neuropathol. Appl. Neurobiol. 2018, 44, 590–605. [Google Scholar] [CrossRef]
- Clark, J.; Zhu, Z.; Chuckowree, J.; Dickson, T.; Blizzard, C. Efficacy of epothilones in central nervous system trauma treatment: What has age got to do with it? Neural Regen. Res. 2021, 16, 618–620. [Google Scholar]
- Zhu, Z.; Chuckowree, J.A.; Musgrove, R.; Dickson, T.C.; Blizzard, C.A. The pathologic outcomes and efficacy of epothilone treatment following traumatic brain injury is determined by age. Neurobiol. Aging 2020, 93, 85–96. [Google Scholar] [CrossRef]
- Dollé, J.; Jayea, A.; Andersonb, S.A.; Ahmadzadehc, H.; Shenoyc, V.B.; Smith, D.H. Newfound sex differences in axonal structure underlie differential outcomes from in vitro traumatic axonal injury. Exp. Neurol. 2018, 300, 121–134. [Google Scholar] [CrossRef]
- Sahenk, Z.; Brady, S.T.; Mendell, J.R. Studies on the Pathogenesis of Vincristine-Induced Neuropathy. Muscle Nerve 1987, 10, 80–84. [Google Scholar] [CrossRef]
- Dumontet, C.; Jordan, M.A. Microtubule-Binding Agents: A Dynamic Field of Cancer Therapeutics. Nat. Rev. Drug Discov. 2010, 9, 790–803. [Google Scholar] [CrossRef] [Green Version]
- Escuin, D.; Kline, E.R.; Giannakakou, P. Both Microtubule-Stabilizing and Microtubule-Destabilizing Drugs Inhibit Hypoxia-Inducible Factor-1α Accumulation and Activity by Disrupting Microtubule Function. Cancer Res. 2005, 65, 9021–9028. [Google Scholar] [CrossRef] [Green Version]
- Jordan, M. Mechanism of Action of Antitumor Drugs That Interact with Microtubules and Tubulin. Curr. Med. Chem. Anticancer Agents 2012, 2, 1–17. [Google Scholar] [CrossRef] [PubMed]
- You, Z.; Zhang, S.; Shen, S.; Yang, J.; Ding, W.; Yang, L.; Lim, G.; Doheny, J.T.; Tate, S.; Chen, L.; et al. Cognitive Impairment in a Rat Model of Neuropathic Pain: Role of Hippocampal Microtubule Stability. Pain 2018, 159, 1518–1528. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, C.; O’Malley, A.; Regan, C.M. Transient, Learning-Induced Ultrastructural Change in Spatially-Clustered Dentate Granule Cells of the Adult Rat Hippocampus. Neuroscience 1996, 76, 55–62. [Google Scholar] [CrossRef]
- Nelson, T.J.; Backlund, P.S.; Alkon, D.L. Hippocampal Protein-Protein Interactions in Spatial Memory. Hippocampus 2004, 14, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Priel, A.; Tuszynski, J.A.; Woolf, N.J. Neural Cytoskeleton Capabilities for Learning and Memory. J. Biol. Phys. 2010, 36, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavallaro, S.; D’Agata, V.; Manickam, P.; Dufour, F.; Alkon, D.L. Memory-Specific Temporal Profiles of Gene Expression in the Hippocampus. Proc. Natl. Acad. Sci. USA 2002, 99, 16279–16284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, S.; Fujii-Taira, I.; Murakami, A.; Hirose, N.; Aoki, N.; Izawa, E.-I.; Fujimoto, Y.; Takano, T.; Matsushima, T.; Homma, K.J. Up-Regulation of Microtubule-Associated Protein 2 Accompanying the Filial Imprinting of Domestic Chicks (Gallus Gallus Domesticus). Brain Res. Bull. 2008, 76, 282–288. [Google Scholar] [CrossRef]
- Woolf, N.J.; Young, S.L.; Johnson, G.V.W.; Fanselow, M.S. Pavlovian Conditioning Alters Cortical Microtubule-Associated Protein-2. NeuroReport 1994, 5, 1045–1048. [Google Scholar] [CrossRef]
- Woolf, N.J. A Structural Basis for Memory Storage in Mammals. Prog. Neurobiol. 1998, 55, 59–77. [Google Scholar] [CrossRef] [Green Version]
- Woolf, N.J.; Zinnerman, M.D.; Johnson, G.V.W. Hippocampal Microtubule-Associated Protein-2 Alterations with Contextual Memory. Brain Res. 1999, 821, 241–249. [Google Scholar] [CrossRef]
- Li, H.; Xu, W.; Wang, D.; Wang, L.; Fang, Q.; Wan, X.; Zhang, J.; Hu, Y.; Li, H.; Zhang, J.; et al. 4R Tau Modulates Cocaine-Associated Memory through Adult Dorsal Hippocampal Neurogenesis. J. Neurosci. 2021, 41, 6753–6774. [Google Scholar] [CrossRef]
- Goedert, M.; Jakes, R. Expression of separate isoforms of human tau protein: Correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. 1990, 9, 4225–4230. [Google Scholar] [CrossRef]
- Mondragón-Rodríguez, S.; Trillaud-Doppia, E.; Dudilot, A.; Bourgeois, C.; Lauzon, M.; Leclerc, N.; Boehm, J. Interaction of Endogenous Tau Protein with Synaptic Proteins Is Regulated by N-Methyl-d-Aspartate Receptor-Dependent Tau Phosphorylation. J. Biol. Chem. 2012, 287, 32040–32053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Zhang, J.; Zheng, P.; Zhang, Y. Altered Expression of MAP-2, GAP-43, and Synaptophysin in the Hippocampus of Rats with Chronic Cerebral Hypoperfusion Correlates with Cognitive Impairment. Mol. Brain Res. 2005, 139, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.; Fone, K.F.C.; Azmi, N.; Heidbreder, C.A.; Hagan, J.J.; Marsden, C.A. Isolation Rearing Induces Recognition Memory Deficits Accompanied by Cytoskeletal Alterations in Rat Hippocampus. Eur. J. Neurosci. 2006, 24, 2894–2902. [Google Scholar] [CrossRef]
- Shimada, A.; Tsuzuki, M.; Keino, H.; Satoh, M.; Chiba, Y.; Saitoh, Y.; Hosokawa, M. Apical Vulnerability to Dendritic Retraction in Prefrontal Neurones of Ageing SAMP10 Mouse: A Model of Cerebral Degeneration. Neuropathol. Appl. Neurobiol. 2006, 32, 1–14. [Google Scholar] [CrossRef]
- Hu, X.; Viesselmann, C.; Nam, S.; Merriam, E.; Dent, E.W. Activity-Dependent Dynamic Microtubule Invasion of Dendritic Spines. J. Neurosci. 2008, 28, 13094–13105. [Google Scholar] [CrossRef]
- Pandey, K.; Sharma, S.K. Activity-Dependent Acetylation of Alpha Tubulin in the Hippocampus. J. Mol. Neurosci. 2011, 45, 1–4. [Google Scholar] [CrossRef]
- Wu, D.; Jin, Y.; Shapiro, T.M.; Hinduja, A.; Baas, P.W.; Tom, V.J. Chronic Neuronal Activation Increases Dynamic Microtubules to Enhance Functional Axon Regeneration after Dorsal Root Crush Injury. Nat. Commun. 2020, 11, 6131. [Google Scholar] [CrossRef]
- Tryba, A.K.; Peña, F.; Ramirez, J.-M. Stabilization of Bursting in Respiratory Pacemaker Neurons. J. Neurosci. 2003, 23, 3538–3546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peña, F.; Tapia, R. Seizures and Neurodegeneration Induced by 4-Aminopyridine in Rat Hippocampus in Vivo: Role of Glutamate- and GABA-Mediated Neurotransmission and of Ion Channels. Neuroscience 2000, 101, 547–561. [Google Scholar] [CrossRef]
- Peña, F.; Tapia, R. Relationships Among Seizures, Extracellular Amino Acid Changes, and Neurodegeneration Induced by 4-Aminopyridine in Rat Hippocampus: A Microdialysis and Electroencephalographic Study. J. Neurochem. 2008, 72, 2006–2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, J.; Ramirez, B.U. Axonal Microtubules: Their Regulation by the Electrical Activity of the Nerve. Neurosci. Lett. 1979, 15, 19–22. [Google Scholar] [CrossRef]
- Halpain, S.; Greengard, P. Activation of NMDA Receptors Induces Rapid Dephosphorylation of the Cytoskeletal Protein MAP2. Neuron 1990, 5, 237–246. [Google Scholar] [CrossRef]
- Montoro, R.J.; Díaz-Nido, J.; Avila, J.; López-Barneo, J. N-Methyl-d-Aspartate Stimulates the Dephosphorylation of the Microtubule-Associated Protein 2 and Potentiates Excitatory Synaptic Pathways in the Rat Hippocampus. Neuroscience 1993, 54, 859–871. [Google Scholar] [CrossRef]
- Quinlan, E.M.; Halpain, S. Postsynaptic Mechanisms for Bidirectional Control of MAP2 Phosphorylation by Glutamate Receptors. Neuron 1996, 16, 357–368. [Google Scholar] [CrossRef] [Green Version]
- Philpot, B.D.; Lim, J.H.; Halpain, S.; Brunjes, P.C. Experience-Dependent Modifications in MAP2 Phosphorylation in Rat Olfactory Bulb. J. Neurosci. 1997, 17, 9596–9604. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, S.; Tanaka, T.; Soeda, Y.; Takashima, A. Enhanced Tau Protein Translation by Hyper-Excitation. Front. Aging Neurosci. 2019, 11, 322. [Google Scholar] [CrossRef] [Green Version]
- Schätzle, P.; Esteves da Silva, M.; Tas, R.P.; Katrukha, E.A.; Hu, H.Y.; Wierenga, C.J.; Kapitein, L.C.; Hoogenraad, C.C. Activity-Dependent Actin Remodeling at the Base of Dendritic Spines Promotes Microtubule Entry. Curr. Biol. 2018, 28, 2081–2093e6. [Google Scholar] [CrossRef] [Green Version]
- Merriam, E.B.; Lumbard, D.C.; Viesselmann, C.; Ballweg, J.; Stevenson, M.; Pietila, L.; Hu, X.; Dent, E.W. Dynamic Microtubules Promote Synaptic NMDA Receptor-Dependent Spine Enlargement. PLoS ONE 2011, 6, e27688. [Google Scholar] [CrossRef] [PubMed]
- Merriam, E.B.; Millette, M.; Lumbard, D.C.; Saengsawang, W.; Fothergill, T.; Hu, X.; Ferhat, L.; Dent, E.W. Synaptic Regulation of Microtubule Dynamics in Dendritic Spines by Calcium, F-Actin, and Drebrin. J. Neurosci. 2013, 33, 16471–16482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsuyama, F.; Niimi, G.; Kato, K.; Hirosawa, K.; Mikoshiba, K.; Okuya, M.; Karagiozov, K.; Kato, Y.; Kanno, T.; Sanoe, H.; et al. Redistribution of Microtubules in Dendrites of Hippocampal CA1 Neurons after Tetanic Stimulation during Long-Term Potentiation. Arch. Ital. Anat. Embriol. 2008, 113, 17–27. [Google Scholar]
- Kapitein, L.C.; Yau, K.W.; Gouveia, S.M.; van der Zwan, W.A.; Wulf, P.S.; Keijzer, N.; Demmers, J.; Jaworski, J.; Akhmanova, A.; Hoogenraad, C.C. NMDA Receptor Activation Suppresses Microtubule Growth and Spine Entry. J. Neurosci. 2011, 31, 8194–8209. [Google Scholar] [CrossRef] [PubMed]
- Weisenberg, R.C. Microtubule Formation in Vitro in Solutions Containing Low Calcium Concentrations. Science 1972, 177, 1104–1105. [Google Scholar] [CrossRef]
- Fuller, G.M.; Brinkley, B.R. Structure and Control of Assembly of Cytoplasmic Microtubules in Normal and Transformed Cells. J. Supramol. Struct. 1976, 5, 497–514. [Google Scholar] [CrossRef]
- Schliwa, M. The Role of Divalent Cations in the Regulation of Microtubule Assembly: In Vivo Studies on Microtubules of the Heliozoan Axopodium Using the Ionophore A23187. J. Cell Biol. 1976, 70, 527–540. [Google Scholar]
- Marcum, J.M.; Dedman, J.R.; Brinkley, B.R.; Means, A.R. Control of Microtubule Assembly-Disassembly by Calcium-Dependent Regulator Protein. Proc. Natl. Acad. Sci. USA 1978, 75, 3771–3775. [Google Scholar] [CrossRef] [Green Version]
- Schliwa, M.; Euteneuer, U.; Bulinski, J.C.; Izant, J.G. Calcium Lability of Cytoplasmic Microtubules and Its Modulation by Microtubule-Associated Proteins. Proc. Natl. Acad. Sci. USA 1981, 78, 1037–1041. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.C.; Wolff, J. Two Opposing Effects of Calmodulin on Microtubule Assembly Depend on the Presence of Microtubule-Associated Proteins. J. Biol. Chem. 1982, 257, 6306–6310. [Google Scholar] [CrossRef]
- Deery, W.J.; Means, A.R.; Brinkley, B.R. Calmodulin-Microtubule Association in Cultured Mammalian Cells. J. Cell Biol. 1984, 98, 904–910. [Google Scholar] [CrossRef] [Green Version]
- Adamec, E.; Mercken, M.; Beermann, M.L.; Didier, M.; Nixon, R.A. Acute Rise in the Concentration of Free Cytoplasmic Calcium Leads to Dephosphorylation of the Microtubule-Associated Protein Tau. Brain Res. 1997, 757, 93–101. [Google Scholar] [CrossRef]
- Maas, C.; Belgardt, D.; Lee, H.K.; Heisler, F.F.; Lappe-Siefke, C.; Magiera, M.M.; van Dijk, J.; Hausrat, T.J.; Janke, C.; Kneussel, M. Synaptic Activation Modifies Microtubules Underlying Transport of Postsynaptic Cargo. Proc. Natl. Acad. Sci. USA 2009, 106, 8731–8736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Hu, Y.; Xiong, Y.; Li, Z.; Wang, W.; Du, C.; Yang, Y.; Zhang, Y.; Xiao, F.; Wang, X. Association of Microtubule Dynamics with Chronic Epilepsy. Mol. Neurobiol. 2016, 53, 5013–5024. [Google Scholar] [CrossRef]
- Wu, J.W.; Hussaini, S.A.; Bastille, I.M.; Rodriguez, G.A.; Mrejeru, A.; Rilett, K.; Sanders, D.W.; Cook, C.; Fu, H.; Boonen, R.A.C.M.; et al. Neuronal Activity Enhances Tau Propagation and Tau Pathology In Vivo. Nat. Neurosci. 2016, 19, 1085–1092. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Holth, J.K.; Liao, F.; Stewart, F.R.; Mahan, T.E.; Jiang, H.; Cirrito, J.R.; Patel, T.K.; Hochgräfe, K.; Mandelkow, E.-M.; et al. Neuronal Activity Regulates Extracellular Tau In Vivo. J. Exp. Med. 2014, 211, 387–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pooler, A.M.; Phillips, E.C.; Lau, D.H.W.; Noble, W.; Hanger, D.P. Physiological Release of Endogenous Tau Is Stimulated by Neuronal Activity. EMBO Rep. 2013, 14, 389–394. [Google Scholar] [CrossRef]
- Froehner, S.C. Regulation of Ion Channel Distribution at Synapses. Ann. Rev. Neurosci. 1993, 16, 347–368. [Google Scholar] [CrossRef]
- Casini, S.; Tan, H.L.; Demirayak, I.; Remme, C.A.; Amin, A.S.; Scicluna, B.P.; Chatyan, H.; Ruijter, J.M.; Bezzina, C.R.; van Ginneken, A.C.G.; et al. Tubulin Polymerization Modifies Cardiac Sodium Channel Expression and Gating. Cardiovasc. Res. 2010, 85, 691–700. [Google Scholar] [CrossRef] [Green Version]
- Goswami, C.; Islam, M.S. Transient Receptor Potential Channels: What Is Happening? Reflections in the Wake of the 2009 TRP Meeting, Karolinska Institutet, Stockholm. Channels 2010, 4, 124–135. [Google Scholar]
- Johnson, B.D.; Byerly, L. A Cytoskeletal Mechanism for Ca2+ Channel Metabolic Dependence and Inactivation by Intracellular Ca2+. Neuron 1993, 10, 797–804. [Google Scholar] [CrossRef]
- Johnson, B.D.; Byerly, L. Ca2+ Channel Ca2+-Dependent Inactivation in a Mammalian Central Neuron Involves the Cytoskeleton. Pflug. Arch. 1994, 429, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Pascarel, C.; Brette, F.; Cazorla, O.; le Guennec, J.-Y. Effects on L-Type Calcium Current of Agents Interfering with the Cytoskeleton of Isolated Guinea-Pig Ventricular Myocytes. Exp. Physiol. 1999, 84, 1043–1050. [Google Scholar] [CrossRef] [PubMed]
- Malan, D. Microtubules Mobility Affects the Modulation of L-Type ICa by Muscarinic and β-Adrenergic Agonists in Guinea-Pig Cardiac Myocytes. J. Mol. Cell. Cardiol. 2003, 35, 195–206. [Google Scholar] [CrossRef]
- Li, Z.; Hall, A.M.; Kelinske, M.; Roberson, E.D. Seizure Resistance without Parkinsonism in Aged Mice after Tau Reduction. Neurobiol. Aging 2014, 35, 2617–2624. [Google Scholar] [CrossRef] [Green Version]
- DeVos, S.L.; Goncharoff, D.K.; Chen, G.; Kebodeaux, C.S.; Yamada, K.; Stewart, F.R.; Schuler, D.R.; Maloney, S.E.; Wozniak, D.F.; Rigo, F.; et al. Antisense Reduction of Tau in Adult Mice Protects against Seizures. J. Neurosci. 2013, 33, 12887–12897. [Google Scholar] [CrossRef] [Green Version]
- Gheyara, A.L.; Ponnusamy, R.; Djukic, B.; Craft, R.J.; Ho, K.; Guo, W.; Finucane, M.M.; Sanchez, P.E.; Mucke, L. Tau Reduction Prevents Disease in a Mouse Model of Dravet Syndrome. Ann. Neurol. 2014, 76, 443–456. [Google Scholar] [CrossRef] [Green Version]
- Holth, J.K.; Bomben, V.C.; Reed, J.G.; Inoue, T.; Younkin, L.; Younkin, S.G.; Pautler, R.G.; Botas, J.; Noebels, J.L. Tau Loss Attenuates Neuronal Network Hyperexcitability in Mouse and Drosophila Genetic Models of Epilepsy. J. Neurosci. 2013, 33, 1651–1659. [Google Scholar] [CrossRef]
- Cloyd, R.A.; Koren, J.; Abisambra, J.F.; Smith, B.N. Effects of Altered Tau Expression on Dentate Granule Cell Excitability in Mice. Exp. Neurol. 2021, 343, 113766. [Google Scholar] [CrossRef]
- Hall, A.M.; Throesch, B.T.; Buckingham, S.C.; Markwardt, S.J.; Peng, Y.; Wang, Q.; Hoffman, D.A.; Roberson, E.D. Tau-Dependent Kv4.2 Depletion and Dendritic Hyperexcitability in a Mouse Model of Alzheimer’s Disease. J. Neurosci. 2015, 35, 6221–6230. [Google Scholar]
- Crimins, J.L.; Rocher, A.B.; Luebke, J.I. Electrophysiological Changes Precede Morphological Changes to Frontal Cortical Pyramidal Neurons in the RTg4510 Mouse Model of Progressive Tauopathy. Acta Neuropathol. 2012, 124, 777–795. [Google Scholar] [CrossRef] [PubMed]
- Crimins, J.L.; Rocher, A.B.; Peters, A.; Shultz, P.; Lewis, J.; Luebke, J.I. Homeostatic Responses by Surviving Cortical Pyramidal Cells in Neurodegenerative Tauopathy. Acta Neuropathol. 2011, 122, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Rocher, A.B.; Crimins, J.L.; Amatrudo, J.M.; Kinson, M.S.; Todd-Brown, M.A.; Lewis, J.; Luebke, J.I. Structural and Functional Changes in Tau Mutant Mice Neurons Are Not Linked to the Presence of NFTs. Exp. Neurol. 2010, 223, 385–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Cabrero, A.M.; Guerrero-López, R.; Giráldez, B.G.; Llorens-Martín, M.; Ávila, J.; Serratosa, J.M.; Sánchez, M.P. Hyperexcitability and Epileptic Seizures in a Model of Frontotemporal Dementia. Neurobiol. Dis. 2013, 58, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ou, S.; Yin, M.; Xu, T.; Wang, T.; Liu, Y.; Ding, X.; Yu, X.; Yuan, J.; Huang, H.; et al. N-Methyl-D-Aspartate Receptors Mediate Epilepsy-Induced Axonal Impairment and Tau Phosphorylation via Activating Glycogen Synthase Kinase-3β and Cyclin-Dependent Kinase 5. Discov. Med. 2017, 23, 221–234. [Google Scholar] [PubMed]
- Pandis, D.; Scarmeas, N. Seizures in Alzheimer Disease: Clinical and Epidemiological Data. Epilepsy Curr. 2012, 12, 184–187. [Google Scholar] [CrossRef] [PubMed]
- Vossel, K.A.; Beagle, A.J.; Rabinovici, G.D.; Shu, H.; Lee, S.E.; Naasan, G.; Hegde, M.; Cornes, S.B.; Henry, M.L.; Nelson, A.B.; et al. Seizures and Epileptiform Activity in the Early Stages of Alzheimer Disease. JAMA Neurol. 2013, 70, 1158–1166. [Google Scholar] [CrossRef]
- Vossel, K.A.; Ranasinghe, K.G.; Beagle, A.J.; Mizuiri, D.; Honma, S.M.; Dowling, A.F.; Darwish, S.M.; van Berlo, V.; Barnes, D.E.; Mantle, M.; et al. Incidence and Impact of Subclinical Epileptiform Activity in Alzheimer’s Disease. Ann. Neurol. 2016, 80, 858–870. [Google Scholar] [CrossRef]
- Peña-Ortega, F. Brain Arrhythmias Induced by Amyloid Beta and Inflammation: Involvement in Alzheimer’s Disease and Other Inflammation-Related Pathologies. Curr. Alzheimer Res. 2019, 16, 1108–1131. [Google Scholar] [CrossRef]
- Menkes-Caspi, N.; Yamin, H.G.; Kellner, V.; Spires-Jones, T.L.; Cohen, D.; Stern, E.A. Pathological Tau Disrupts Ongoing Network Activity. Neuron 2015, 85, 959–966. [Google Scholar] [CrossRef] [Green Version]
- Hatch, R.J.; Wei, Y.; Xia, D.; Götz, J. Hyperphosphorylated Tau Causes Reduced Hippocampal CA1 Excitability by Relocating the Axon Initial Segment. Acta Neuropathol. 2017, 133, 717–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busche, M.A.; Wegmann, S.; Dujardin, S.; Commins, C.; Schiantarelli, J.; Klickstein, N.; Kamath, T.V.; Carlson, G.A.; Nelken, I.; Hyman, B.T. Tau Impairs Neural Circuits, Dominating Amyloid-β Effects, in Alzheimer Models In Vivo. Nat. Neurosci. 2019, 22, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Kopach, O.; Esteras, N.; Wray, S.; Rusakov, D.A.; Abramov, A.Y. Maturation and Phenotype of Pathophysiological Neuronal Excitability of Human Cells in Tau-Related Dementia. J. Cell Sci. 2020, 133, jcs241687. [Google Scholar] [CrossRef] [PubMed]
- Zempel, H.; Dennissen, F.J.A.; Kumar, Y.; Luedtke, J.; Biernat, J.; Mandelkow, E.-M.; Mandelkow, E. Axodendritic Sorting and Pathological Missorting of Tau Are Isoform-Specific and Determined by Axon Initial Segment Architecture. J. Biol. Chem. 2017, 292, 12192–12207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schappacher, K.A.; Xie, W.; Zhang, J.-M.; Baccei, M.L. Neonatal Vincristine Administration Modulates Intrinsic Neuronal Excitability in the Rat Dorsal Root Ganglion and Spinal Dorsal Horn during Adolescence. Pain 2019, 160, 645–657. [Google Scholar] [CrossRef]
- Sun, S.; Zhang, H.; Liu, J.; Popugaeva, E.; Xu, N.-J.; Feske, S.; White, C.L.; Bezprozvanny, I. Reduced Synaptic STIM2 Expression and Impaired Store-Operated Calcium Entry Cause Destabilization of Mature Spines in Mutant Presenilin Mice. Neuron 2014, 82, 79–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsushima, H.; Emanuele, M.; Polenghi, A.; Esposito, A.; Vassalli, M.; Barberis, A.; Difato, F.; Chieregatti, E. HDAC6 and RhoA Are Novel Players in Abeta-Driven Disruption of Neuronal Polarity. Nat. Commun. 2015, 6, 7781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Kumar, Y.; Zempel, H.; Mandelkow, E.-M.; Biernat, J.; Mandelkow, E. Novel Diffusion Barrier for Axonal Retention of Tau in Neurons and Its Failure in Neurodegeneration. EMBO J. 2011, 30, 4825–4837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Yang, J.; Wang, H.; Shan, B.; Yin, C.; Yu, H.; Zhang, X.; Dong, Z.; Yu, Y.; Zhao, R.; et al. Fibroblast Growth Factor 13 Stabilizes Microtubules to Promote Na+ Channel Function in Nociceptive DRG Neurons and Modulates Inflammatory Pain. J. Adv. Res. 2021, 31, 97–111. [Google Scholar] [CrossRef]
- Akin, E.J.; Alsaloum, M.; Higerd, G.P.; Liu, S.; Zhao, P.; Dib-Hajj, F.B.; Waxman, S.G.; Dib-Hajj, S.D. Paclitaxel Increases Axonal Localization and Vesicular Trafficking of Nav1.7. Brain 2021, 144, 1727–1737. [Google Scholar] [CrossRef]
- Illán-Gala, I.; Díaz de Terán, F.J.; Alonso, P.; Aguilar-Amat, M.-J. Nonconvulsive Status Epilepticus Secondary to Paclitaxel Administration. Epilepsy Behav. Case Rep. 2015, 4, 20–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carletti, F.; Sardo, P.; Gambino, G.; Liu, X.-A.; Ferraro, G.; Rizzo, V. Hippocampal Hyperexcitability Is Modulated by Microtubule-Active Agent: Evidence from In Vivo and In Vitro Epilepsy Models in the Rat. Front. Cell. Neurosci. 2016, 10, 29. [Google Scholar] [CrossRef] [Green Version]
- Sohn, P.D.; Huang, C.T.L.; Yan, R.; Fan, L.; Tracy, T.E.; Camargo, C.M.; Montgomery, K.M.; Arhar, T.; Mok, S.A.; Freilich, R.; et al. Pathogenic Tau Impairs Axon Initial Segment Plasticity and Excitability Homeostasis. Neuron 2019, 104, 458–470.e5. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.D.; Sammons, R.P.; Lebron, S.; Dumitrescu, A.S.; Watkins, T.B.K.; Uebele, V.N.; Renger, J.J.; Grubb, M.S. Calcineurin Signaling Mediates Activity-Dependent Relocation of the Axon Initial Segment. J. Neurosci. 2013, 33, 6950–6963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, M.D.; Dumitrescu, A.S.; Kruijssen, D.L.H.; Taylor, S.E.; Grubb, M.S. Rapid Modulation of Axon Initial Segment Length Influences Repetitive Spike Firing. Cell Rep. 2015, 13, 1233–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuba, H.; Oichi, Y.; Ohmori, H. Presynaptic Activity Regulates Na+ Channel Distribution at the Axon Initial Segment. Nature 2010, 465, 1075–1078. [Google Scholar] [CrossRef]
- Yamada, R.; Kuba, H. Structural and Functional Plasticity at the Axon Initial Segment. Front. Cell. Neurosci. 2016, 10, 250. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, Y.; Rasband, M.N. The Functional Organization and Assembly of the Axon Initial Segment. Curr. Opin. Neurobiol. 2008, 18, 307–313. [Google Scholar] [CrossRef]
- Jones, S.L.; Korobova, F.; Svitkina, T. Axon Initial Segment Cytoskeleton Comprises a Multiprotein Submembranous Coat Containing Sparse Actin Filaments. J. Cell Biol. 2014, 205, 67–81. [Google Scholar] [CrossRef] [Green Version]
- Leterrier, C.; Potier, J.; Caillol, G.; Debarnot, C.; Rueda Boroni, F.; Dargent, B. Nanoscale Architecture of the Axon Initial Segment Reveals an Organized and Robust Scaffold. Cell Rep. 2015, 13, 2781–2793. [Google Scholar] [CrossRef] [Green Version]
- Rasband, M.N. The Axon Initial Segment and the Maintenance of Neuronal Polarity. Nat. Rev. Neurosci. 2010, 11, 552–562. [Google Scholar] [CrossRef] [PubMed]
- Palay, S.L.; Sotelo, C.; Peters, A.; Orkand, P.M. The Axon Hillock And The Axon Initial Segment. J. Cell Biol. 1968, 38, 193–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winckler, B.; Forscher, P.; Mellman, I. A Diffusion Barrier Maintains Distribution of Membrane Proteins in Polarized Neurons. Nature 1999, 397, 698–701. [Google Scholar] [CrossRef] [PubMed]
- Song, A.; Wang, D.; Chen, G.; Li, Y.; Luo, J.; Duan, S.; Poo, M. A Selective Filter for Cytoplasmic Transport at the Axon Initial Segment. Cell 2009, 136, 1148–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakada, C.; Ritchie, K.; Oba, Y.; Nakamura, M.; Hotta, Y.; Iino, R.; Kasai, R.S.; Yamaguchi, K.; Fujiwara, T.; Kusumi, A. Accumulation of Anchored Proteins Forms Membrane Diffusion Barriers during Neuronal Polarization. Nat. Cell Biol. 2003, 5, 626–632. [Google Scholar] [CrossRef]
- Jenkins, S.M.; Bennett, V. Ankyrin-G Coordinates Assembly of the Spectrin-Based Membrane Skeleton, Voltage-Gated Sodium Channels, and L1 CAMs at Purkinje Neuron Initial Segments. J. Cell Biol. 2001, 155, 739–746. [Google Scholar] [CrossRef]
- Yang, Y.; Ogawa, Y.; Hedstrom, K.L.; Rasband, M.N. ΒIV Spectrin Is Recruited to Axon Initial Segments and Nodes of Ranvier by AnkyrinG. J. Cell Biol. 2007, 176, 509–519. [Google Scholar] [CrossRef]
- Bender, K.J.; Trussell, L.O. Axon Initial Segment Ca2+ Channels Influence Action Potential Generation and Timing. Neuron 2009, 61, 259–271. [Google Scholar] [CrossRef] [Green Version]
- Naundorf, B.; Wolf, F.; Volgushev, M. Unique Features of Action Potential Initiation in Cortical Neurons. Nature 2006, 440, 1060–1063. [Google Scholar] [CrossRef]
- Grubb, M.S.; Burrone, J. Activity-Dependent Relocation of the Axon Initial Segment Fine-Tunes Neuronal Excitability. Nature 2010, 465, 1070–1074. [Google Scholar] [CrossRef] [Green Version]
- Chand, A.N.; Galliano, E.; Chesters, R.A.; Grubb, M.S. A Distinct Subtype of Dopaminergic Interneuron Displays Inverted Structural Plasticity at the Axon Initial Segment. J. Neurosci. 2015, 35, 1573–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Wu, Y.; Gu, M.; Liu, Z.; Ma, Y.; Li, J.; Zhang, Y. Selective Filtering Defect at the Axon Initial Segment in Alzheimer’s Disease Mouse Models. Proc. Natl. Acad. Sci. USA 2014, 111, 14271–14276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohn, P.D.; Tracy, T.E.; Son, H.I.; Zhou, Y.; Leite, R.E.P.; Miller, B.L.; Seeley, W.W.; Grinberg, L.T.; Gan, L. Acetylated Tau Destabilizes the Cytoskeleton in the Axon Initial Segment and Is Mislocalized to the Somatodendritic Compartment. Mol. Neurodegener. 2016, 11, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez, J.-M.; Tryba, A.K.; Peña, F. Pacemaker Neurons and Neuronal Networks: An Integrative View. Curr. Opin. Neurobiol. 2004, 14, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Morales, M.; Colicos, M.A.; Goda, Y. Actin-Dependent Regulation of Neurotransmitter Release at Central Synapses. Neuron 2000, 27, 539–550. [Google Scholar] [CrossRef] [Green Version]
- Lepicard, S.; Franco, B.; de Bock, F.; Parmentier, M.-L. A Presynaptic Role of Microtubule-Associated Protein 1/Futsch in Drosophila: Regulation of Active Zone Number and Neurotransmitter Release. J. Neurosci. 2014, 34, 6759–6771. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, S.; Rost, B.R.; Camacho-Pérez, M.; Davis, M.W.; Söhl-Kielczynski, B.; Rosenmund, C.; Jorgensen, E.M. Ultrafast Endocytosis at Mouse Hippocampal Synapses. Nature 2013, 504, 242–247. [Google Scholar] [CrossRef] [Green Version]
- Delvendahl, I.; Vyleta, N.P.; von Gersdorff, H.; Hallermann, S. Fast, Temperature-Sensitive and Clathrin-Independent Endocytosis at Central Synapses. Neuron 2016, 90, 492–498. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.-S.; Lee, S.H.; Sheng, J.; Zhang, Z.; Zhao, W.-D.; Wang, D.; Jin, Y.; Charnay, P.; Ervasti, J.M.; Wu, L.-G. Actin Is Crucial for All Kinetically Distinguishable Forms of Endocytosis at Synapses. Neuron 2016, 92, 1020–1035. [Google Scholar] [CrossRef] [Green Version]
- Sakaba, T.; Neher, E. Involvement of Actin Polymerization in Vesicle Recruitment at the Calyx of Held Synapse. J. Neurosci. 2003, 23, 837–846. [Google Scholar] [CrossRef] [Green Version]
- Cole, J.C.; Villa, B.R.S.; Wilkinson, R.S. Disruption of Actin Impedes Transmitter Release in Snake Motor Terminals. J. Physiol. 2000, 525, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Lipstein, N.; Sakaba, T.; Cooper, B.H.; Lin, K.-H.; Strenzke, N.; Ashery, U.; Rhee, J.-S.; Taschenberger, H.; Neher, E.; Brose, N. Dynamic Control of Synaptic Vesicle Replenishment and Short-Term Plasticity by Ca2+-Calmodulin-Munc13-1 Signaling. Neuron 2013, 79, 82–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosoi, N.; Holt, M.; Sakaba, T. Calcium Dependence of Exo- and Endocytotic Coupling at a Glutamatergic Synapse. Neuron 2009, 63, 216–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.S.; Ho, W.-K.; Lee, S.-H. Actin-Dependent Rapid Recruitment of Reluctant Synaptic Vesicles into a Fast-Releasing Vesicle Pool. Proc. Natl. Acad. Sci. USA 2012, 109, E765–E774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Jung, K.J.; Jung, H.S.; Chang, S. Dynamics of Multiple Trafficking Behaviors of Individual Synaptic Vesicles Revealed by Quantum-Dot Based Presynaptic Probe. PLoS ONE 2012, 7, e38045. [Google Scholar] [CrossRef]
- Hirokawa, N.; Niwa, S.; Tanaka, Y. Molecular Motors in Neurons: Transport Mechanisms and Roles in Brain Function, Development, and Disease. Neuron 2010, 68, 610–638. [Google Scholar] [CrossRef] [Green Version]
- Melkov, A.; Abdu, U. Regulation of Long-Distance Transport of Mitochondria along Microtubules. Cell. Mol. Life Sci. 2018, 75, 163–176. [Google Scholar] [CrossRef]
- Gray, G. Synaptic Vesicles and Microtubules in Frog Motor Endplates. Proc. R. Soc. Lond. Ser. B Biol. Sci. 1978, 203, 219–227. [Google Scholar]
- Gray, G. Neurotransmitter Release Mechanisms and Microtubules. Proc. R. Soc. Lond. Ser. B Biol. Sci. 1983, 218, 253–258. [Google Scholar]
- Hirokawa, N.; Sobue, K.; Kanda, K.; Harada, A.; Yorifuji, H. The Cytoskeletal Architecture of the Presynaptic Terminal and Molecular Structure of Synapsin 1. J. Cell Biol. 1989, 108, 111–126. [Google Scholar] [CrossRef] [Green Version]
- Hummel, T.; Krukkert, K.; Roos, J.; Davis, G.; Klämbt, C. Drosophila Futsch/22C10 Is a MAP1B-like Protein Required for Dendritic and Axonal Development. Neuron 2000, 26, 357–370. [Google Scholar] [CrossRef] [Green Version]
- Perkins, G.A.; Tjong, J.; Brown, J.M.; Poquiz, P.H.; Scott, R.T.; Kolson, D.R.; Ellisman, M.H.; Spirou, G.A. The Micro-Architecture of Mitochondria at Active Zones: Electron Tomography Reveals Novel Anchoring Scaffolds and Cristae Structured for High-Rate Metabolism. J. Neurosci. 2010, 30, 1015–1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillaud, L.; Dimitrov, D.; Takahashi, T. Presynaptic Morphology and Vesicular Composition Determine Vesicle Dynamics in Mouse Central Synapses. eLife 2017, 6, e24845. [Google Scholar] [CrossRef] [PubMed]
- Guedes-Dias, P.; Nirschl, J.J.; Abreu, N.; Tokito, M.K.; Janke, C.; Magiera, M.M.; Holzbaur, E.L.F. Kinesin-3 Responds to Local Microtubule Dynamics to Target Synaptic Cargo Delivery to the Presynapse. Curr. Biol. 2019, 29, 268–282.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grigoriev, I.; Gouveia, S.M.; van der Vaart, B.; Demmers, J.; Smyth, J.T.; Honnappa, S.; Splinter, D.; Steinmetz, M.O.; Putney, J.W.; Hoogenraad, C.C.; et al. STIM1 Is a MT-Plus-End-Tracking Protein Involved in Remodeling of the ER. Curr. Biol. 2008, 18, 177–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honnappa, S.; Gouveia, S.M.; Weisbrich, A.; Damberger, F.F.; Bhavesh, N.S.; Jawhari, H.; Grigoriev, I.; van Rijssel, F.J.A.; Buey, R.M.; Lawera, A.; et al. An EB1-Binding Motif Acts as a Microtubule Tip Localization Signal. Cell 2009, 138, 366–376. [Google Scholar] [CrossRef] [Green Version]
- Asanov, A.; Sherry, R.; Sampieri, A.; Vaca, L. A Relay Mechanism between EB1 and APC Facilitate STIM1 Puncta Assembly at Endoplasmic Reticulum-Plasma Membrane Junctions. Cell Calcium 2013, 54, 246–256. [Google Scholar] [CrossRef]
- Decker, J.M.; Krüger, L.; Sydow, A.; Dennissen, F.J.; Siskova, Z.; Mandelkow, E.; Mandelkow, E. The Tau/A152T Mutation, a Risk Factor for Frontotemporal-spectrum Disorders, Leads to NR 2B Receptor-mediated Excitotoxicity. EMBO Rep. 2016, 17, 552–569. [Google Scholar] [CrossRef] [Green Version]
- Hunsberger, H.C.; Rudy, C.C.; Batten, S.R.; Gerhardt, G.A.; Reed, M.N. P301L Tau Expression Affects Glutamate Release and Clearance in the Hippocampal Trisynaptic Pathway. J. Neurochem. 2015, 132, 169–182. [Google Scholar] [CrossRef] [Green Version]
- Roberson, E.D.; Halabisky, B.; Yoo, J.W.; Yao, J.; Chin, J.; Yan, F.; Wu, T.; Hamto, P.; Devidze, N.; Yu, G.Q.; et al. Amyloid-β/Fyn-Induced Synaptic, Network, and Cognitive Impairments Depend on Tau Levels in Multiple Mouse Models of Alzheimer’s Disease. J. Neurosci. 2011, 31, 700–711. [Google Scholar] [CrossRef] [Green Version]
- Maeda, S.; Djukic, B.; Taneja, P.; Yu, G.; Lo, I.; Davis, A.; Craft, R.; Guo, W.; Wang, X.; Kim, D.; et al. Expression of A152T Human Tau Causes Age-dependent Neuronal Dysfunction and Loss in Transgenic Mice. EMBO Rep. 2016, 17, 530–551. [Google Scholar] [CrossRef] [PubMed]
- Sydow, A.; van der Jeugd, A.; Zheng, F.; Ahmed, T.; Balschun, D.; Petrova, O.; Drexler, D.; Zhou, L.; Rune, G.; Mandelkow, E.; et al. Tau-Induced Defects in Synaptic Plasticity, Learning, and Memory Are Reversible in Transgenic Mice after Switching Off the Toxic Tau Mutant. J. Neurosci. 2011, 31, 2511–2525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.-M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M.-Y. Synapse Loss and Microglial Activation Precede Tangles in a P301S Tauopathy Mouse Model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes, A.T.; Hausrat, T.J.; Heisler, F.F.; Gromova, K.V.; Lombino, F.L.; Fischer, T.; Ruschkies, L.; Breiden, P.; Thies, E.; Hermans-Borgmeyer, I.; et al. Spastin Depletion Increases Tubulin Polyglutamylation and Impairs Kinesin-Mediated Neuronal Transport, Leading to Working and Associative Memory Deficits. PLoS Biol. 2020, 18, e3000820. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.-D.; Chen, S.-R.; Chen, H.; Zeng, W.-A.; Pan, H.-L. Presynaptic N-Methyl-d-Aspartate (NMDA) Receptor Activity Is Increased Through Protein Kinase C in Paclitaxel-Induced Neuropathic Pain. J. Biol. Chem. 2016, 291, 19364–19373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graffe, M.; Zenisek, D.; Taraska, J.W. A Marginal Band of Microtubules Transports and Organizes Mitochondria in Retinal Bipolar Synaptic Terminals. J. Gen. Physiol. 2015, 146, 109–117. [Google Scholar] [CrossRef]
- Yuen, E.Y.; Jiang, Q.; Chen, P.; Gu, Z.; Feng, J.; Yan, Z. Serotonin 5-HT1A Receptors Regulate NMDA Receptor Channels through a Microtubule-Dependent Mechanism. J. Neurosci. 2005, 25, 5488–5501. [Google Scholar] [CrossRef] [Green Version]
- Setou, M.; Seog, D.-H.; Tanaka, Y.; Kanai, Y.; Takei, Y.; Kawagishi, M.; Hirokawa, N. Glutamate-Receptor-Interacting Protein GRIP1 Directly Steers Kinesin to Dendrites. Nature 2002, 417, 83–87. [Google Scholar] [CrossRef]
- Setou, M.; Nakagawa, T.; Seog, D.-H.; Hirokawa, N. Kinesin Superfamily Motor Protein KIF17 and MLin-10 in NMDA Receptor-Containing Vesicle Transport. Science 2000, 288, 1796–1802. [Google Scholar] [CrossRef]
- Salgado-Puga, K.; Pena-Ortega, F. Cellular and Network Mechanisms Underlying Memory Impairment Induced by Amyloid β Protein. Protein Pept. Lett. 2015, 22, 303–321. [Google Scholar] [CrossRef]
- Wagner, W.; Brenowitz, S.D.; Hammer, J.A. Myosin-Va Transports the Endoplasmic Reticulum into the Dendritic Spines of Purkinje Neurons. Nat. Cell Biol. 2011, 13, 40–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McVicker, D.P.; Awe, A.M.; Richters, K.E.; Wilson, R.L.; Cowdrey, D.A.; Hu, X.; Chapman, E.R.; Dent, E.W. Transport of a Kinesin-Cargo Pair along Microtubules into Dendritic Spines Undergoing Synaptic Plasticity. Nat. Commun. 2016, 7, 12741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shumyatsky, G.P.; Malleret, G.; Shin, R.-M.; Takizawa, S.; Tully, K.; Tsvetkov, E.; Zakharenko, S.S.; Joseph, J.; Vronskaya, S.; Yin, D.; et al. Stathmin, a Gene Enriched in the Amygdala, Controls Both Learned and Innate Fear. Cell 2005, 123, 697–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Y.-Y.; Lu, B.; Li, M.; Liu, Y.; Chen, J.; Chi, Z.-Q.; Liu, J.-G. Involvement of Actin Rearrangements within the Amygdala and the Dorsal Hippocampus in Aversive Memories of Drug Withdrawal in Acute Morphine-Dependent Rats. J. Neurosci. 2009, 29, 12244–12254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, T.; van der Jeugd, A.; Blum, D.; Galas, M.-C.; D’Hooge, R.; Buee, L.; Balschun, D. Cognition and Hippocampal Synaptic Plasticity in Mice with a Homozygous Tau Deletion. Neurobiol. Aging 2014, 35, 2474–2478. [Google Scholar] [CrossRef]
- Shipton, O.A.; Leitz, J.R.; Dworzak, J.; Acton, C.E.J.; Tunbridge, E.M.; Denk, F.; Dawson, H.N.; Vitek, M.P.; Wade-Martins, R.; Paulsen, O.; et al. Tau Protein Is Required for Amyloid-Induced Impairment of Hippocampal Long-Term Potentiation. J. Neurosci. 2011, 31, 1688–1692. [Google Scholar] [CrossRef] [Green Version]
- Fá, M.; Puzzo, D.; Piacentini, R.; Staniszewski, A.; Zhang, H.; Baltrons, M.A.; Li Puma, D.D.; Chatterjee, I.; Li, J.; Saeed, F.; et al. Extracellular Tau Oligomers Produce An Immediate Impairment of LTP and Memory. Sci. Rep. 2016, 6, 19393. [Google Scholar] [CrossRef] [Green Version]
- Hill, E.; Karikari, T.K.; Moffat, K.G.; Richardson, M.J.E.; Wall, M.J. Introduction of Tau Oligomers into Cortical Neurons Alters Action Potential Dynamics and Disrupts Synaptic Transmission and Plasticity. eNeuro 2019, 6. [Google Scholar] [CrossRef]
- Boekhoorn, K.; Terwel, D.; Biemans, B.; Borghgraef, P.; Wiegert, O.; Ramakers, G.J.A.; de Vos, K.; Krugers, H.; Tomiyama, T.; Mori, H.; et al. Improved Long-Term Potentiation and Memory in Young Tau-P301L Transgenic Mice before Onset of Hyperphosphorylation and Tauopathy. J. Neurosci. 2006, 26, 3514–3523. [Google Scholar] [CrossRef] [Green Version]
- Ondrejcak, T.; Hu, N.W.; Qi, Y.; Klyubin, I.; Corbett, G.T.; Fraser, G.; Perkinton, M.S.; Walsh, D.M.; Billinton, A.; Rowan, M.J. Soluble Tau Aggregates Inhibit Synaptic Long-Term Depression and Amyloid β-Facilitated LTD in Vivo. Neurobiol. Dis. 2019, 127, 582–590. [Google Scholar] [CrossRef]
- Barnes, S.J.; Opitz, T.; Merkens, M.; Kelly, T.; von der Brelie, C.; Krueppel, R.; Beck, H. Stable Mossy Fiber Long-Term Potentiation Requires Calcium Influx at the Granule Cell Soma, Protein Synthesis, and Microtubule-Dependent Axonal Transport. J. Neurosci. 2010, 30, 12996–13004. [Google Scholar] [CrossRef]
- Vickers, C.A.; Wyllie, D.J.A. Late-Phase, Protein Synthesis-Dependent Long-Term Potentiation in Hippocampal CA1 Pyramidal Neurones with Destabilized Microtubule Networks. Br. J. Pharmacol. 2007, 151, 1071–1077. [Google Scholar] [CrossRef] [Green Version]
- Regan, P.; Piers, T.; Yi, J.H.; Kim, D.H.; Huh, S.; Park, S.J.; Ryu, J.H.; Whitcomb, D.J.; Cho, K. Tau Phosphorylation at Serine 396 Residue Is Required for Hippocampal LTD. J. Neurosci. 2015, 35, 4804–4812. [Google Scholar] [CrossRef] [Green Version]
- Kimura, T.; Whitcomb, D.J.; Jo, J.; Regan, P.; Piers, T.; Heo, S.; Brown, C.; Hashikawa, T.; Murayama, M.; Seok, H.; et al. Microtubule-Associated Protein Tau Is Essential for Long-Term Depression in the Hippocampus. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130144. [Google Scholar] [CrossRef] [Green Version]
- Scheuss, V.; Bonhoeffer, T. Function of Dendritic Spines on Hippocampal Inhibitory Neurons. Cereb. Cortex 2014, 24, 3142–3153. [Google Scholar] [CrossRef] [Green Version]
- Brandt, R.; Paululat, A. Microcompartments in the Drosophila Heart and the Mammalian Brain: General Features and Common Principles. Biol. Chem. 2013, 394, 217–230. [Google Scholar] [CrossRef]
- Matus, A.; Ackermann, M.; Pehling, G.; Byers, H.R.; Fujiwara, K. High Actin Concentrations in Brain Dendritic Spines and Postsynaptic Densities. Proc. Natl. Acad. Sci. USA 1982, 79, 7590–7594. [Google Scholar] [CrossRef] [Green Version]
- Gray, E.G.; Westrum, L.E.; Burgoyne, R.D.; Barron, J. Synaptic Organisation and Neuron Microtubule Distribution. Cell Tissue Res. 1982, 226, 579–588. [Google Scholar] [CrossRef]
- Westrum, L.E.; Jones, D.H.; Gray, E.G.; Barron, J. Microtubules, Dendritic Spines and Spine Apparatuses. Cell Tissue Res. 1980, 208, 171–181. [Google Scholar] [CrossRef]
- Conde, C.; Cáceres, A. Microtubule Assembly, Organization and Dynamics in Axons and Dendrites. Nat. Rev. Neurosci. 2009, 10, 319–332. [Google Scholar] [CrossRef]
- Hoogenraad, C.C.; Bradke, F. Control of Neuronal Polarity and Plasticity—A Renaissance for Microtubules? Trends Cell Biol. 2009, 19, 669–676. [Google Scholar] [CrossRef]
- Westrum, L.E.; Gray, E.G.; Burgoyne, R.D.; Barron, J. Synaptic Development and Microtubule Organization. Cell Tissue Res. 1983, 231, 93–102. [Google Scholar] [CrossRef]
- Müller-Thomsen, L.; Borgmann, D.; Morcinek, K.; Schröder, S.; Dengler, B.; Moser, N.; Neumaier, F.; Schneider, T.; Schröder, H.; Huggenberger, S. Consequences of Hyperphosphorylated Tau on the Morphology and Excitability of Hippocampal Neurons in Aged Tau Transgenic Mice. Neurobiol. Aging 2020, 93, 109–123. [Google Scholar] [CrossRef]
- Gu, J.; Firestein, B.L.; Zheng, J.Q. Microtubules in Dendritic Spine Development. J. Neurosci. 2008, 28, 12120–12124. [Google Scholar] [CrossRef]
- Geraldo, S.; Khanzada, U.K.; Parsons, M.; Chilton, J.K.; Gordon-Weeks, P.R. Targeting of the F-Actin-Binding Protein Drebrin by the Microtubule plus-Tip Protein EB3 Is Required for Neuritogenesis. Nat. Cell Biol. 2008, 10, 1181–1189. [Google Scholar] [CrossRef]
- Gordon-Weeks, P.R. The Role of the Drebrin/EB3/Cdk5 Pathway in Dendritic Spine Plasticity, Implications for Alzheimer’s Disease. Brain Res. Bull. 2016, 126, 293–299. [Google Scholar] [CrossRef] [Green Version]
- Pchitskaya, E.; Kraskovskaya, N.; Chernyuk, D.; Popugaeva, E.; Zhang, H.; Vlasova, O.; Bezprozvanny, I. Stim2-Eb3 Association and Morphology of Dendritic Spines in Hippocampal Neurons. Sci. Rep. 2017, 7, 17625. [Google Scholar] [CrossRef] [Green Version]
- Matsuzaki, M.; Honkura, N.; Ellis-Davies, G.C.R.; Kasai, H. Structural Basis of Long-Term Potentiation in Single Dendritic Spines. Nature 2004, 429, 761–766. [Google Scholar] [CrossRef]
- Okamoto, K.-I.; Nagai, T.; Miyawaki, A.; Hayashi, Y. Rapid and Persistent Modulation of Actin Dynamics Regulates Postsynaptic Reorganization Underlying Bidirectional Plasticity. Nat. Neurosci. 2004, 7, 1104–1112. [Google Scholar] [CrossRef]
- Hu, X.; Ballo, L.; Pietila, L.; Viesselmann, C.; Ballweg, J.; Lumbard, D.; Stevenson, M.; Merriam, E.; Dent, E.W. BDNF-Induced Increase of PSD-95 in Dendritic Spines Requires Dynamic Microtubule Invasions. J. Neurosci. 2011, 31, 15597–15603. [Google Scholar] [CrossRef] [Green Version]
- Caceres, A.; Payne, M.R.; Binder, L.I.; Steward, O. Immunocytochemical Localization of Actin and Microtubule-Associated Protein MAP2 in Dendritic Spines. Proc. Natl. Acad. Sci. USA 1983, 80, 1738–1742. [Google Scholar] [CrossRef] [Green Version]
- Buddle, M.; Eberhardt, E.; Ciminello, L.H.; Levin, T.; Wing, R.; DiPasquale, K.; Raley-Susman, K.M. Microtubule-Associated Protein 2 Associates with the NMDA Receptor and Is Spatially Redistributed within Rat Hippocampal Neurons after Oxygen-Glucose Deprivation. Brain Res. 2003, 978, 38–50. [Google Scholar] [CrossRef]
- Gertz, H.J.; Cervos-Navarro, J.; Ewald, V. The Septo-Hippocampal Pathway in Patients Suffering from Senile Dementia of Alzheimer’s Type: Evidence for Neuronal Plasticity? Neurosci. Lett. 1987, 76, 228–232. [Google Scholar] [CrossRef]
- Androuin, A.; Potier, B.; Nägerl, U.V.; Cattaert, D.; Danglot, L.; Thierry, M.; Youssef, I.; Triller, A.; Duyckaerts, C.; el Hachimi, K.H.; et al. Evidence for Altered Dendritic Spine Compartmentalization in Alzheimer’s Disease and Functional Effects in a Mouse Model. Acta Neuropathol. 2018, 135, 839–854. [Google Scholar] [CrossRef]
- Esteves da Silva, M.; Adrian, M.; Schätzle, P.; Lipka, J.; Watanabe, T.; Cho, S.; Futai, K.; Wierenga, C.J.; Kapitein, L.C.; Hoogenraad, C.C. Positioning of AMPA Receptor-Containing Endosomes Regulates Synapse Architecture. Cell Rep. 2015, 13, 933–943. [Google Scholar] [CrossRef] [Green Version]
- Glantz, L.A.; Lewis, D.A. Decreased Dendritic Spine Density on Prefrontal Cortical Pyramidal Neurons in Schizophrenia. Arch. Gen. Psychiatry 2000, 57, 65–73. [Google Scholar] [CrossRef] [Green Version]
- Irwin, S.A.; Patel, B.; Idupulapati, M.; Harris, J.B.; Crisostomo, R.A.; Larsen, B.P.; Kooy, F.; Willems, P.J.; Cras, P.; Kozlowski, P.B.; et al. Abnormal Dendritic Spine Characteristics in the Temporal and Visual Cortices of Patients with Fragile-X Syndrome: A Quantitative Examination. Am. J. Med. Genet. 2001, 98, 161–167. [Google Scholar] [CrossRef]
- Penazzi, L.; Tackenberg, C.; Ghori, A.; Golovyashkina, N.; Niewidok, B.; Selle, K.; Ballatore, C.; Smith, A.B.; Bakota, L.; Brandt, R. Aβ-Mediated Spine Changes in the Hippocampus Are Microtubule-Dependent and Can Be Reversed by a Subnanomolar Concentration of the Microtubule-Stabilizing Agent Epothilone D. Neuropharmacology 2016, 105, 84–95. [Google Scholar] [CrossRef] [Green Version]
- Chuckowree, J.A.; Vickers, J.C. Cytoskeletal and Morphological Alterations Underlying Axonal Sprouting after Localized Transection of Cortical Neuron Axons In Vitro. J. Neurosci. 2003, 23, 3715–3725. [Google Scholar] [CrossRef] [Green Version]
- Tang-Schomer, M.D.; Johnson, V.E.; Baas, P.W.; Stewart, W.; Smith, D.H. Partial Interruption of Axonal Transport Due to Microtubule Breakage Accounts for the Formation of Periodic Varicosities after Traumatic Axonal Injury. Exp. Neurol. 2012, 233, 364–372. [Google Scholar] [CrossRef] [Green Version]
- Qu, X.; Yuan, F.N.; Corona, C.; Pasini, S.; Pero, M.E.; Gundersen, G.G.; Shelanski, M.L.; Bartolini, F. Stabilization of Dynamic Microtubules by MDia1 Drives Tau-Dependent Aβ1–42 Synaptotoxicity. J. Cell Biol. 2017, 216, 3161–3178. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.; Zheng, J.Q. Microtubules in Dendritic Spine Development and Plasticity. Open Neurosci. J. 2009, 3, 128–133. [Google Scholar] [CrossRef]
- Briones, T.L.; Woods, J. Chemotherapy-Induced Cognitive Impairment Is Associated with Decreases in Cell Proliferation and Histone Modifications. BMC Neurosci. 2011, 12, 124. [Google Scholar] [CrossRef] [Green Version]
- Winocur, G.; Wojtowicz, J.M.; Tannock, I.F. Memory Loss in Chemotherapy-Treated Rats Is Exacerbated in High-Interference Conditions and Related to Suppression of Hippocampal Neurogenesis. Behav. Brain Res. 2015, 281, 239–244. [Google Scholar] [CrossRef]
- Muallaoglu, S.; Disel, U.; Mertsoylu, H.; Besen, A.; Karadeniz, C.; Sumbul, A.T.; Abali, H.; Ozyilkan, O. Acute Infusion Reactions to Chemotherapeutic Drugs: A Single Institute Experience. J. BUON 2013, 18, 261–267. [Google Scholar]
- Perry, J.R.; Warner, E. Transient Encephalopathy after Paclitaxel (Taxol) Infusion. Neurology 1996, 46, 1596–1599. [Google Scholar] [CrossRef]
- Yousefzadeh, S.A.; Youngkin, A.E.; Lusk, N.A.; Wen, S.; Meck, W.H. Bidirectional Role of Microtubule Dynamics in the Acquisition and Maintenance of Temporal Information in Dorsolateral Striatum. Neurobiol. Learn. Mem. 2021, 183, 107468. [Google Scholar] [CrossRef]
- Cassar, M.; Law, A.D.; Chow, E.S.; Giebultowicz, J.M.; Kretzschmar, D. Disease-Associated Mutant Tau Prevents Circadian Changes in the Cytoskeleton of Central Pacemaker Neurons. Front. Neurosci. 2020, 14, 232. [Google Scholar] [CrossRef]
- Mershin, A.; Pavlopoulos, E.; Fitch, O.; Braden, B.C.; Nanopoulos, D.V.; Skoulakis, E.M.C. Learning and Memory Deficits Upon TAU Accumulation in Drosophila Mushroom Body Neurons. Learn. Mem. 2004, 11, 277–287. [Google Scholar] [CrossRef] [Green Version]
- Pennanen, L.; Wolfer, D.P.; Nitsch, R.M.; Gotz, J. Impaired Spatial Reference Memory and Increased Exploratory Behavior in P301L Tau Transgenic Mice. Genes Brain Behav. 2006, 5, 369–379. [Google Scholar] [CrossRef]
- Uchida, S.; Teubner, B.J.W.; Hevi, C.; Hara, K.; Kobayashi, A.; Dave, R.M.; Shintaku, T.; Jaikhan, P.; Yamagata, H.; Suzuki, T.; et al. CRTC1 Nuclear Translocation Following Learning Modulates Memory Strength via Exchange of Chromatin Remodeling Complexes on the Fgf1 Gene. Cell Rep. 2017, 18, 352–366. [Google Scholar] [CrossRef]
- Muhia, M.; Thies, E.; Labonté, D.; Ghiretti, A.E.; Gromova, K.V.; Xompero, F.; Lappe-Siefke, C.; Hermans-Borgmeyer, I.; Kuhl, D.; Schweizer, M.; et al. The Kinesin KIF21B Regulates Microtubule Dynamics and Is Essential for Neuronal Morphology, Synapse Function, and Learning and Memory. Cell Rep. 2016, 15, 968–977. [Google Scholar] [CrossRef] [Green Version]
- Ballatore, C.; Brunden, K.R.; Huryn, D.M.; Trojanowski, J.Q.; Lee, V.M.Y.; Smith, A.B. Microtubule Stabilizing Agents as Potential Treatment for Alzheimers Disease and Related Neurodegenerative Tauopathies. J. Med. Chem. 2012, 55, 8979–8996. [Google Scholar] [CrossRef] [Green Version]
- Guo, B.; Huang, Y.; Gao, Q.; Zhou, Q. Stabilization of Microtubules Improves Cognitive Functions and Axonal Transport of Mitochondria in Alzheimer’s Disease Model Mice. Neurobiol. Aging 2020, 96, 223–232. [Google Scholar] [CrossRef]
- Cross, D.J.; Meabon, J.S.; Cline, M.M.; Richards, T.L.; Stump, A.J.; Cross, C.G.; Minoshima, S.; Banks, W.A.; Cook, D.G. Paclitaxel Reduces Brain Injury from Repeated Head Trauma in Mice. J. Alzheimers Dis. 2019, 67, 859–874. [Google Scholar] [CrossRef]
- Chang, A.; Chung, N.-C.; Lawther, A.J.; Ziegler, A.I.; Shackleford, D.M.; Sloan, E.K.; Walker, A.K. The Anti-Inflammatory Drug Aspirin Does Not Protect Against Chemotherapy-Induced Memory Impairment by Paclitaxel in Mice. Front. Oncol. 2020, 10, 564965. [Google Scholar] [CrossRef]
- Fardell, J.E.; Vardy, J.; Johnston, I.N. The Short and Long Term Effects of Docetaxel Chemotherapy on Rodent Object Recognition and Spatial Reference Memory. Life Sci. 2013, 93, 596–604. [Google Scholar] [CrossRef]
- Callaghan, C.K.; O’Mara, S.M. Long-Term Cognitive Dysfunction in the Rat Following Docetaxel Treatment Is Ameliorated by the Phosphodiesterase-4 Inhibitor, Rolipram. Behav. Brain Res. 2015, 290, 84–89. [Google Scholar] [CrossRef]
- Atarod, D.; Eskandari-Sedighi, G.; Pazhoohi, F.; Karimian, S.M.; Khajeloo, M.; Riazi, G.H. Microtubule Dynamicity Is More Important than Stability in Memory Formation: An In Vivo Study. J. Mol. Neurosci. 2015, 56, 313–319. [Google Scholar] [CrossRef]
- Panoz-Brown, D.; Carey, L.M.; Smith, A.E.; Gentry, M.; Sluka, C.M.; Corbin, H.E.; Wu, J.-E.; Hohmann, A.G.; Crystal, J.D. The Chemotherapeutic Agent Paclitaxel Selectively Impairs Reversal Learning While Sparing Prior Learning, New Learning and Episodic Memory. Neurobiol. Learn. Mem. 2017, 144, 259–270. [Google Scholar] [CrossRef] [Green Version]
- Huehnchen, P.; Boehmerle, W.; Springer, A.; Freyer, D.; Endres, M. A Novel Preventive Therapy for Paclitaxel-Induced Cognitive Deficits: Preclinical Evidence from C57BL/6 Mice. Transl. Psychiatry 2017, 7, e1185. [Google Scholar] [CrossRef] [Green Version]
- Fardell, J.E.; Zhang, J.; de Souza, R.; Vardy, J.; Johnston, I.; Allen, C.; Henderson, J.; Piquette-Miller, M. The Impact of Sustained and Intermittent Docetaxel Chemotherapy Regimens on Cognition and Neural Morphology in Healthy Mice. Psychopharmacology 2014, 231, 841–852. [Google Scholar] [CrossRef]
- Li, Z.; Zhao, S.; Zhang, H.-L.; Liu, P.; Liu, F.-F.; Guo, Y.-X.; Wang, X.-L. Proinflammatory Factors Mediate Paclitaxel-Induced Impairment of Learning and Memory. Mediat. Inflamm. 2018, 2018, 3941840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.D.; Fischer, T.T.; Ehrlich, B.E. Pharmacological Rescue of Cognitive Function in a Mouse Model of Chemobrain. Mol. Neurodegener. 2021, 16, 41. [Google Scholar] [CrossRef]
- Seigers, R.; Loos, M.; van Tellingen, O.; Boogerd, W.; Smit, A.B.; Schagen, S.B. Cognitive Impact of Cytotoxic Agents in Mice. Psychopharmacology 2015, 232, 17–37. [Google Scholar] [CrossRef]
- Bensimon, G.; Chermat, R. Microtubule Disruption and Cognitive Defects: Effect of Colchicine on Learning Behavior in Rats. Pharmacol. Biochem. Behav. 1991, 38, 141–145. [Google Scholar] [CrossRef]
- Nakayama, T.; Sawada, T. Involvement of Microtubule Integrity in Memory Impairment Caused by Colchicine. Pharmacol. Biochem. Behav. 2002, 71, 119–138. [Google Scholar] [CrossRef]
- Di Patre, P.L.; Oh, J.D.; Simmons, J.M.; Butcher, L.L. Intrafimbrial Colchicine Produces Transient Impairment of Radial-Arm Maze Performance Correlated with Morphologic Abnormalities of Septohippocampal Neurons Expressing Cholinergic Markers and Nerve Growth Factor Receptor. Brain Res. 1990, 523, 316–320. [Google Scholar] [CrossRef]
- Mileusnic, R.; Lancashire, C.L.; Rose, S.P.R. Recalling an Aversive Experience by Day-Old Chicks Is Not Dependent on Somatic Protein Synthesis. Learn. Mem. 2005, 12, 615–619. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Seghal, N.; S Naidu, P.S.; Padi, S.S.; Goyal, R. Colchicines-Induced Neurotoxicity as an Animal Model of Sporadic Dementia of Alzheimer’s Type. Pharmacol. Rep. 2007, 59, 274–283. [Google Scholar]
- Tilson, H.A.; Rogers, B.C.; Grimes, L.; Harry, G.J.; Peterson, N.J.; Hong, J.S.; Dyer, R.S. Time-Dependent Neurobiological Effects of Colchicine Administered Directly into the Hippocampus of Rats. Brain Res. 1987, 408, 163–172. [Google Scholar] [CrossRef]
- Tilson, H.A.; Peterson, N.J. Colchicine as an Investigative Tool in Neurobiology. Toxicology 1987, 46, 159–175. [Google Scholar] [CrossRef]
- Walsh, T.J.; Schulz, D.W.; Tilson, H.A.; Schmechel, D.E. Cochicine-Induced Granule Cell Loss in Rat Hippocampus: Selective Behavioral and Histological Alterations. Brain Res. 1986, 398, 23–36. [Google Scholar] [CrossRef]
- Goldschmidt, R.B.; Steward, O. Neurotoxic Effects of Colchicine: Differential Susceptibility of CNS Neuronal Populations. Neuroscience 1982, 7, 695–714. [Google Scholar] [CrossRef]
- Lothman, E.W.; Stein, D.A.; Wooten, G.F.; Zucker, D.K. Potential Mechanisms Underlying the Destruction of Dentate Gyrus Granule Cells by Colchicine. Exp. Neurol. 1982, 78, 293–302. [Google Scholar] [CrossRef]
- Jarrard, L.E.; Okaichi, H.; Steward, O.; Goldschmidt, R.B. On the Role of Hippocampal Connections in the Performance of Place and Cue Tasks: Comparisons with Damage to Hippocampus. Behav. Neurosci. 1984, 98, 946–954. [Google Scholar] [CrossRef]
- Ionescu, T. Exploring the nature of cognitive flexibility. New Ideas Psychol. 2012, 30, 190–200. [Google Scholar] [CrossRef]
- Tanimura, Y.; Yang, M.C.; Lewis, M.H. Procedural learning and cognitive flexibility in a mouse model of restricted, repetitive behaviour. Behav. Brain Res. 2008, 189, 250–256. [Google Scholar] [CrossRef]
- Cowen, S.L.; Phelps, C.E.; Navratilova, E.; McKinzie, D.L.; Okun, A.; Husain, O.; Gleason, S.D.; Witkin, J.M.; Porreca, F. Chronic pain impairs cognitive flexibility and engages novel learning strategies in rats. Pain 2018, 159, 1403–1412. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peña-Ortega, F.; Robles-Gómez, Á.A.; Xolalpa-Cueva, L. Microtubules as Regulators of Neural Network Shape and Function: Focus on Excitability, Plasticity and Memory. Cells 2022, 11, 923. https://doi.org/10.3390/cells11060923
Peña-Ortega F, Robles-Gómez ÁA, Xolalpa-Cueva L. Microtubules as Regulators of Neural Network Shape and Function: Focus on Excitability, Plasticity and Memory. Cells. 2022; 11(6):923. https://doi.org/10.3390/cells11060923
Chicago/Turabian StylePeña-Ortega, Fernando, Ángel Abdiel Robles-Gómez, and Lorena Xolalpa-Cueva. 2022. "Microtubules as Regulators of Neural Network Shape and Function: Focus on Excitability, Plasticity and Memory" Cells 11, no. 6: 923. https://doi.org/10.3390/cells11060923