
Modulation of Intestinal Epithelial Permeability via Protease-Activated Receptor-2-Induced Autophagy

 ,
,
Abstract
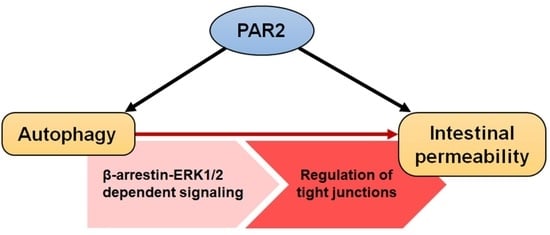
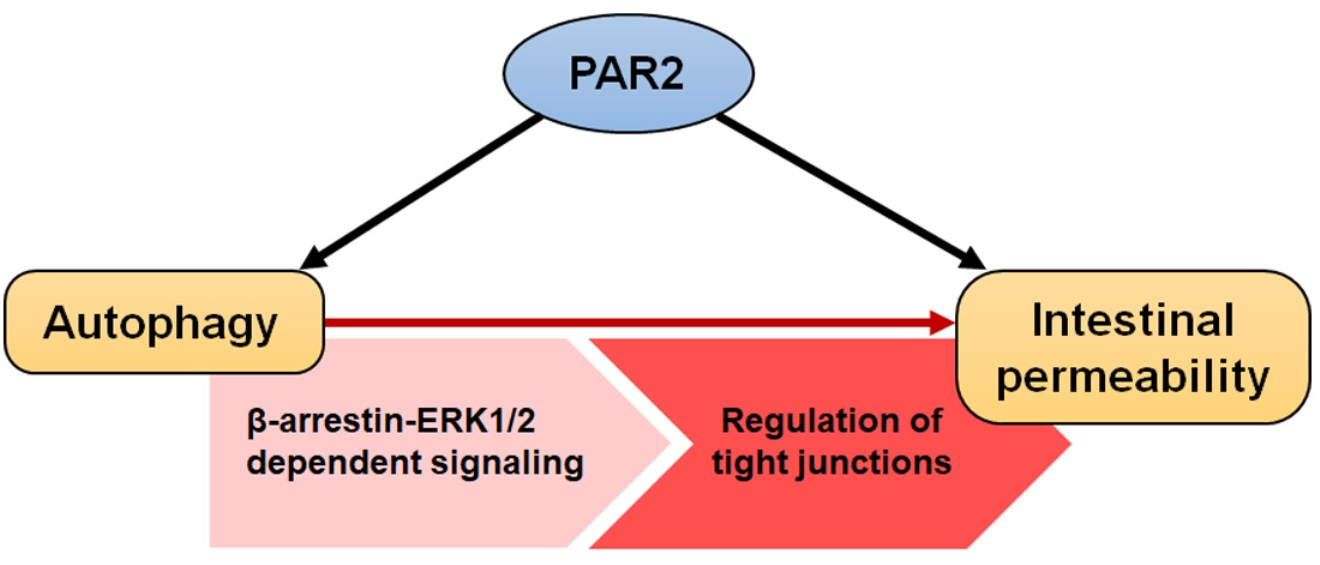
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatment
2.2. Transfection
2.3. Reverse-Transcription Polymerase Chain Reaction (RT-PCR)
2.4. Western Blot Analysis
2.5. Immunoprecipitation (IP) Assay
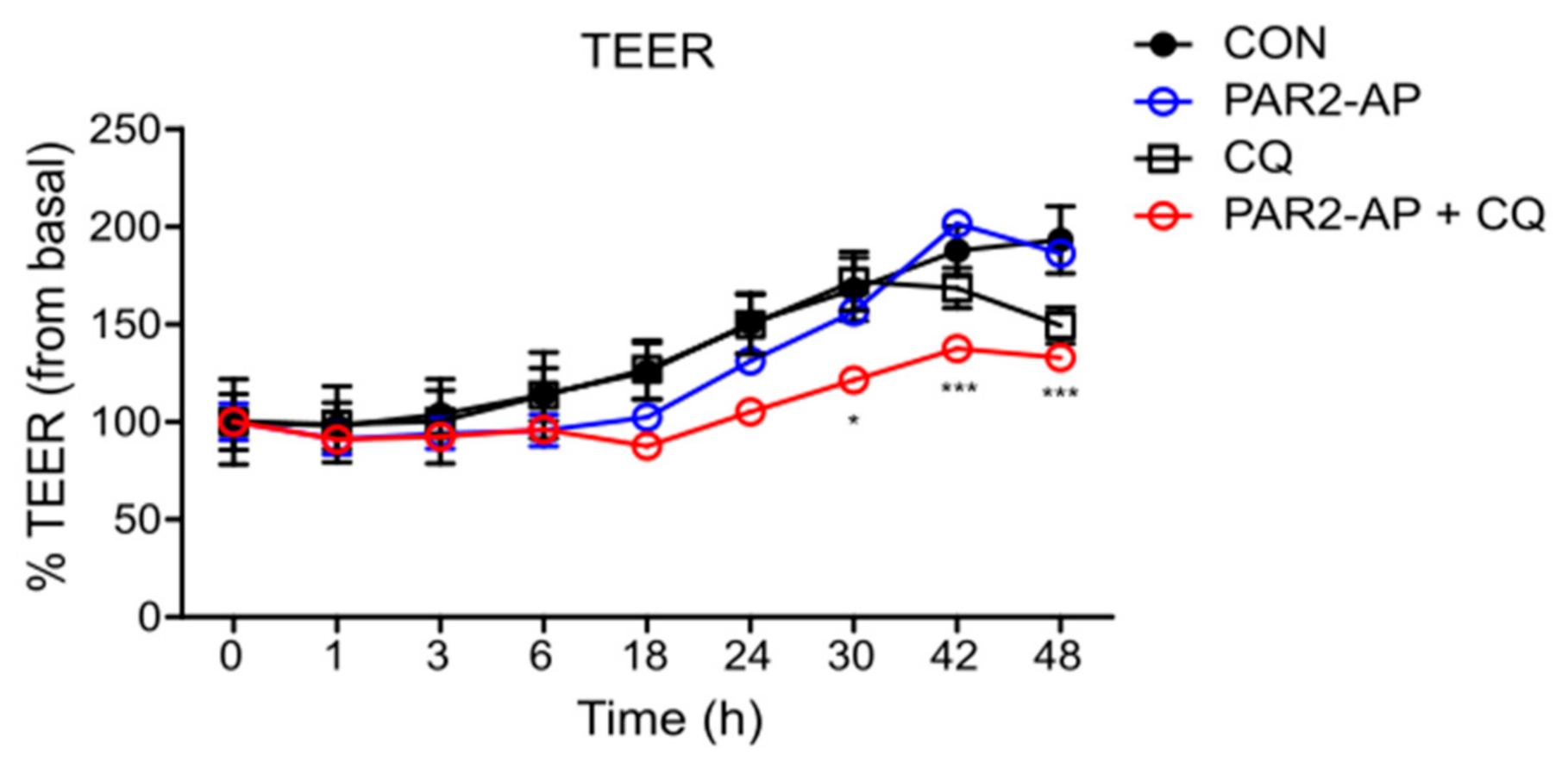
2.6. Transepithelial Electrical Resistance (TEER) Measurement
3. Results
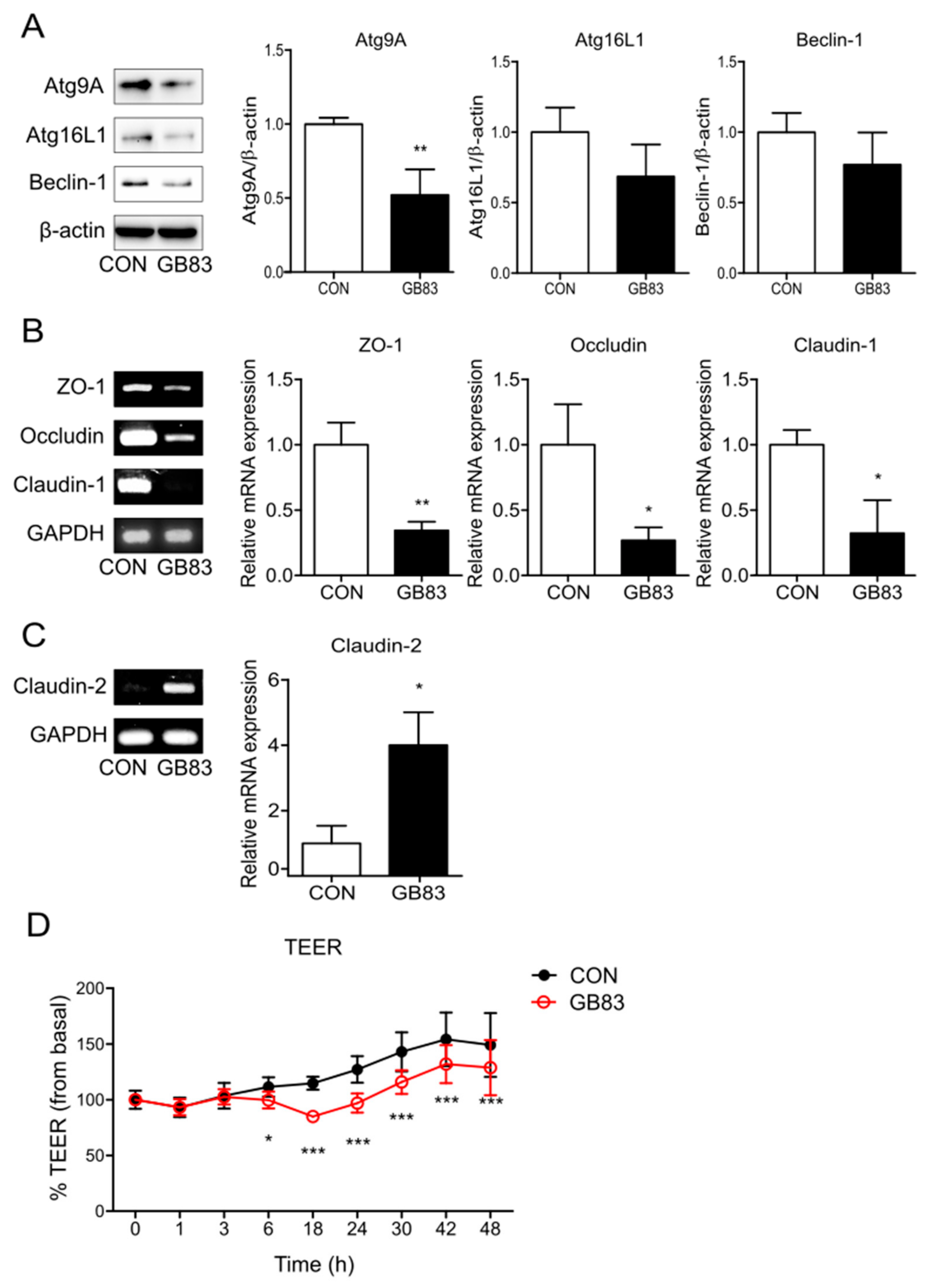
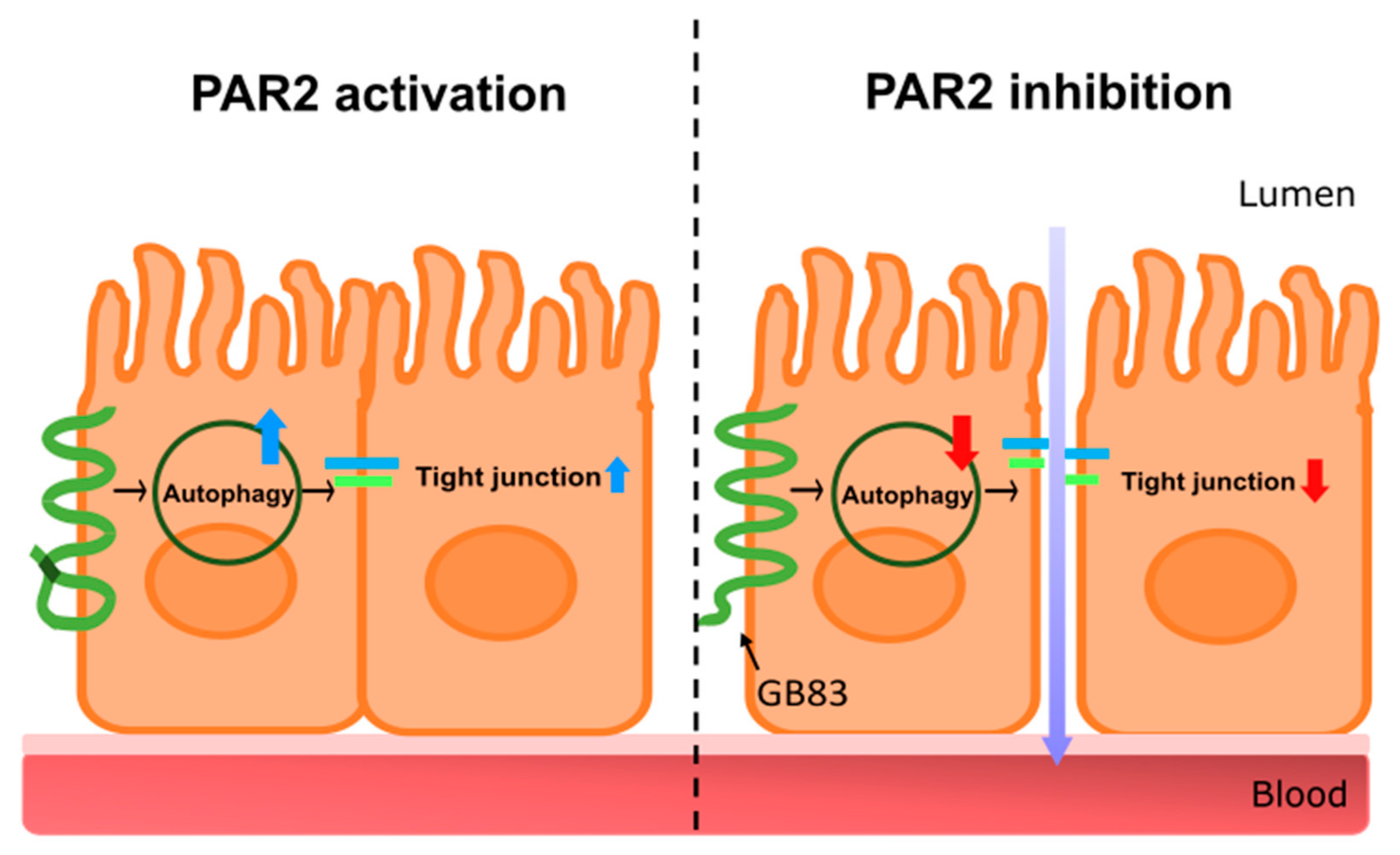
3.1. PAR2 Inhibition Downregulated Autophagy and Reduced Intestinal Barrier Function in Caco-2 Cells
3.2. PAR2 Prolonged ERK1/2 Phosphorylation through the Assembly of the PAR2-ERK1/2-β-Arrestin Complex
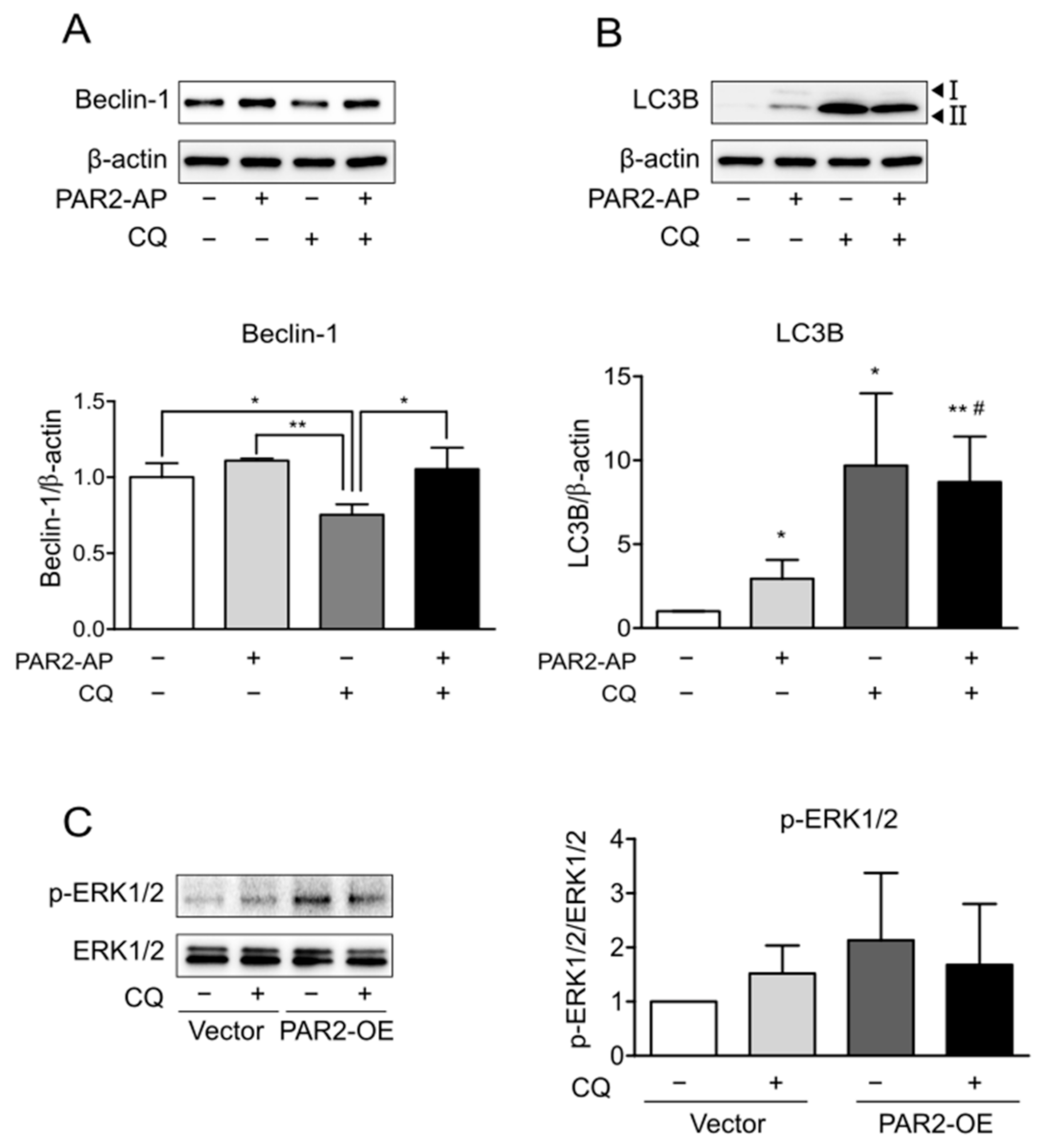
3.3. Enhanced Autophagy Signaling by PAR2 Was Related to ERK1/2 Phosphorylation
3.4. PAR2 Regulated Epithelial Permeability by Autophagy
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, P.; Metcalf, M.; Bunnett, N.W. Biased signaling of protease-activated receptors. Front. Endocrinol. 2014, 5, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Randall-Demllo, S.; Chieppa, M.; Eri, R. Intestinal epithelium and autophagy: Partners in gut homeostasis. Front. Immunol. 2013, 4, 301. [Google Scholar] [CrossRef] [Green Version]
- El-Khider, F.; McDonald, C. Links of Autophagy Dysfunction to Inflammatory Bowel Disease Onset. Dig. Dis. 2016, 34, 27–34. [Google Scholar] [CrossRef] [Green Version]
- Haq, S.; Grondin, J.; Banskota, S.; Khan, W.I. Autophagy: Roles in intestinal mucosal homeostasis and inflammation. J. Biomed. Sci. 2019, 26, 19. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.; Moon, K.M.; Kim, C.Y. Tight Junction in the Intestinal Epithelium: Its Association with Diseases and Regulation by Phytochemicals. J. Immunol. Res. 2018, 2018, 2645465. [Google Scholar] [CrossRef] [Green Version]
- Ulluwishewa, D.; Anderson, R.C.; McNabb, W.C.; Moughan, P.J.; Wells, J.M.; Roy, N.C. Regulation of tight junction permeability by intestinal bacteria and dietary components. J. Nutr. 2011, 141, 769–776. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T. Regulation of the intestinal barrier by nutrients: The role of tight junctions. Anim. Sci. J. 2020, 91, e13357. [Google Scholar] [CrossRef] [Green Version]
- Bischoff, S.C.; Barbara, G.; Buurman, W.; Ockhuizen, T.; Schulzke, J.D.; Serino, M.; Tilg, H.; Watson, A.; Wells, J.M. Intestinal permeability—A new target for disease prevention and therapy. BMC Gastroenterol. 2014, 14, 189. [Google Scholar] [CrossRef] [Green Version]
- Bhat, A.A.; Uppada, S.; Achkar, I.W.; Hashem, S.; Yadav, S.K.; Shanmugakonar, M.; Al-Naemi, H.A.; Haris, M.; Uddin, S. Tight Junction Proteins and Signaling Pathways in Cancer and Inflammation: A Functional Crosstalk. Front. Physiol. 2019, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Günzel, D.; Yu, A.S. Claudins and the modulation of tight junction permeability. Physiol. Rev. 2013, 93, 525–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saitou, M.; Fujimoto, K.; Doi, Y.; Itoh, M.; Fujimoto, T.; Furuse, M.; Takano, H.; Noda, T.; Tsukita, S. Occludin-deficient embryonic stem cells can differentiate into polarized epithelial cells bearing tight junctions. J. Cell Biol. 1998, 141, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Chelakkot, C.; Ghim, J.; Ryu, S.H. Mechanisms regulating intestinal barrier integrity and its pathological implications. Exp. Mol. Med. 2018, 50, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Fukui, H. Increased Intestinal Permeability and Decreased Barrier Function: Does It Really Influence the Risk of Inflammation? Inflamm. Intest. Dis. 2016, 1, 135–145. [Google Scholar] [CrossRef]
- Clayburgh, D.R.; Barrett, T.A.; Tang, Y.; Meddings, J.B.; Van Eldik, L.J.; Watterson, D.M.; Clarke, L.L.; Mrsny, R.J.; Turner, J.R. Epithelial myosin light chain kinase-dependent barrier dysfunction mediates T cell activation-induced diarrhea in vivo. J. Clin. Investig. 2005, 115, 2702–2715. [Google Scholar] [CrossRef]
- Groschwitz, K.R.; Hogan, S.P. Intestinal barrier function: Molecular regulation and disease pathogenesis. J. Allergy Clin. Immunol. 2009, 124, 3–20. [Google Scholar] [CrossRef] [Green Version]
- Coughlin, S.R. Protease-activated receptors start a family. Proc. Natl. Acad. Sci. USA 1994, 91, 9200–9202. [Google Scholar] [CrossRef] [Green Version]
- Vergnolle, N. Clinical relevance of proteinase activated receptors (pars) in the gut. Gut 2005, 54, 867–874. [Google Scholar] [CrossRef] [Green Version]
- Sebert, M.; Sola-Tapias, N.; Mas, E.; Barreau, F.; Ferrand, A. Protease-Activated Receptors in the Intestine: Focus on Inflammation and Cancer. Front. Endocrinol. 2019, 10, 717. [Google Scholar] [CrossRef]
- Lau, C.; Lytle, C.; Straus, D.S.; DeFea, K.A. Apical and basolateral pools of proteinase-activated receptor-2 direct distinct signaling events in the intestinal epithelium. Am. J. Physiol. Cell Physiol. 2011, 300, C113–C123. [Google Scholar] [CrossRef] [PubMed]
- Rothmeier, A.S.; Ruf, W. Protease-activated receptor 2 signaling in inflammation. Semin. Immunopathol. 2012, 34, 133–149. [Google Scholar] [CrossRef] [PubMed]
- DeFea, K.A.; Zalevsky, J.; Thoma, M.S.; Déry, O.; Mullins, R.D.; Bunnett, N.W. Beta-arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2. J. Cell Biol. 2000, 148, 1267–1281. [Google Scholar] [CrossRef] [PubMed]
- Her, J.Y.; Lee, Y.; Kim, S.J.; Heo, G.; Choo, J.; Kim, Y.; Howe, C.; Rhee, S.H.; Yu, H.S.; Chung, H.Y.; et al. Blockage of protease-activated receptor 2 exacerbates inflammation in high-fat environment partly through autophagy inhibition. Am. J. Physiol. Gastrointest. Liver Physiol. 2021, 320, G30–G42. [Google Scholar] [CrossRef]
- Jacob, C.; Yang, P.C.; Darmoul, D.; Amadesi, S.; Saito, T.; Cottrell, G.S.; Coelho, A.M.; Singh, P.; Grady, E.F.; Perdue, M.; et al. Mast cell tryptase controls paracellular permeability of the intestine. Role of protease-activated receptor 2 and beta-arrestins. J. Biol. Chem. 2005, 280, 31936–31948. [Google Scholar] [CrossRef] [Green Version]
- Enjoji, S.; Ohama, T.; Sato, K. Regulation of epithelial cell tight junctions by protease-activated receptor 2. J. Vet. Med. Sci. 2014, 76, 1225–1229. [Google Scholar] [CrossRef] [Green Version]
- Nighot, P.K.; Hu, C.A.; Ma, T.Y. Autophagy enhances intestinal epithelial tight junction barrier function by targeting claudin-2 protein degradation. J. Biol. Chem. 2015, 290, 7234–7246. [Google Scholar] [CrossRef] [Green Version]
- Bueno, L.; Fioramonti, J. Protease-activated receptor 2 and gut permeability: A review. Neurogastroenterol. Motil. 2008, 20, 580–587. [Google Scholar] [CrossRef]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Kawabata, A.; Matsunami, M.; Sekiguchi, F. Gastrointestinal roles for proteinase-activated receptors in health and disease. Br. J. Pharmacol. 2008, 153 (Suppl. 1), S230–S240. [Google Scholar] [CrossRef] [Green Version]
- Luettig, J.; Rosenthal, R.; Barmeyer, C.; Schulzke, J.D. Claudin-2 as a mediator of leaky gut barrier during intestinal inflammation. Tissue Barriers 2015, 3, e977176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.; Wang, S.; Zhou, C.; He, M.; Wang, J.; Ladds, M.; Lianoudaki, D.; Sedimbi, S.K.; Lane, D.P.; Westerberg, L.S.; et al. CD36 and LC3B initiated autophagy in B cells regulates the humoral immune response. Autophagy 2021, 17, 3577–3591. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wang, S.; Yu, L.; Mueller, J.; Fortunato, F.; Rausch, V.; Mueller, S. H2O2-mediated autophagy during ethanol metabolism. Redox Biol. 2021, 46, 102081. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Chung, S.W. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis. 2019, 10, 822. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.; Lee, Y.; Heo, G.; Jeong, S.; Park, S.; Yoo, J.-W.; Jung, Y.; Im, E. Modulation of Intestinal Epithelial Permeability via Protease-Activated Receptor-2-Induced Autophagy. Cells 2022, 11, 878. https://doi.org/10.3390/cells11050878
Kim Y, Lee Y, Heo G, Jeong S, Park S, Yoo J-W, Jung Y, Im E. Modulation of Intestinal Epithelial Permeability via Protease-Activated Receptor-2-Induced Autophagy. Cells. 2022; 11(5):878. https://doi.org/10.3390/cells11050878
Chicago/Turabian StyleKim, Yuju, Yunna Lee, Gwangbeom Heo, Sihyun Jeong, Soyeong Park, Jin-Wook Yoo, Yunjin Jung, and Eunok Im. 2022. "Modulation of Intestinal Epithelial Permeability via Protease-Activated Receptor-2-Induced Autophagy" Cells 11, no. 5: 878. https://doi.org/10.3390/cells11050878