
BTK Inhibitors Impair Platelet-Mediated Antifungal Activity

 , , , , ,
, , , , ,  , , , ,
, , , ,
Abstract
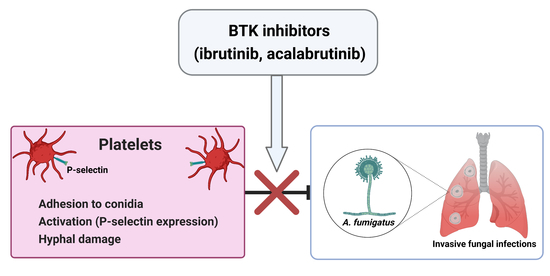
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Patients and Samples
2.2. Platelets–Conidia Adherence Assay
2.3. P-Selectin Expression Assay
2.4. XTT Assay
2.5. Statistical Analyses
3. Results
3.1. Ibrutinib Inhibits the Ability of Platelets to Adhere to Conidia
3.2. Ibrutinib and Acalabrutinib Reduce P-Selectin Expression on Platelets in Response to A. fumigatus Conidia
3.3. Ibrutinib and Acalabrutinib Hamper Platelet-Mediated Hyphal Damage
3.4. Platelet-Mediated Antifungal Activity Decreases during Ibrutinib Treatment in CLL Patients
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shirley, M. Bruton Tyrosine Kinase Inhibitors in B-Cell Malignancies: Their Use and Differential Features. Target. Oncol. 2022, 17, 69–84. [Google Scholar] [CrossRef]
- Williams, A.M.; Baran, A.M.; Meacham, P.J.; Feldman, M.M.; Valencia, H.E.; Newsom-Stewart, C.; Gupta, N.; Janelsins, M.C.; Barr, P.M.; Zent, C.S. Analysis of the Risk of Infection in Patients with Chronic Lymphocytic Leukemia in the Era of Novel Therapies. Leuk. Lymphoma 2018, 59, 625–632. [Google Scholar] [CrossRef]
- Tillman, B.F.; Pauff, J.M.; Satyanarayana, G.; Talbott, M.; Warner, J.L. Systematic Review of Infectious Events with the Bruton Tyrosine Kinase Inhibitor Ibrutinib in the Treatment of Hematologic Malignancies. Eur. J. Haematol. 2018, 100, 325–334. [Google Scholar] [CrossRef]
- Visentin, A.; Nasillo, V.; Marchetti, M.; Ferrarini, I.; Paolini, R.; Sancetta, R.; Rigolin, G.M.; Cibien, F.; Riva, M.; Briani, C.; et al. Clinical Characteristics and Outcome of West Nile Virus Infection in Patients with Lymphoid Neoplasms: An Italian Multicentre Study. Hemasphere 2020, 4, e395. [Google Scholar] [CrossRef]
- Ball, S.; Das, A.; Vutthikraivit, W.; Edwards, P.J.; Hardwicke, F.; Short, N.J.; Borthakur, G.; Maiti, A. Risk of Infection Associated With Ibrutinib in Patients With B-Cell Malignancies: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Clin. Lymphoma Myeloma Leuk. 2020, 20, 87–97.e5. [Google Scholar] [CrossRef]
- Mauro, F.R.; Giannarelli, D.; Visentin, A.; Reda, G.; Sportoletti, P.; Frustaci, A.M.; Chiarenza, A.; Ciolli, S.; Vitale, C.; Laurenti, L.; et al. Prognostic Impact and Risk Factors of Infections in Patients with Chronic Lymphocytic Leukemia Treated with Ibrutinib. Cancers 2021, 13, 3240. [Google Scholar] [CrossRef]
- Varughese, T.; Taur, Y.; Cohen, N.; Palomba, M.L.; Seo, S.K.; Hohl, T.M.; Redelman-Sidi, G. Serious Infections in Patients Receiving Ibrutinib for Treatment of Lymphoid Cancer. Clin. Infect. Dis. 2018, 67, 687–692. [Google Scholar] [CrossRef] [Green Version]
- Ghez, D.; Calleja, A.; Protin, C.; Baron, M.; Ledoux, M.-P.; Damaj, G.; Dupont, M.; Dreyfus, B.; Ferrant, E.; Herbaux, C.; et al. Early-Onset Invasive Aspergillosis and Other Fungal Infections in Patients Treated with Ibrutinib. Blood 2018, 131, 1955–1959. [Google Scholar] [CrossRef]
- Ruchlemer, R.; Ben-Ami, R.; Bar-Meir, M.; Brown, J.R.; Malphettes, M.; Mous, R.; Tonino, S.H.; Soussain, C.; Barzic, N.; Messina, J.A.; et al. Ibrutinib-associated Invasive Fungal Diseases in Patients with Chronic Lymphocytic Leukaemia and Non-Hodgkin Lymphoma: An Observational Study. Mycoses 2019, 62, 1140–1147. [Google Scholar] [CrossRef]
- Rogers, K.A.; Mousa, L.; Zhao, Q.; Bhat, S.A.; Byrd, J.C.; El Boghdadly, Z.; Guerrero, T.; Levine, L.B.; Lucas, F.; Shindiapina, P.; et al. Incidence of Opportunistic Infections during Ibrutinib Treatment for B-Cell Malignancies. Leukemia 2019, 33, 2527–2530. [Google Scholar] [CrossRef]
- Teh, B.W.; Chui, W.; Handunnetti, S.; Tam, C.; Worth, L.J.; Thursky, K.A.; Slavin, M.A. High Rates of Proven Invasive Fungal Disease with the Use of Ibrutinib Monotherapy for Relapsed or Refractory Chronic Lymphocytic Leukemia. Leuk. Lymphoma 2019, 60, 1572–1575. [Google Scholar] [CrossRef] [PubMed]
- Fürstenau, M.; Simon, F.; Cornely, O.A.; Hicketier, T.; Eichhorst, B.; Hallek, M.; Mellinghoff, S.C. Invasive Aspergillosis in Patients Treated With Ibrutinib. HemaSphere 2020, 4, e309. [Google Scholar] [CrossRef] [PubMed]
- Frei, M.; Aitken, S.L.; Jain, N.; Thompson, P.; Wierda, W.; Kontoyiannis, D.P.; DiPippo, A.J. Incidence and Characterization of Fungal Infections in Chronic Lymphocytic Leukemia Patients Receiving Ibrutinib. Leuk Lymphoma 2020, 61, 2488–2491. [Google Scholar] [CrossRef] [PubMed]
- Holowka, T.; Cheung, H.; Malinis, M.; Gan, G.; Deng, Y.; Perreault, S.; Isufi, I.; Azar, M.M. Incidence and Associated Risk Factors for Invasive Fungal Infections and Other Serious Infections in Patients on Ibrutinib. J. Infect. Chemotherapy 2021, 27, 1700–1705. [Google Scholar] [CrossRef]
- Little, J.S.; Weiss, Z.F.; Hammond, S.P. Invasive Fungal Infections and Targeted Therapies in Hematological Malignancies. J. Fungi 2021, 7, 1058. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, G.; Slavin, M.; Teh, B.W. Ibrutinib and Invasive Fungal Infections: The Known, the Unknown and the Known Unknowns. Leuk Lymphoma 2020, 61, 2292–2294. [Google Scholar] [CrossRef] [PubMed]
- Maffei, R.; Maccaferri, M.; Arletti, L.; Fiorcari, S.; Benatti, S.; Potenza, L.; Luppi, M.; Marasca, R. Immunomodulatory Effect of Ibrutinib: Reducing the Barrier against Fungal Infections. Blood Rev. 2020, 40, 100635. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Tian, X.; Lee, Y.S.; Gunti, S.; Lipsky, A.; Herman, S.E.M.; Salem, D.; Stetler-Stevenson, M.; Yuan, C.; Kardava, L.; et al. Partial Reconstitution of Humoral Immunity and Fewer Infections in Patients with Chronic Lymphocytic Leukemia Treated with Ibrutinib. Blood 2015, 126, 2213–2219. [Google Scholar] [CrossRef] [PubMed]
- Luppi, M.; Forghieri, F.; Potenza, L. Ibrutinib Is a Newly Recognized Host Factor for the Definition of Probable Invasive Pulmonary Mold Disease, Based on Off-Target Effects, Unrelated to Its B-Cell Immunosuppressant Activity. Clin. Infect. Dis. 2020, 71, 3265–3266. [Google Scholar] [CrossRef]
- Zhu, S.; Gokhale, S.; Jung, J.; Spirollari, E.; Tsai, J.; Arceo, J.; Wu, B.W.; Victor, E.; Xie, P. Multifaceted Immunomodulatory Effects of the BTK Inhibitors Ibrutinib and Acalabrutinib on Different Immune Cell Subsets—Beyond B Lymphocytes. Front. Cell Dev. Biol. 2021, 9, 727531. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.N.R.; Bittner, Z.; Liu, X.; Dang, T.-M.; Radsak, M.P.; Brunner, C. Bruton’s Tyrosine Kinase: An Emerging Key Player in Innate Immunity. Front. Immunol. 2017, 8, 1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, K.L.; Ifrim, D.C.; Quintin, J.; Netea, M.G.; van de Veerdonk, F.L. Antifungal Innate Immunity: Recognition and Inflammatory Networks. Semin. Immunopathol. 2015, 37, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Casadevall, A. Immunity to Invasive Fungal Diseases. Annu. Rev. Immunol. 2022, 40. [Google Scholar] [CrossRef] [PubMed]
- Mircescu, M.M.; Lipuma, L.; van Rooijen, N.; Pamer, E.G.; Hohl, T.M. Essential Role for Neutrophils but Not Alveolar Macrophages at Early Time Points Following Aspergillus Fumigatus Infection. J. Infect. Dis. 2009, 200, 647–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, J.V.; Lionakis, M.S. The Role of Neutrophils in Host Defense against Invasive Fungal Infections. Curr. Clin. Microbiol. Rep. 2018, 5, 181–189. [Google Scholar] [CrossRef]
- Mueller, H.; Stadtmann, A.; Van Aken, H.; Hirsch, E.; Wang, D.; Ley, K.; Zarbock, A. Tyrosine Kinase Btk Regulates E-Selectin–Mediated Integrin Activation and Neutrophil Recruitment by Controlling Phospholipase C (PLC) Γ2 and PI3Kγ Pathways. Blood 2010, 115, 3118–3127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yago, T.; Shao, B.; Miner, J.J.; Yao, L.; Klopocki, A.G.; Maeda, K.; Coggeshall, K.M.; McEver, R.P. E-Selectin Engages PSGL-1 and CD44 through a Common Signaling Pathway to Induce Integrin ALβ2-Mediated Slow Leukocyte Rolling. Blood 2010, 116, 485–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, C.J.; Schroder, K. Pattern Recognition Receptor Function in Neutrophils. Trends Immunol. 2013, 34, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Dubey, L.K.; Moeller, J.B.; Schlosser, A.; Sorensen, G.L.; Holmskov, U. Induction of Innate Immunity by Aspergillus Fumigatus Cell Wall Polysaccharides Is Enhanced by the Composite Presentation of Chitin and Beta-Glucan. Immunobiology 2014, 219, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Patin, E.C.; Thompson, A.; Orr, S.J. Pattern Recognition Receptors in Fungal Immunity. Semin. Cell Dev. Biol. 2019, 89, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Branzk, N.; Lubojemska, A.; Hardison, S.E.; Wang, Q.; Gutierrez, M.G.; Brown, G.D.; Papayannopoulos, V. Neutrophils Sense Microbe Size and Selectively Release Neutrophil Extracellular Traps in Response to Large Pathogens. Nat. Immunol. 2014, 15, 1017–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangla, A.; Khare, A.; Vineeth, V.; Panday, N.N.; Mukhopadhyay, A.; Ravindran, B.; Bal, V.; George, A.; Rath, S. Pleiotropic Consequences of Bruton Tyrosine Kinase Deficiency in Myeloid Lineages Lead to Poor Inflammatory Responses. Blood 2004, 104, 1191–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiedler, K.; Sindrilaru, A.; Terszowski, G.; Kokai, E.; Feyerabend, T.B.; Bullinger, L.; Rodewald, H.-R.; Brunner, C. Neutrophil Development and Function Critically Depend on Bruton Tyrosine Kinase in a Mouse Model of X-Linked Agammaglobulinemia. Blood 2011, 117, 1329–1339. [Google Scholar] [CrossRef] [Green Version]
- Blez, D.; Blaize, M.; Soussain, C.; Boissonnas, A.; Meghraoui-Kheddar, A.; Menezes, N.; Portalier, A.; Combadière, C.; Leblond, V.; Ghez, D.; et al. Ibrutinib Induces Multiple Functional Defects in the Neutrophil Response against Aspergillus fumigatus. Haematologica 2020, 105, 478–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbst, S.; Shah, A.; Mazon Moya, M.; Marzola, V.; Jensen, B.; Reed, A.; Birrell, M.A.; Saijo, S.; Mostowy, S.; Shaunak, S.; et al. Phagocytosis-dependent Activation of a TLR 9–BTK–Calcineurin–NFAT Pathway Co-ordinates Innate Immunity to Aspergillus fumigatus. EMBO Mol. Med. 2015, 7, 240–258. [Google Scholar] [CrossRef]
- Fiorcari, S.; Maffei, R.; Audrito, V.; Martinelli, S.; ten Hacken, E.; Zucchini, P.; Grisendi, G.; Potenza, L.; Luppi, M.; Burger, J.A.; et al. Ibrutinib Modifies the Function of Monocyte/Macrophage Population in Chronic Lymphocytic Leukemia. Oncotarget 2016, 7, 65968–65981. [Google Scholar] [CrossRef]
- Fiorcari, S.; Maffei, R.; Atene, C.G.; Potenza, L.; Luppi, M.; Marasca, R. Nurse-Like Cells and Chronic Lymphocytic Leukemia B Cells: A Mutualistic Crosstalk inside Tissue Microenvironments. Cells 2021, 10, 217. [Google Scholar] [CrossRef]
- Bercusson, A.; Colley, T.; Shah, A.; Warris, A.; Armstrong-James, D. Ibrutinib Blocks Btk-Dependent NF-ĸB and NFAT Responses in Human Macrophages during Aspergillus Fumigatus Phagocytosis. Blood 2018, 132, 1985–1988. [Google Scholar] [CrossRef] [Green Version]
- Ren, L.; Campbell, A.; Fang, H.; Gautam, S.; Elavazhagan, S.; Fatehchand, K.; Mehta, P.; Stiff, A.; Reader, B.F.; Mo, X.; et al. Analysis of the Effects of the Bruton’s Tyrosine Kinase (Btk) Inhibitor Ibrutinib on Monocyte Fcγ Receptor (FcγR) Function. J. Biol. Chem. 2016, 291, 3043–3052. [Google Scholar] [CrossRef] [Green Version]
- Fiorcari, S.; Maffei, R.; Vallerini, D.; Scarfò, L.; Barozzi, P.; Maccaferri, M.; Potenza, L.; Ghia, P.; Luppi, M.; Marasca, R. BTK Inhibition Impairs the Innate Response Against Fungal Infection in Patients With Chronic Lymphocytic Leukemia. Front. Immunol. 2020, 11, 2158. [Google Scholar] [CrossRef]
- Cox, D.; Kerrigan, S.W.; Watson, S.P. Platelets and the Innate Immune System: Mechanisms of Bacterial-Induced Platelet Activation. J. Thromb. Haemost. 2011, 9, 1097–1107. [Google Scholar] [CrossRef] [PubMed]
- Lam, F.W.; Vijayan, K.V.; Rumbaut, R.E. Platelets and Their Interactions with Other Immune Cells. Compr. Physiol. 2015, 5, 1265–1280. [Google Scholar] [CrossRef] [Green Version]
- Dib, P.R.B.; Quirino-Teixeira, A.C.; Merij, L.B.; Pinheiro, M.B.M.; Rozini, S.V.; Andrade, F.B.; Hottz, E.D. Innate Immune Receptors in Platelets and Platelet-Leukocyte Interactions. J. Leukoc. Biol. 2020, 108, 1157–1182. [Google Scholar] [CrossRef]
- Gros, A.; Ollivier, V.; Ho-Tin-Noé, B. Platelets in Inflammation: Regulation of Leukocyte Activities and Vascular Repair. Front. Immunol. 2015, 5, 678. [Google Scholar] [CrossRef] [PubMed]
- Jenne, C.N.; Urrutia, R.; Kubes, P. Platelets: Bridging Hemostasis, Inflammation, and Immunity. Int. Jnl. Lab. Hem. 2013, 35, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Garraud, O. Editorial: Platelets as Immune Cells in Physiology and Immunopathology. Front. Immunol. 2015, 6, 274. [Google Scholar] [CrossRef] [Green Version]
- Stocker, T.J.; Ishikawa-Ankerhold, H.; Massberg, S.; Schulz, C. Small but Mighty: Platelets as Central Effectors of Host Defense. Thromb. Haemost. 2017, 117, 651–661. [Google Scholar] [CrossRef]
- Martinod, K.; Deppermann, C. Immunothrombosis and Thromboinflammation in Host Defense and Disease. Platelets 2021, 32, 314–324. [Google Scholar] [CrossRef]
- Perkhofer, S.; Kainzner, B.; Kehrel, B.E.; Dierich, M.P.; Nussbaumer, W.; Lass-Flörl, C. Potential Antifungal Effects of Human Platelets against Zygomycetes in Vitro. J. Infect. Dis. 2009, 200, 1176–1179. [Google Scholar] [CrossRef]
- Rødland, E.K.; Ueland, T.; Pedersen, T.M.; Halvorsen, B.; Muller, F.; Aukrust, P.; Frøland, S.S. Activation of Platelets by Aspergillus Fumigatus and Potential Role of Platelets in the Immunopathogenesis of Aspergillosis. Infect. Immun. 2010, 78, 1269–1275. [Google Scholar] [CrossRef] [Green Version]
- Perkhofer, S.; Kehrel, B.E.; Dierich, M.P.; Donnelly, J.P.; Nussbaumer, W.; Hofmann, J.; von Eiff, C.; Lass-Flörl, C. Human Platelets Attenuate Aspergillus Species via Granule-Dependent Mechanisms. J. Infect. Dis. 2008, 198, 1243–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perkhofer, S.; Striessnig, B.; Sartori, B.; Hausott, B.; Ott, H.W.; Lass-Flörl, C. Interaction of Platelets and Anidulafungin against Aspergillus Fumigatus. Antimicrob. Agents Chemother. 2013, 57, 626–628. [Google Scholar] [CrossRef] [Green Version]
- Speth, C.; Löffler, J.; Krappmann, S.; Lass-Flörl, C.; Rambach, G. Platelets as Immune Cells in Infectious Diseases. Future Microbiol. 2013, 8, 1431–1451. [Google Scholar] [CrossRef] [PubMed]
- Bruns, S.; Kniemeyer, O.; Hasenberg, M.; Aimanianda, V.; Nietzsche, S.; Thywissen, A.; Jeron, A.; Latgé, J.-P.; Brakhage, A.A.; Gunzer, M. Production of Extracellular Traps against Aspergillus Fumigatus in Vitro and in Infected Lung Tissue Is Dependent on Invading Neutrophils and Influenced by Hydrophobin RodA. PLoS Pathog. 2010, 6, e1000873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCormick, A.; Heesemann, L.; Wagener, J.; Marcos, V.; Hartl, D.; Loeffler, J.; Heesemann, J.; Ebel, F. NETs Formed by Human Neutrophils Inhibit Growth of the Pathogenic Mold Aspergillus Fumigatus. Microbes Infect. 2010, 12, 928–936. [Google Scholar] [CrossRef] [PubMed]
- Fréalle, E.; Gosset, P.; Leroy, S.; Delattre, C.; Wacrenier, A.; Zenzmaier, C.; Zawadzki, C.; Aliouat, E.M.; Perkhofer, S. In Vitro Coagulation Triggers Anti-Aspergillus Fumigatus Neutrophil Response. Future Microbiol. 2018, 13, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Chang, F.Y.; Singh, N.; Gayowski, T.; Wagener, M.M.; Mietzner, S.M.; Stout, J.E.; Marino, I.R. Thrombocytopenia in Liver Transplant Recipients: Predictors, Impact on Fungal Infections, and Role of Endogenous Thrombopoietin. Transplantation 2000, 69, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Nouér, S.A.; Nucci, M.; Kumar, N.S.; Grazziutti, M.; Restrepo, A.; Anaissie, E. Baseline Platelet Count and Creatinine Clearance Rate Predict the Outcome of Neutropenia-Related Invasive Aspergillosis. Clin. Infect. Dis. 2012, 54, e173–e183. [Google Scholar] [CrossRef] [PubMed]
- Series, J.; Garcia, C.; Levade, M.; Viaud, J.; Sié, P.; Ysebaert, L.; Payrastre, B. Differences and Similarities in the Effects of Ibrutinib and Acalabrutinib on Platelet Functions. Haematologica 2019, 104, 2292–2299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Series, J.; Ribes, A.; Garcia, C.; Souleyreau, P.; Bauters, A.; Morschhauser, F.; Jürgensmeier, J.M.; Sié, P.; Ysebaert, L.; Payrastre, B. Effects of Novel Btk and Syk Inhibitors on Platelet Functions Alone and in Combination in Vitro and in Vivo. J. Thromb. Haemost. 2020, 18, 3336–3351. [Google Scholar] [CrossRef] [PubMed]
- Colado, A.; Marín Franco, J.L.; Elías, E.E.; Amondarain, M.; Vergara Rubio, M.; Sarapura Martínez, V.; Cordini, G.; Fuentes, F.; Balboa, L.; Fernandez Grecco, H.; et al. Second Generation BTK Inhibitors Impair the Anti-Fungal Response of Macrophages and Neutrophils. Am. J. Hematol. 2020, 95, E174–E178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estupiñán, H.Y.; Berglöf, A.; Zain, R.; Smith, C.I.E. Comparative Analysis of BTK Inhibitors and Mechanisms Underlying Adverse Effects. Front. Cell Dev. Biol. 2021, 9, 630942. [Google Scholar] [CrossRef] [PubMed]
- Palma, M.; Mulder, T.A.; Österborg, A. BTK Inhibitors in Chronic Lymphocytic Leukemia: Biological Activity and Immune Effects. Front. Immunol. 2021, 12, 686768. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, J.P.; Chen, S.C.; Kauffman, C.A.; Steinbach, W.J.; Baddley, J.W.; Verweij, P.E.; Clancy, C.J.; Wingard, J.R.; Lockhart, S.R.; Groll, A.H.; et al. Revision and Update of the Consensus Definitions of Invasive Fungal Disease From the European Organization for Research and Treatment of Cancer and the Mycoses Study Group Education and Research Consortium. Clin. Infect. Dis. 2020, 71, 1367–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speth, C.; Rambach, G.; Lass-Flörl, C. Platelet Immunology in Fungal Infections. Thromb. Haemost. 2014, 112, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Von Hundelshausen, P.; Siess, W. Bleeding by Bruton Tyrosine Kinase-Inhibitors: Dependency on Drug Type and Disease. Cancers 2021, 13, 1103. [Google Scholar] [CrossRef] [PubMed]
- Naylor-Adamson, L.; Chacko, A.R.; Booth, Z.; Caserta, S.; Jarvis, J.; Khan, S.; Hart, S.P.; Rivero, F.; Allsup, D.J.; Arman, M. Bruton’s Tyrosine Kinase Inhibitors Impair FcγRIIA-Driven Platelet Responses to Bacteria in Chronic Lymphocytic Leukemia. Front. Immunol. 2021, 12, 766272. [Google Scholar] [CrossRef]
- Zhao, D.; Qiu, G.; Luo, Z.; Zhang, Y. Platelet Parameters and (1,3)-β-D-Glucan as a Diagnostic and Prognostic Marker of Invasive Fungal Disease in Preterm Infants. PLoS ONE 2015, 10, e0123907. [Google Scholar] [CrossRef] [Green Version]
- Izzi, B.; Gialluisi, A.; Gianfagna, F.; Orlandi, S.; De Curtis, A.; Magnacca, S.; Costanzo, S.; Di Castelnuovo, A.; Donati, M.B.; de Gaetano, G.; et al. Platelet Distribution Width Is Associated with P-Selectin Dependent Platelet Function: Results from the Moli-Family Cohort Study. Cells 2021, 10, 2737. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nasillo, V.; Lagreca, I.; Vallerini, D.; Barozzi, P.; Riva, G.; Maccaferri, M.; Paolini, A.; Forghieri, F.; Fiorcari, S.; Maffei, R.; et al. BTK Inhibitors Impair Platelet-Mediated Antifungal Activity. Cells 2022, 11, 1003. https://doi.org/10.3390/cells11061003
Nasillo V, Lagreca I, Vallerini D, Barozzi P, Riva G, Maccaferri M, Paolini A, Forghieri F, Fiorcari S, Maffei R, et al. BTK Inhibitors Impair Platelet-Mediated Antifungal Activity. Cells. 2022; 11(6):1003. https://doi.org/10.3390/cells11061003
Chicago/Turabian StyleNasillo, Vincenzo, Ivana Lagreca, Daniela Vallerini, Patrizia Barozzi, Giovanni Riva, Monica Maccaferri, Ambra Paolini, Fabio Forghieri, Stefania Fiorcari, Rossana Maffei, and et al. 2022. "BTK Inhibitors Impair Platelet-Mediated Antifungal Activity" Cells 11, no. 6: 1003. https://doi.org/10.3390/cells11061003