
Direct Interaction of ATP7B and LC3B Proteins Suggests a Cooperative Role of Copper Transportation and Autophagy

 , , ,
, , ,  and
and
Abstract
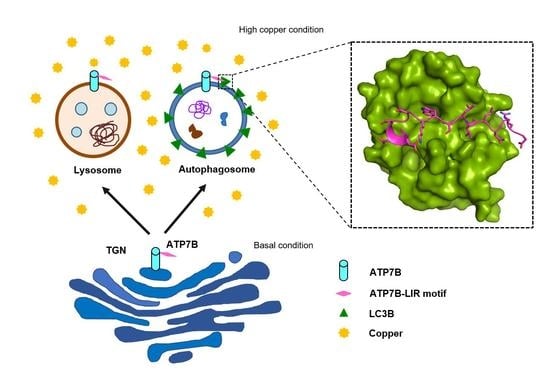
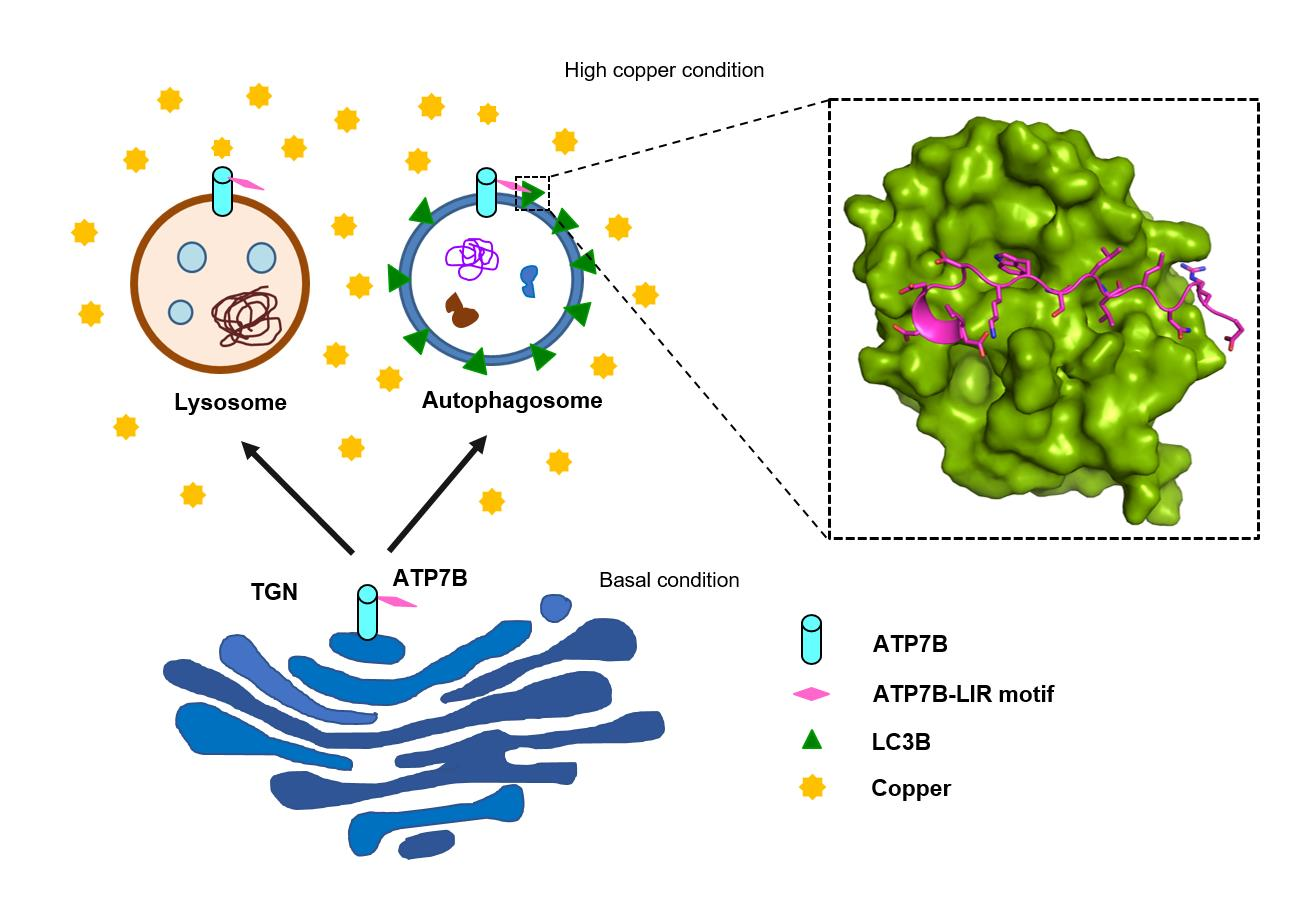
:
{kind=link}
{kind=link}
{kind=link}
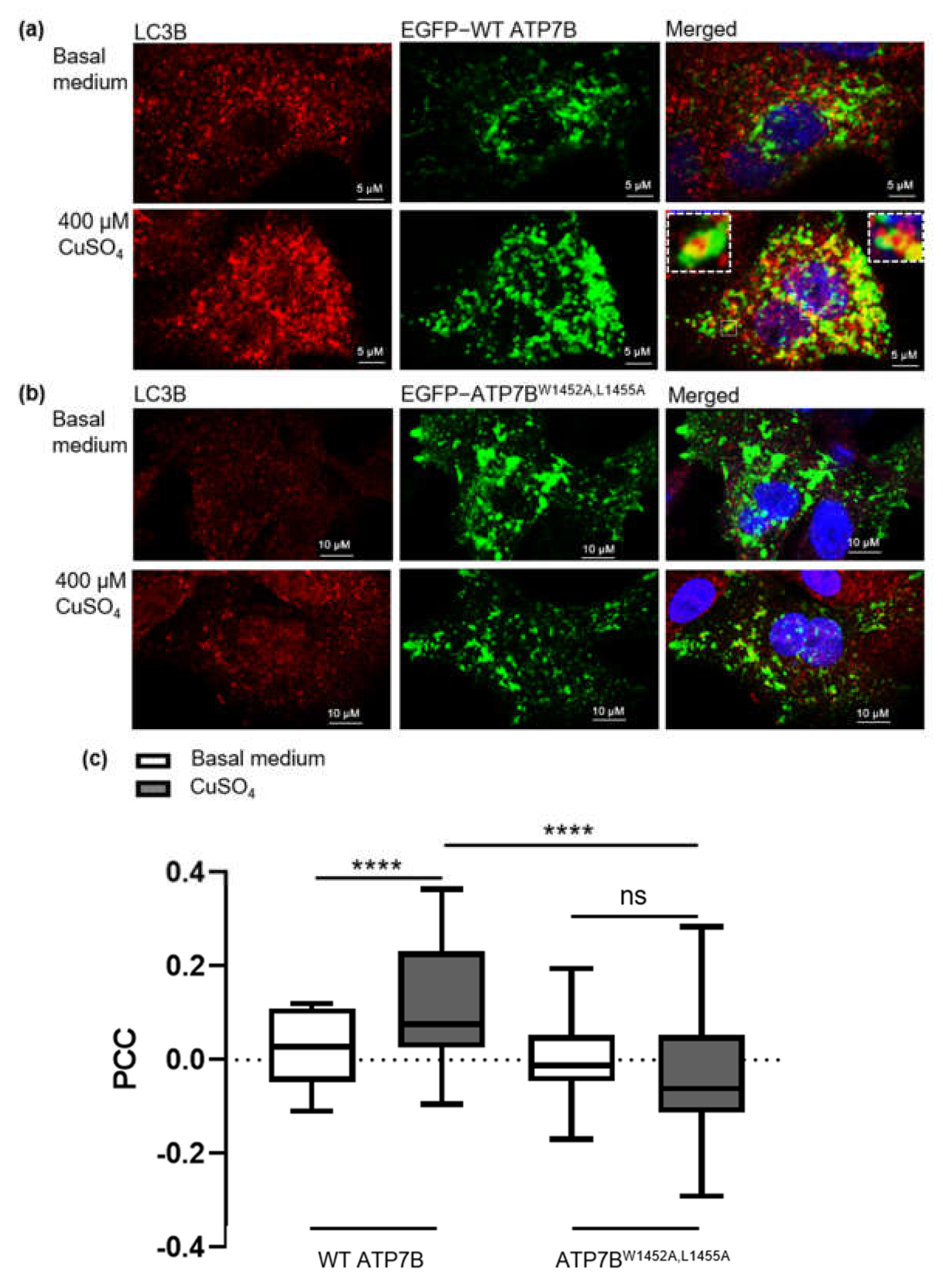{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines, Antibodies and Subcellular Markers
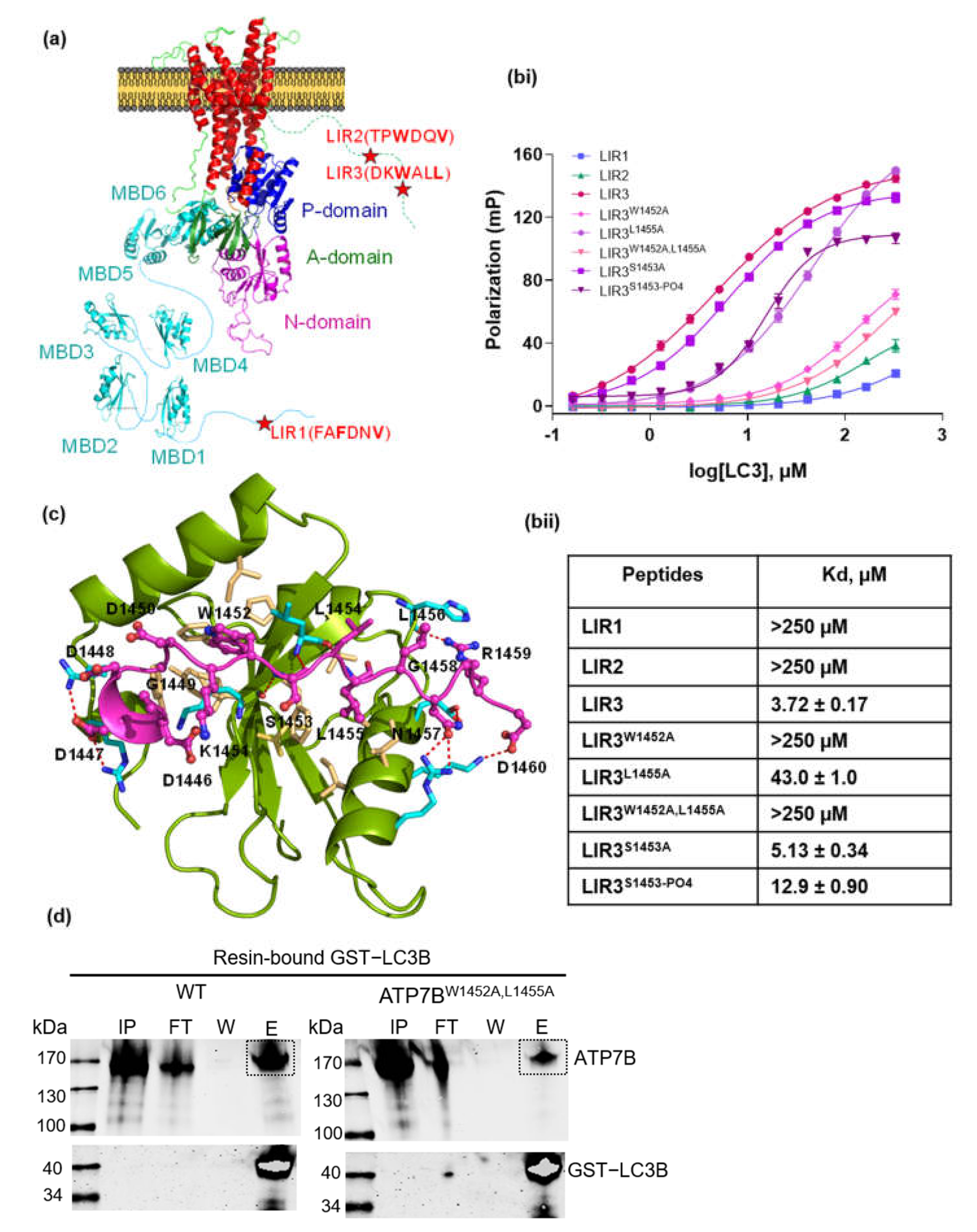
2.2. Identification of Canonical LIR Motifs on ATP7B
2.3. Peptide Synthesis
2.4. Cloning and Purification of LC3B
2.5. Fluorescent Polarization
2.6. Pull-Down Experiment with GST Affinity Tag
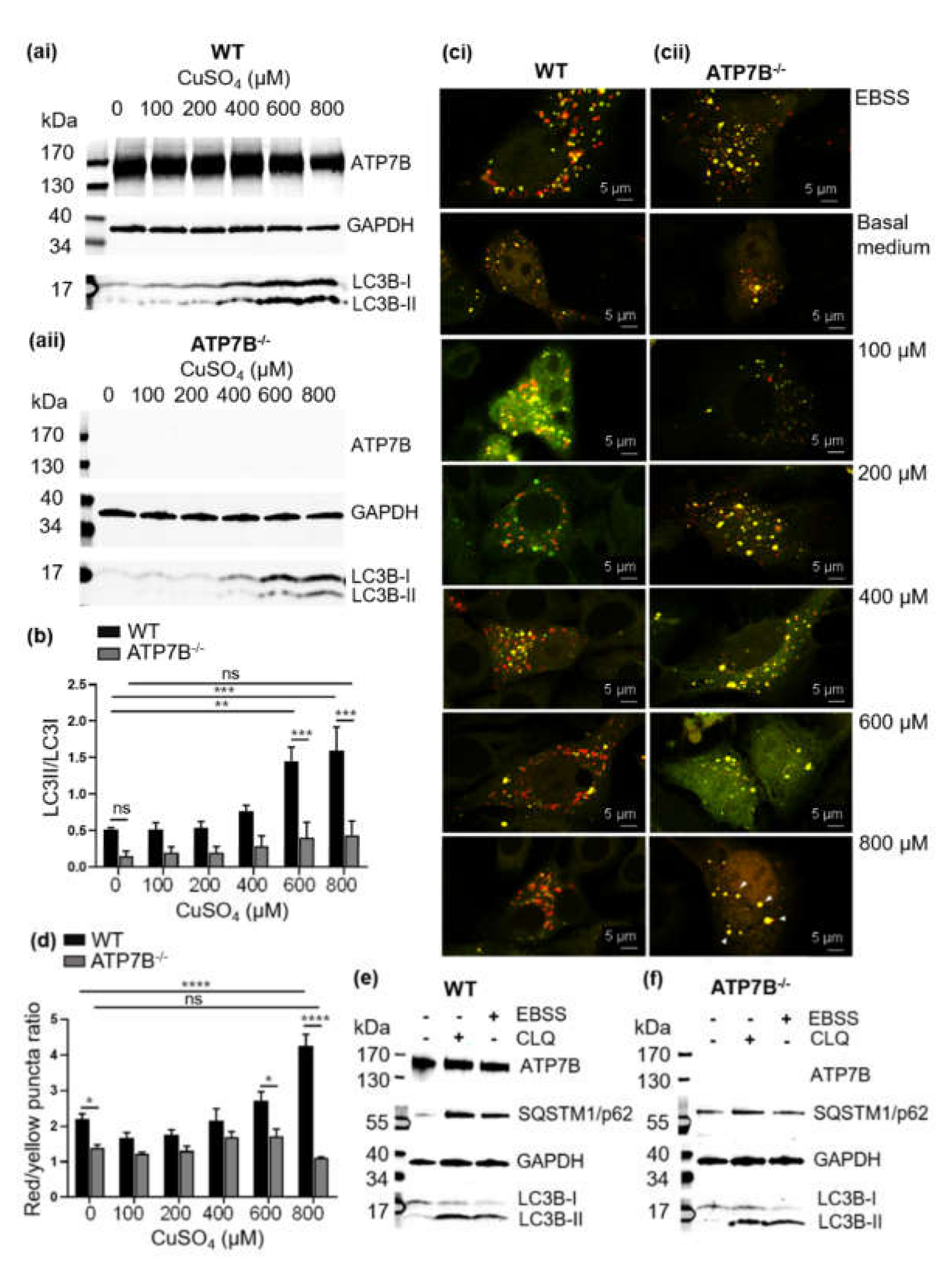
2.7. Autophagy Detection
2.8. ATP7B Complementation of HepG2 ATPB−/− Cells
2.9. Colocalization Experiments
2.10. Statistical Analysis
3. Results
3.1. Investigation of LC3-ATP7B Interaction
3.2. Copper Induces Autophagy
3.3. Impairment of Copper-Induced Autophagy in HepG2 Cells Deficient of the ATP7B Copper Transporter (HepG2 ATP7B−/−)
3.4. Copper-Dependent Cellular Trafficking of ATP7B in HepG2 Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Camakaris, J.; Voskoboinik, I.; Mercer, J.F. Molecular mechanisms of copper homeostasis. Biochem. Biophys. Res. Commun. 1999, 261, 225–232. [Google Scholar] [CrossRef]
- Lutsenko, S. Human copper homeostasis: A network of interconnected pathways. Curr. Opin. Chem. Biol. 2010, 14, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Blaby-Haas, C.E.; Merchant, S.S. Lysosome-related organelles as mediators of metal homeostasis. J. Biol. Chem. 2014, 289, 28129–28136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polishchuk, E.V.; Concilli, M.; Iacobacci, S.; Chesi, G.; Pastore, N.; Piccolo, P.; Paladino, S.; Baldantoni, D.; van IJzendoorn, S.C.D.; Chan, J.; et al. Wilson disease protein ATP7B utilizes lysosomal exocytosis to maintain copper homeostasis. Dev. Cell 2014, 29, 686–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, A.; Lutsenko, S. Human copper transporters: Mechanism, role in human diseases and therapeutic potential. Future Med. Chem. 2009, 1, 1125–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartee, M.Y.; Lutsenko, S. Hepatic copper-transporting ATPase ATP7B: Function and inactivation at the molecular and cellular level. Biometals 2007, 20, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Hellman, N.E.; Kono, S.; Mancini, G.M.; Hoogeboom, A.J.; de Jong, G.J.; Gitlin, J.D. Mechanisms of copper incorporation into human ceruloplasmin. J. Biol. Chem. 2002, 277, 46632–46638. [Google Scholar] [CrossRef] [Green Version]
- Masaldan, S.; Clatworthy, S.A.S.; Gamell, C.; Smith, Z.M.; Francis, P.S.; Denoyer, D.; Meggyesy, P.M.; La Fontaine, S.; Cater, M.A. Copper accumulation in senescent cells: Interplay between copper transporters and impaired autophagy. Redox Biol. 2018, 16, 322–331. [Google Scholar] [CrossRef]
- Polishchuk, E.V.; Merolla, A.; Lichtmannegger, J.; Romano, A.; Indrieri, A.; Ilyechova, E.Y.; Concilli, M.; de Cegli, R.; Crispino, R.; Mariniello, M.; et al. Activation of Autophagy, Observed in Liver Tissues from Patients with Wilson Disease and from ATP7B-Deficient Animals, Protects Hepatocytes from Copper-Induced Apoptosis. Gastroenterology 2019, 156, 1173–1189.e5. [Google Scholar] [CrossRef] [Green Version]
- Stolz, A.; Ernst, A.; Dikic, I. Cargo recognition and trafficking in selective autophagy. Nat. Cell Biol. 2014, 16, 495–501. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [Green Version]
- Lamb, C.A.; Yoshimori, T.; Tooze, S.A. The autophagosome: Origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 759–774. [Google Scholar] [CrossRef]
- Xie, Z.; Nair, U.; Klionsky, D.J. Dissecting autophagosome formation: The missing pieces. Autophagy 2008, 4, 920–922. [Google Scholar] [CrossRef] [Green Version]
- Sou, Y.-s.; Waguri, S.; Iwata, J.-i.; Ueno, T.; Fujimura, T.; Hara, T.; Sawada, N.; Yamada, A.; Mizushima, N.; Uchiyama, Y.; et al. The Atg8 conjugation system is indispensable for proper development of autophagic isolation membranes in mice. Mol. Biol. Cell 2008, 19, 4762–4775. [Google Scholar] [CrossRef] [Green Version]
- Kirisako, T.; Ichimura, Y.; Okada, H.; Kabeya, Y.; Mizushima, N.; Yoshimori, T.; Ohsumi, M.; Takao, T.; Noda, T.; Ohsumi, Y. The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J. Cell Biol. 2000, 151, 263–276. [Google Scholar] [CrossRef]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.-A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [Green Version]
- Kirkin, V.; McEwan, D.G.; Novak, I.; Dikic, I. A role for ubiquitin in selective autophagy. Mol. Cell 2009, 34, 259–269. [Google Scholar] [CrossRef]
- Zheng, Y.T.; Shahnazari, S.; Brech, A.; Lamark, T.; Johansen, T.; Brumell, J.H. The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J. Immunol. 2009, 183, 5909–5916. [Google Scholar] [CrossRef] [Green Version]
- Von Muhlinen, N.; Akutsu, M.; Ravenhill, B.J.; Foeglein, Á.; Bloor, S.; Rutherford, T.J.; Freund, S.M.V.; Komander, D.; Randow, F. LC3C, bound selectively by a noncanonical LIR motif in NDP52, is required for antibacterial autophagy. Mol. Cell 2012, 48, 329–342. [Google Scholar] [CrossRef] [Green Version]
- Johansen, T.; Lamark, T. Selective Autophagy: ATG8 Family Proteins, LIR Motifs and Cargo Receptors. J. Mol. Biol. 2020, 432, 80–103. [Google Scholar] [CrossRef]
- Satoo, K.; Noda, N.N.; Kumeta, H.; Fujioka, Y.; Mizushima, N.; Ohsumi, Y.; Inagaki, F. The structure of Atg4B-LC3 complex reveals the mechanism of LC3 processing and delipidation during autophagy. EMBO J. 2009, 28, 1341–1350. [Google Scholar] [CrossRef]
- Alemu, E.A.; Lamark, T.; Torgersen, K.M.; Birgisdottir, A.B.; Larsen, K.B.; Jain, A.; Olsvik, H.; Øvervatn, A.; Kirkin, V.; Johansen, T. ATG8 family proteins act as scaffolds for assembly of the ULK complex: Sequence requirements for LC3-interacting region (LIR) motifs. J. Biol. Chem. 2012, 287, 39275–39290. [Google Scholar] [CrossRef] [Green Version]
- Yang, A.; Pantoom, S.; Wu, Y.-W. Elucidation of the anti-autophagy mechanism of the Legionella effector RavZ using semisynthetic LC3 proteins. Elife 2017, 6, e23905. [Google Scholar] [CrossRef]
- Pantoom, S.; Yang, A.; Wu, Y.-W. Lift and cut: Anti-host autophagy mechanism of Legionella pneumophila. Autophagy 2017, 13, 1467–1469. [Google Scholar] [CrossRef] [Green Version]
- Kalvari, I.; Tsompanis, S.; Mulakkal, N.C.; Osgood, R.; Johansen, T.; Nezis, I.P.; Promponas, V.J. iLIR: A web resource for prediction of Atg8-family interacting proteins. Autophagy 2014, 10, 913–925. [Google Scholar] [CrossRef] [Green Version]
- Bohl, C.; Pomorski, A.; Seemann, S.; Knospe, A.-M.; Zheng, C.; Krężel, A.; Rolfs, A.; Lukas, J. Fluorescent probes for selective protein labeling in lysosomes: A case of α-galactosidase A. FASEB J. 2017, 31, 5258–5267. [Google Scholar] [CrossRef] [Green Version]
- Noda, N.N.; Kumeta, H.; Nakatogawa, H.; Satoo, K.; Adachi, W.; Ishii, J.; Fujioka, Y.; Ohsumi, Y.; Inagaki, F. Structural basis of target recognition by Atg8/LC3 during selective autophagy. Genes Cells 2008, 13, 1211–1218. [Google Scholar] [CrossRef]
- Ichimura, Y.; Kumanomidou, T.; Sou, Y.-s.; Mizushima, T.; Ezaki, J.; Ueno, T.; Kominami, E.; Yamane, T.; Tanaka, K.; Komatsu, M. Structural basis for sorting mechanism of p62 in selective autophagy. J. Biol. Chem. 2008, 283, 22847–22857. [Google Scholar] [CrossRef] [Green Version]
- Popelka, H.; Klionsky, D.J. Analysis of the native conformation of the LIR/AIM motif in the Atg8/LC3/GABARAP-binding proteins. Autophagy 2015, 11, 2153–2159. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Heo, L.; Lee, M.S.; Seok, C. GalaxyPepDock: A protein-peptide docking tool based on interaction similarity and energy optimization. Nucleic Acids Res. 2015, 43, W431–W435. [Google Scholar] [CrossRef] [Green Version]
- Harada, M.; Sakisaka, S.; Terada, K.; Kimura, R.; Kawaguchi, T.; Koga, H.; Taniguchi, E.; Sasatomi, K.; Miura, N.; Suganuma, T.; et al. Role of ATP7B in biliary copper excretion in a human hepatoma cell line and normal rat hepatocytes. Gastroenterology 2000, 118, 921–928. [Google Scholar] [CrossRef]
- Tan, X.; Guan, H.; Yang, Y.; Luo, S.; Hou, L.; Chen, H.; Li, J. Cu(II) disrupts autophagy-mediated lysosomal degradation of oligomeric Aβ in microglia via mTOR-TFEB pathway. Toxicol. Appl. Pharmacol. 2020, 401, 115090. [Google Scholar] [CrossRef] [PubMed]
- Frudd, K.; Burgoyne, T.; Burgoyne, J.R. Oxidation of Atg3 and Atg7 mediates inhibition of autophagy. Nat. Commun. 2018, 9, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Zhou, P.; Zhang, J.; Zhang, Y.; Zhang, G.; Liu, Y.; Cheng, X. Generation of reactive oxygen species by promoting the Cu(II)/Cu(I) redox cycle with reducing agents in aerobic aqueous solution. Water Sci. Technol. 2018, 78, 1390–1399. [Google Scholar] [CrossRef] [Green Version]
- Sahani, M.H.; Itakura, E.; Mizushima, N. Expression of the autophagy substrate SQSTM1/p62 is restored during prolonged starvation depending on transcriptional upregulation and autophagy-derived amino acids. Autophagy 2014, 10, 431–441. [Google Scholar] [CrossRef] [Green Version]
- Polishchuk, E.V.; Polishchuk, R.S. The emerging role of lysosomes in copper homeostasis. Metallomics 2016, 8, 853–862. [Google Scholar] [CrossRef]
- Polishchuk, R.S.; Polishchuk, E.V. From and to the Golgi—Defining the Wilson disease protein road map. FEBS Lett. 2019, 593, 2341–2350. [Google Scholar] [CrossRef] [Green Version]
- Nyasae, L.K.; Schell, M.J.; Hubbard, A.L. Copper directs ATP7B to the apical domain of hepatic cells via basolateral endosomes. Traffic 2014, 15, 1344–1365. [Google Scholar] [CrossRef] [Green Version]
- Braulke, T.; Bonifacino, J.S. Sorting of lysosomal proteins. Biochim. Biophys. Acta 2009, 1793, 605–614. [Google Scholar] [CrossRef] [Green Version]
- Lalioti, V.; Hernandez-Tiedra, S.; Sandoval, I.V. DKWSLLL, a versatile DXXXLL-type signal with distinct roles in the Cu(+)-regulated trafficking of ATP7B. Traffic 2014, 15, 839–860. [Google Scholar] [CrossRef]
- Jain, S.; Farías, G.G.; Bonifacino, J.S. Polarized sorting of the copper transporter ATP7B in neurons mediated by recognition of a dileucine signal by AP-1. Mol. Biol. Cell 2015, 26, 218–228. [Google Scholar] [CrossRef] [Green Version]
- Staudt, C.; Puissant, E.; Boonen, M. Subcellular Trafficking of Mammalian Lysosomal Proteins: An Extended View. Int. J. Mol. Sci. 2016, 18, 47. [Google Scholar] [CrossRef] [Green Version]
- Braiterman, L.T.; Gupta, A.; Chaerkady, R.; Cole, R.N.; Hubbard, A.L. Communication between the N and C termini is required for copper-stimulated Ser/Thr phosphorylation of Cu(I)-ATPase (ATP7B). J. Biol. Chem. 2015, 290, 8803–8819. [Google Scholar] [CrossRef] [Green Version]
- Pilankatta, R.; Lewis, D.; Adams, C.M.; Inesi, G. High yield heterologous expression of wild-type and mutant Cu+-ATPase (ATP7B, Wilson disease protein) for functional characterization of catalytic activity and serine residues undergoing copper-dependent phosphorylation. J. Biol. Chem. 2009, 284, 21307–21316. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pantoom, S.; Pomorski, A.; Huth, K.; Hund, C.; Petters, J.; Krężel, A.; Hermann, A.; Lukas, J. Direct Interaction of ATP7B and LC3B Proteins Suggests a Cooperative Role of Copper Transportation and Autophagy. Cells 2021, 10, 3118. https://doi.org/10.3390/cells10113118
Pantoom S, Pomorski A, Huth K, Hund C, Petters J, Krężel A, Hermann A, Lukas J. Direct Interaction of ATP7B and LC3B Proteins Suggests a Cooperative Role of Copper Transportation and Autophagy. Cells. 2021; 10(11):3118. https://doi.org/10.3390/cells10113118
Chicago/Turabian StylePantoom, Supansa, Adam Pomorski, Katharina Huth, Christina Hund, Janine Petters, Artur Krężel, Andreas Hermann, and Jan Lukas. 2021. "Direct Interaction of ATP7B and LC3B Proteins Suggests a Cooperative Role of Copper Transportation and Autophagy" Cells 10, no. 11: 3118. https://doi.org/10.3390/cells10113118