
Optimization of Thymidine Kinase-Based Safety Switch for Neural Cell Therapy


Abstract
:1. Introduction
2. Results
2.1. Generation of Cell Lines That Stably Express Thymidine Kinase
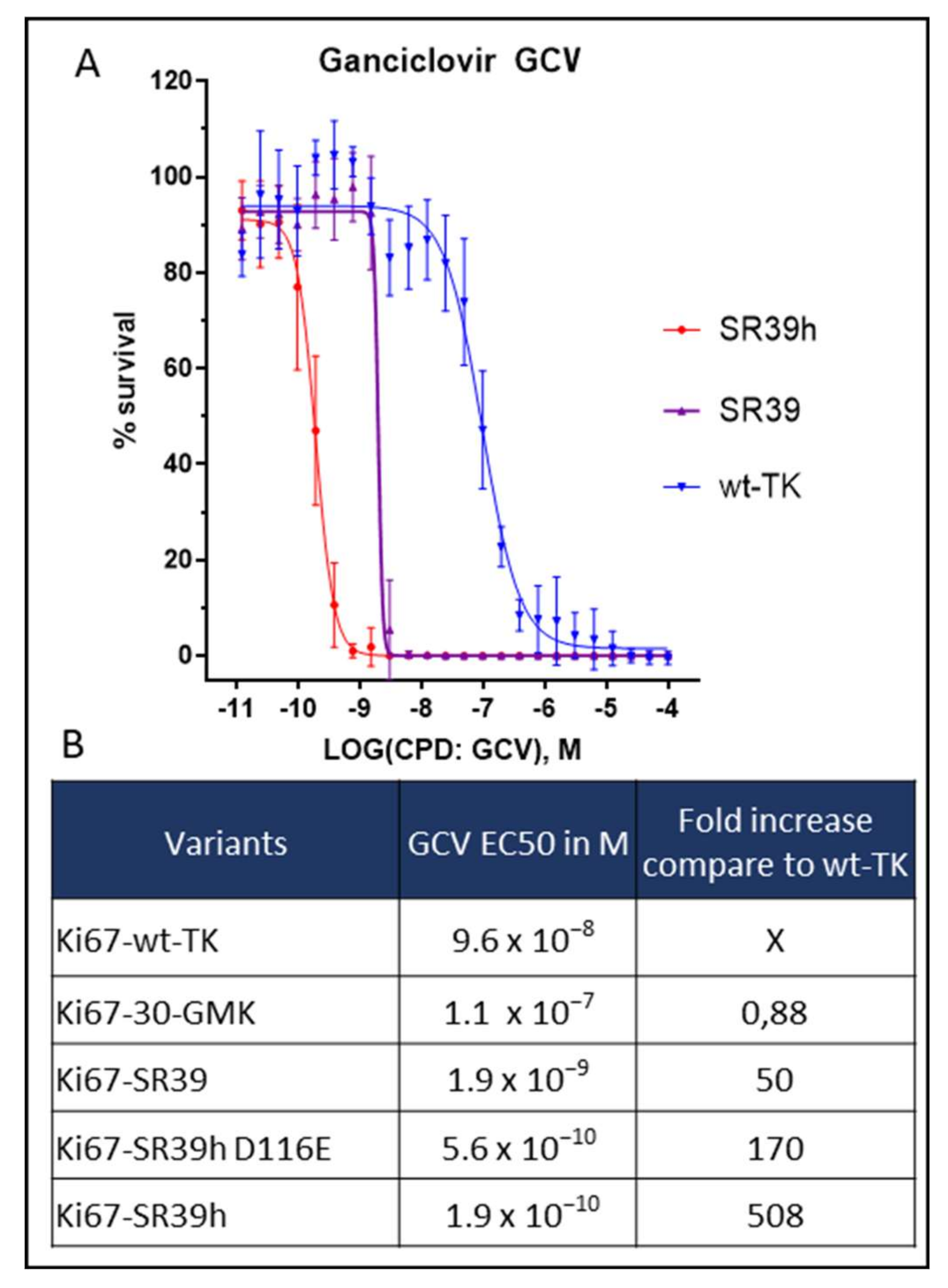
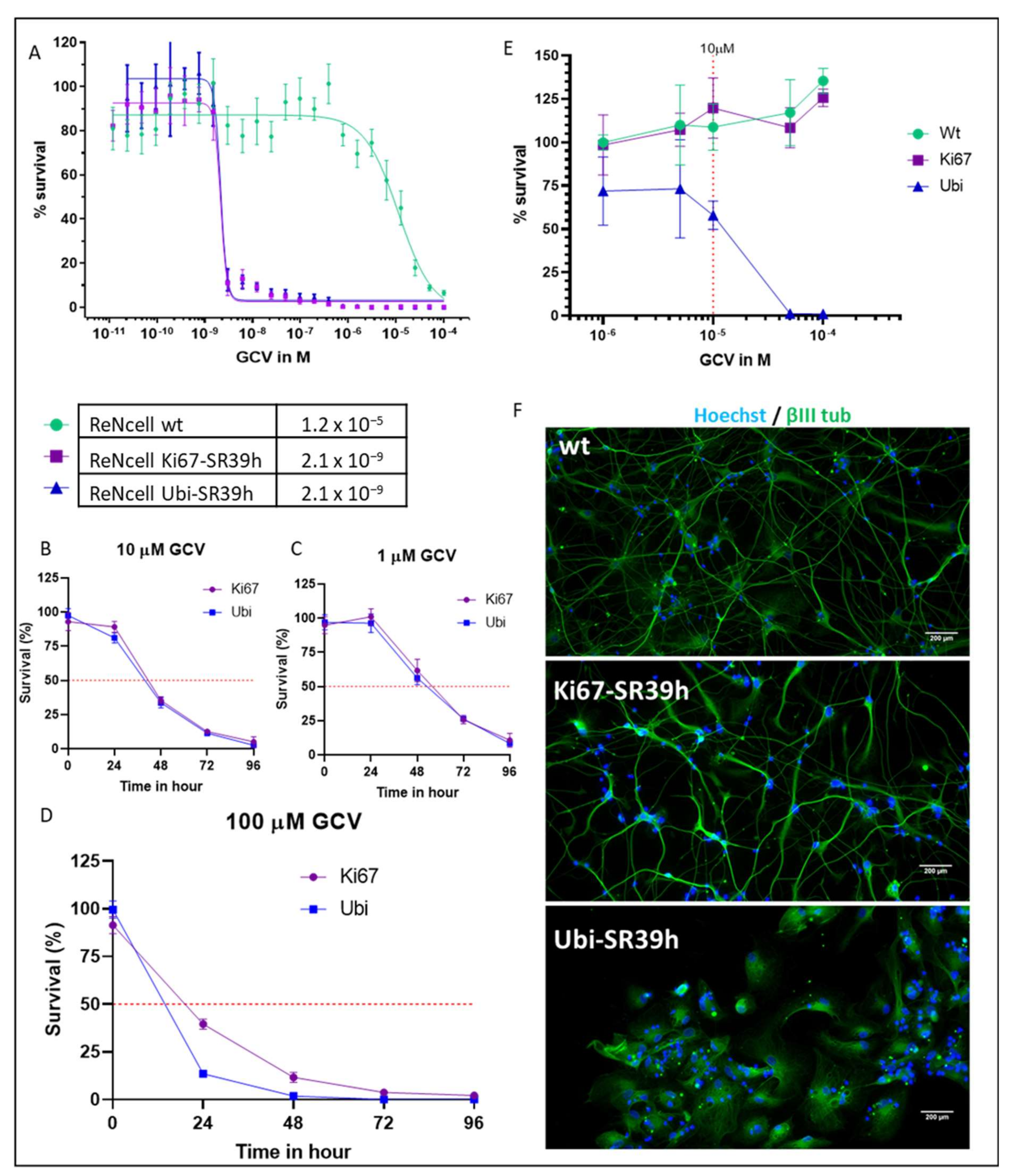
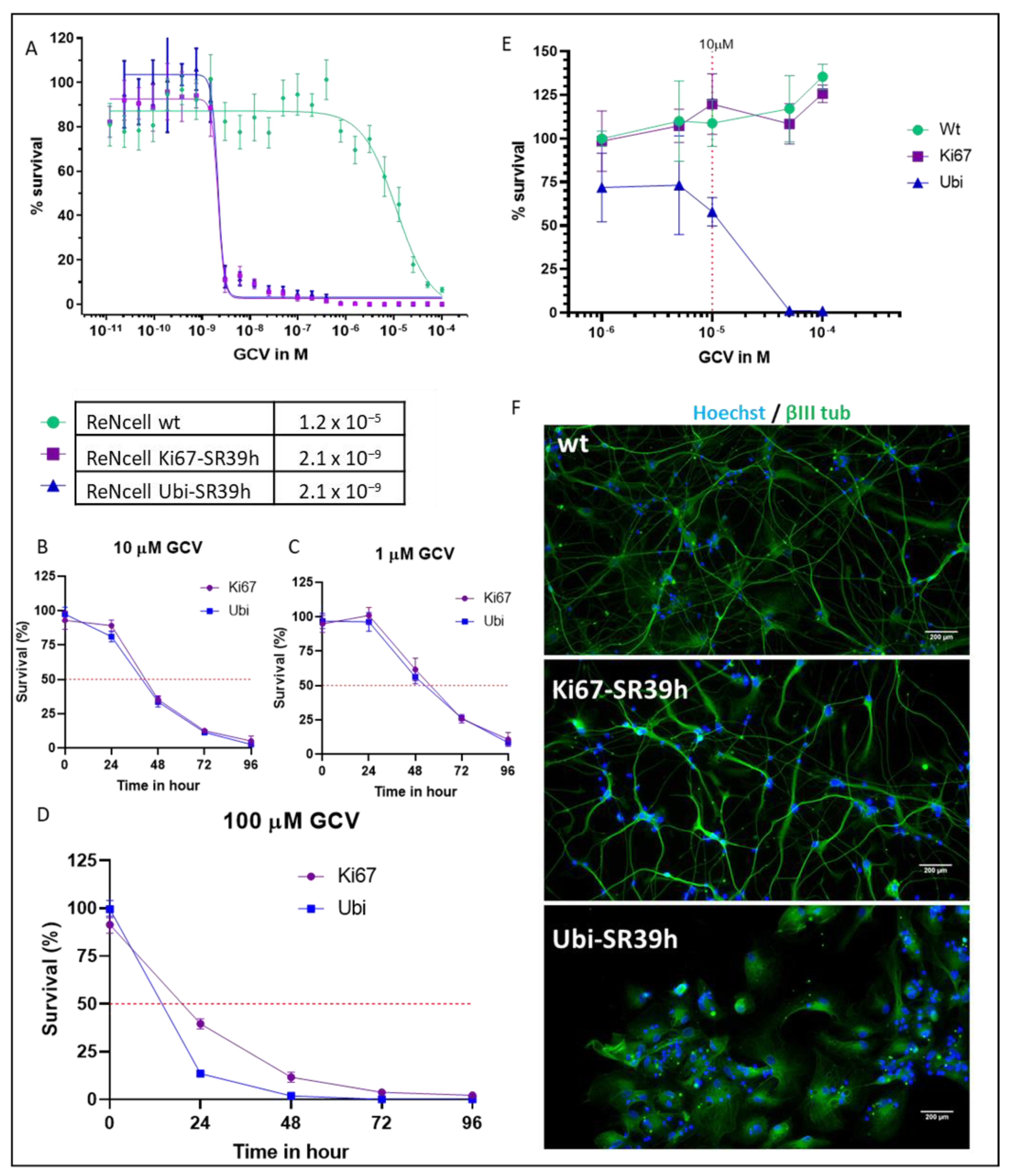
2.2. Thymidine Kinase Variants and Ganciclovir-Induced Cells Death
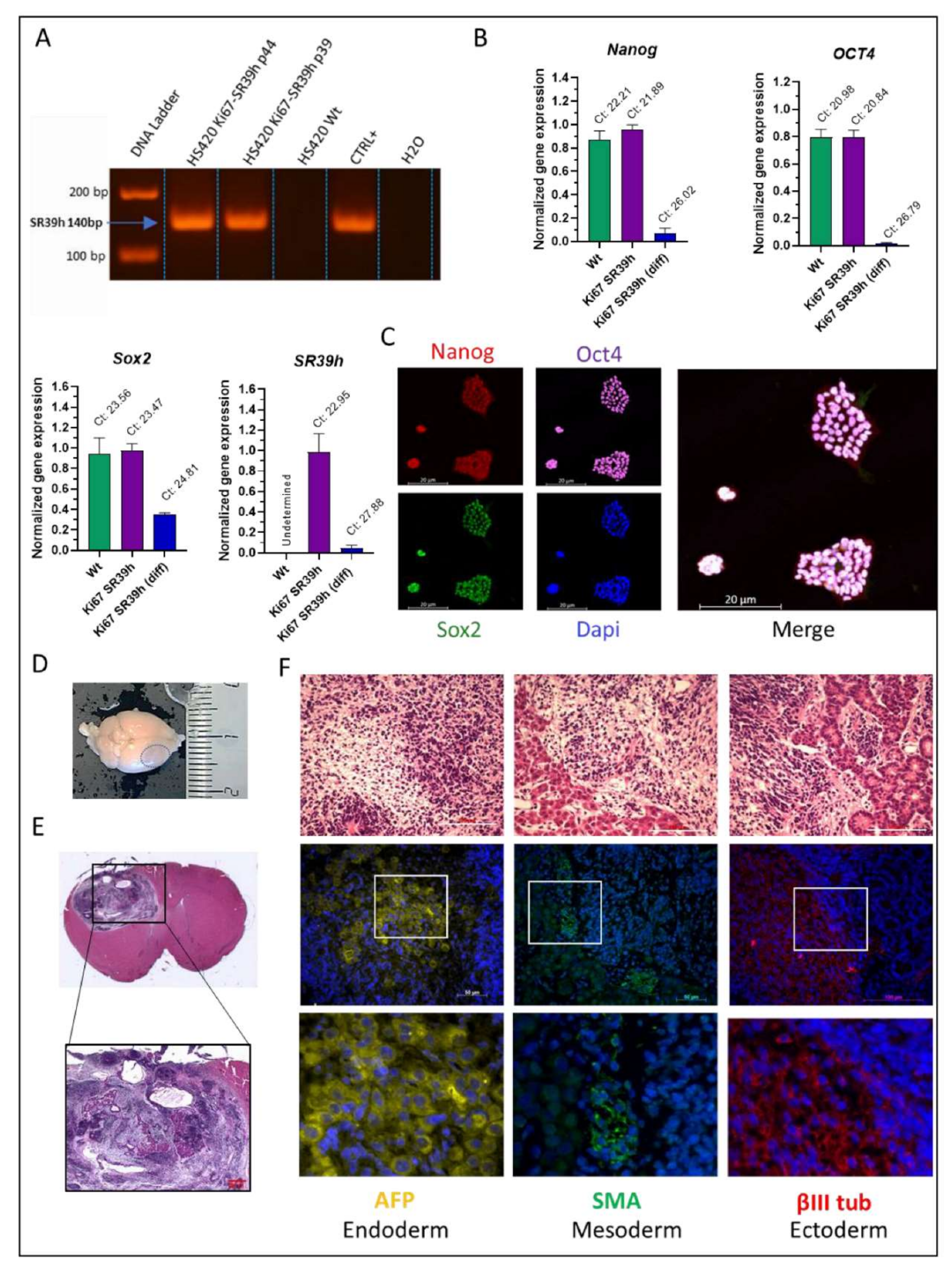
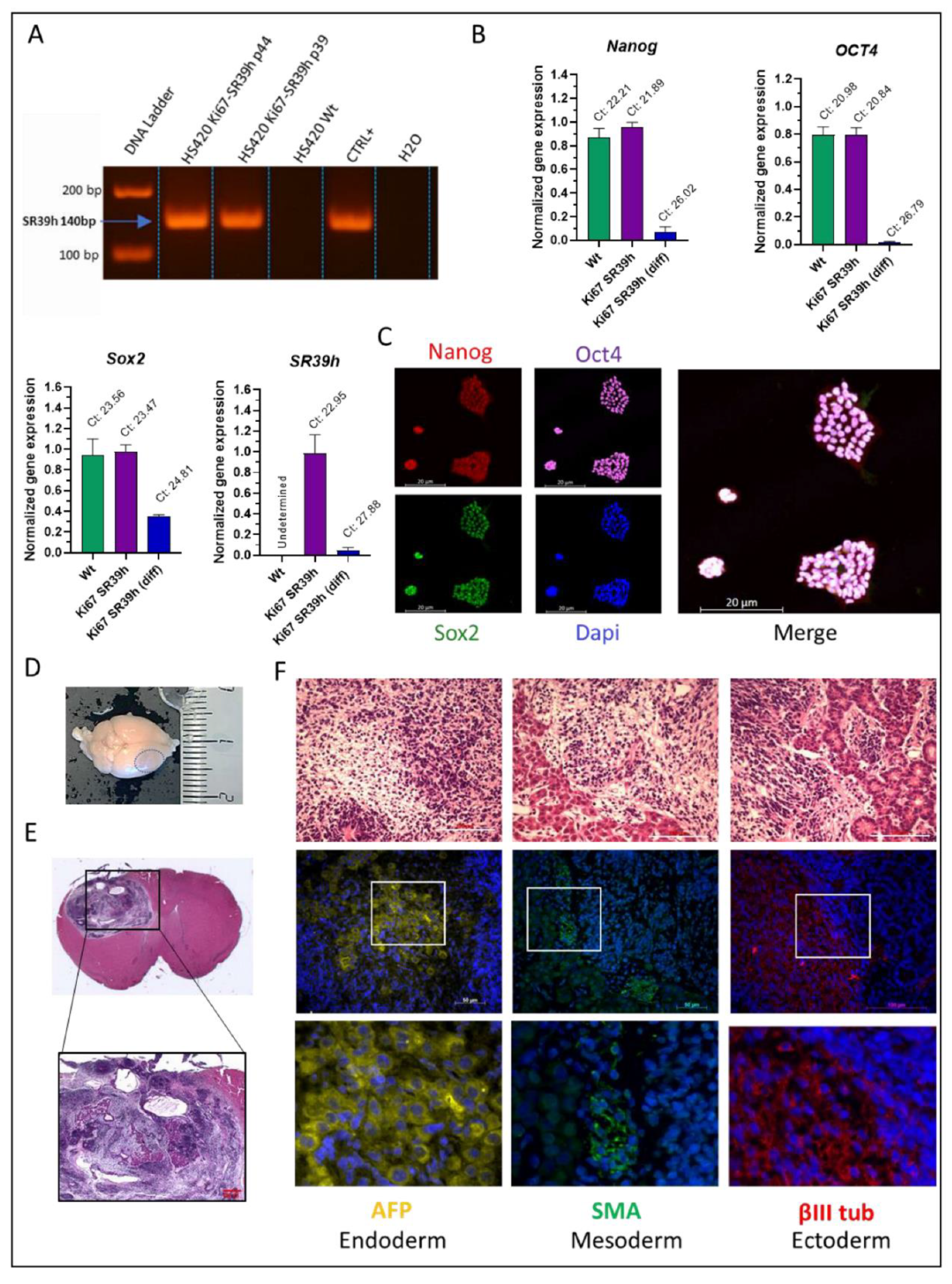
2.3. Characterization of the SR39h-Expressing HS420 Line
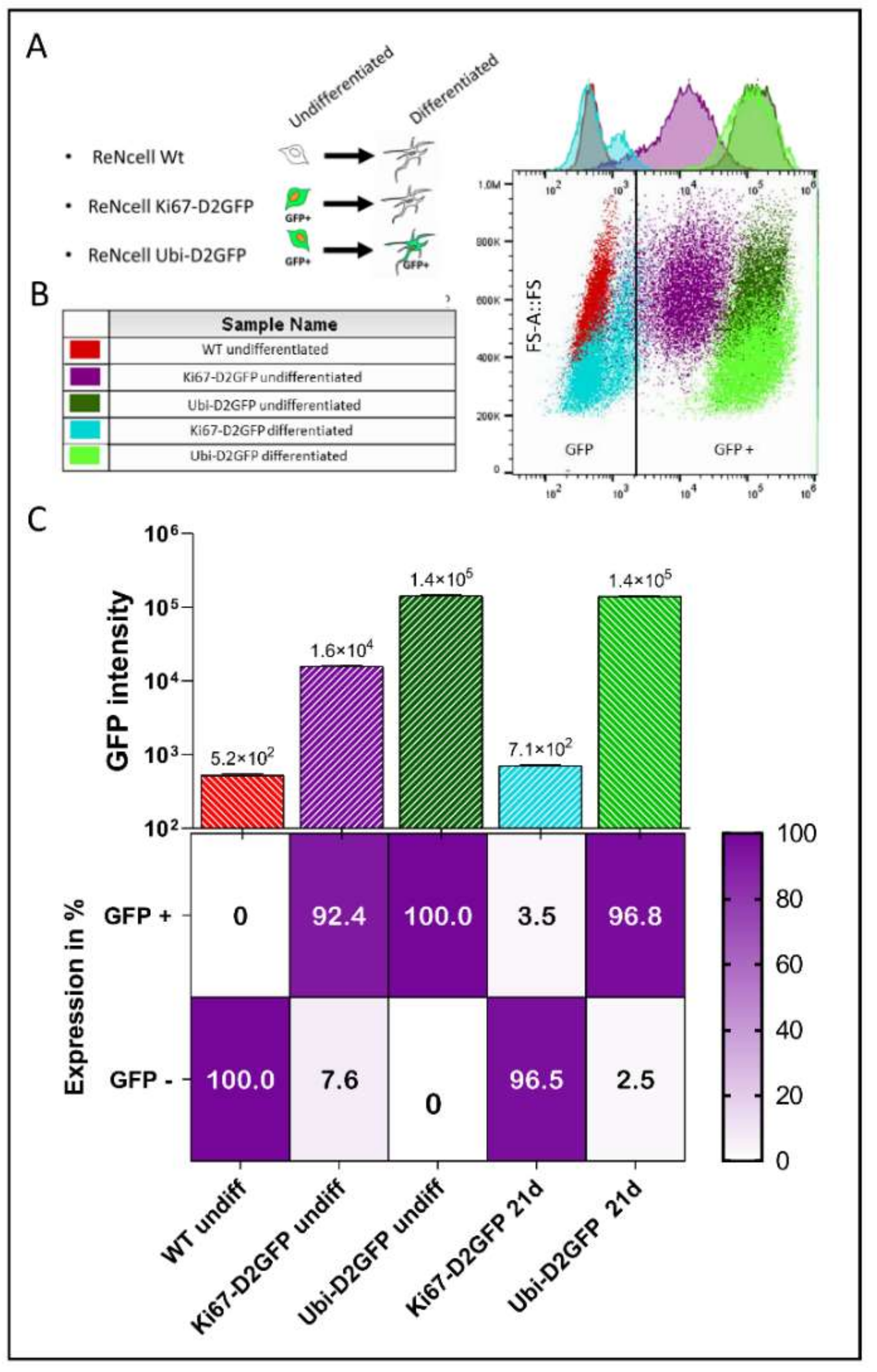
2.4. Cell Cycle-Dependent vs. Ubiquitous Promoter: Impact on Cell Death Induction
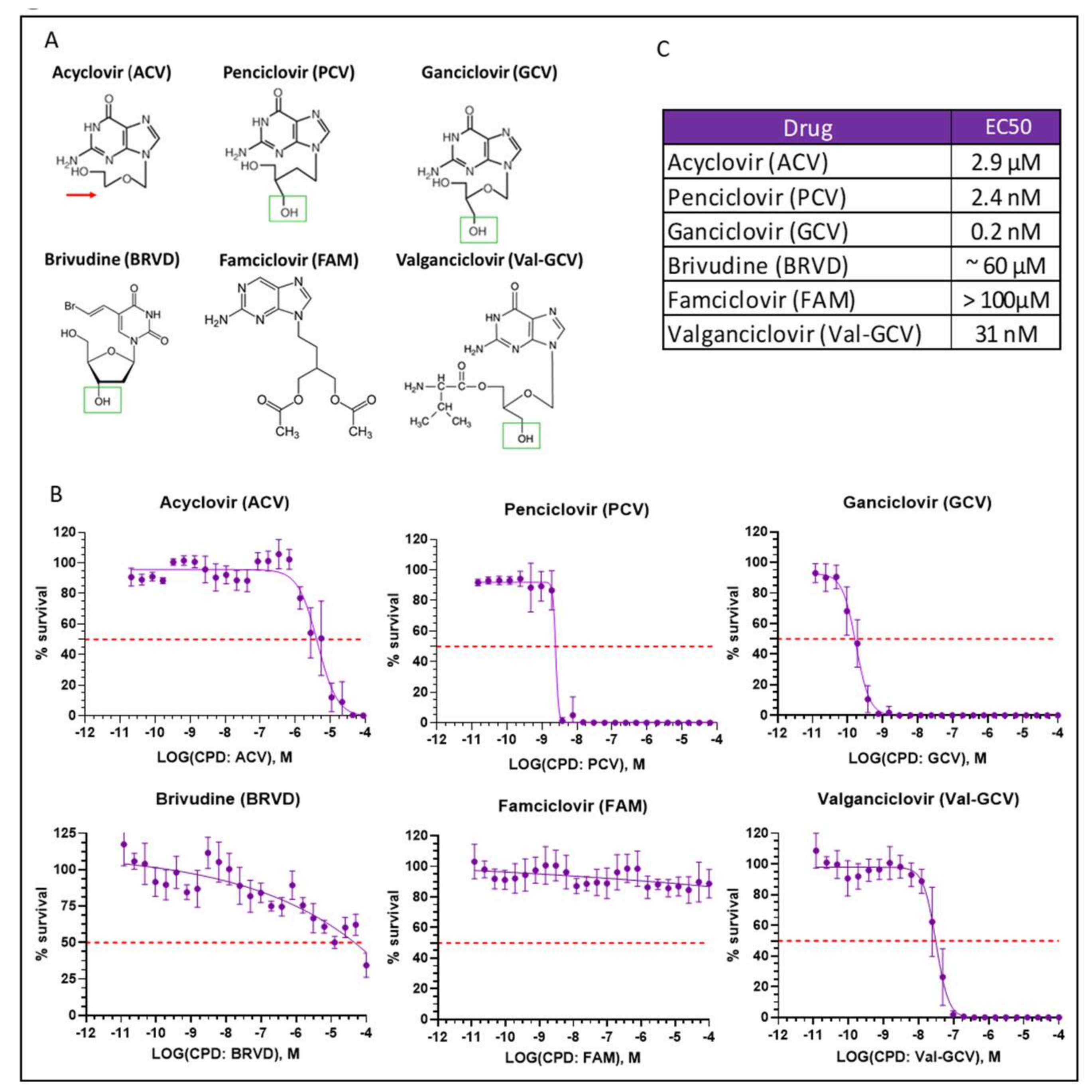
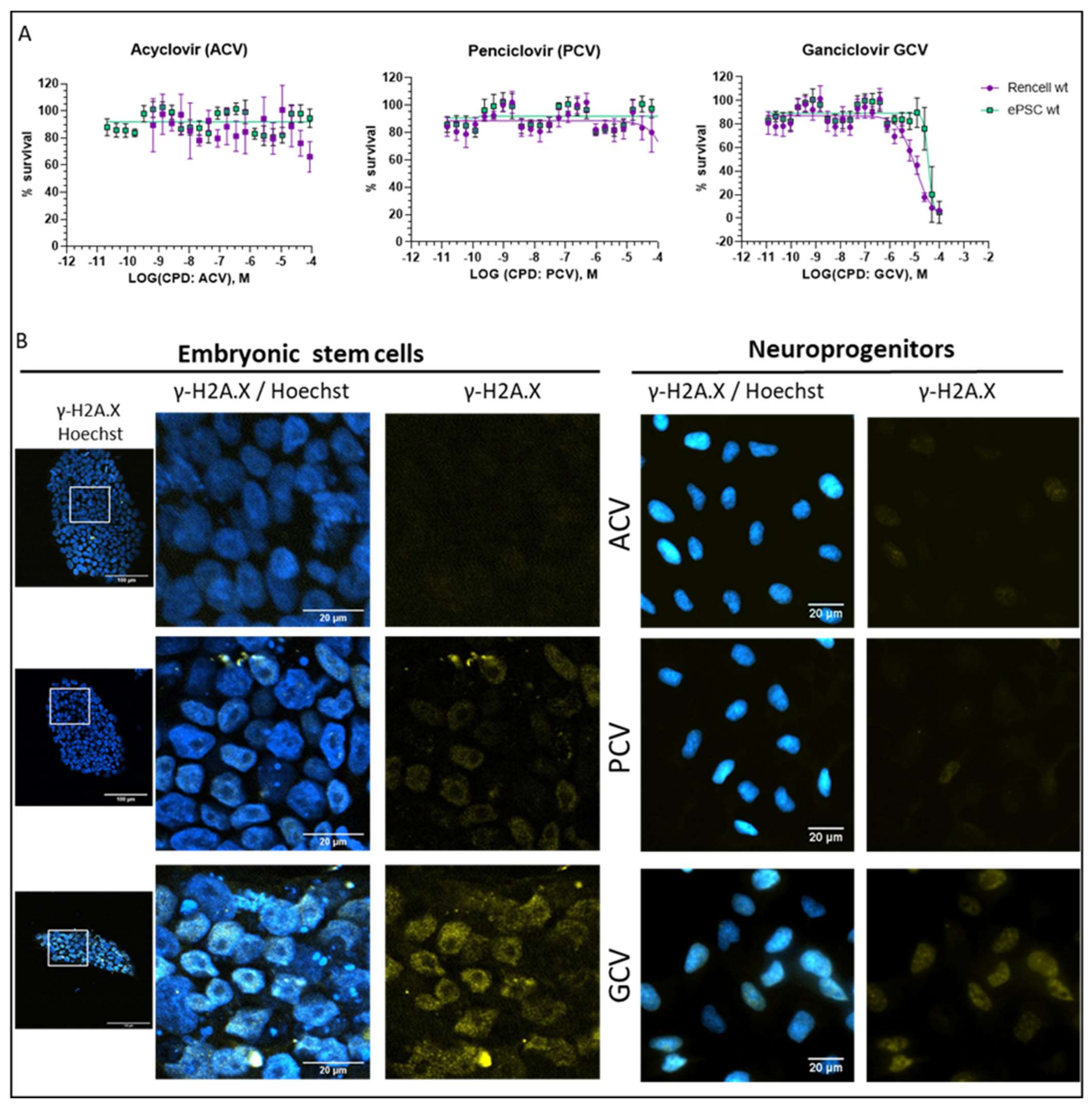
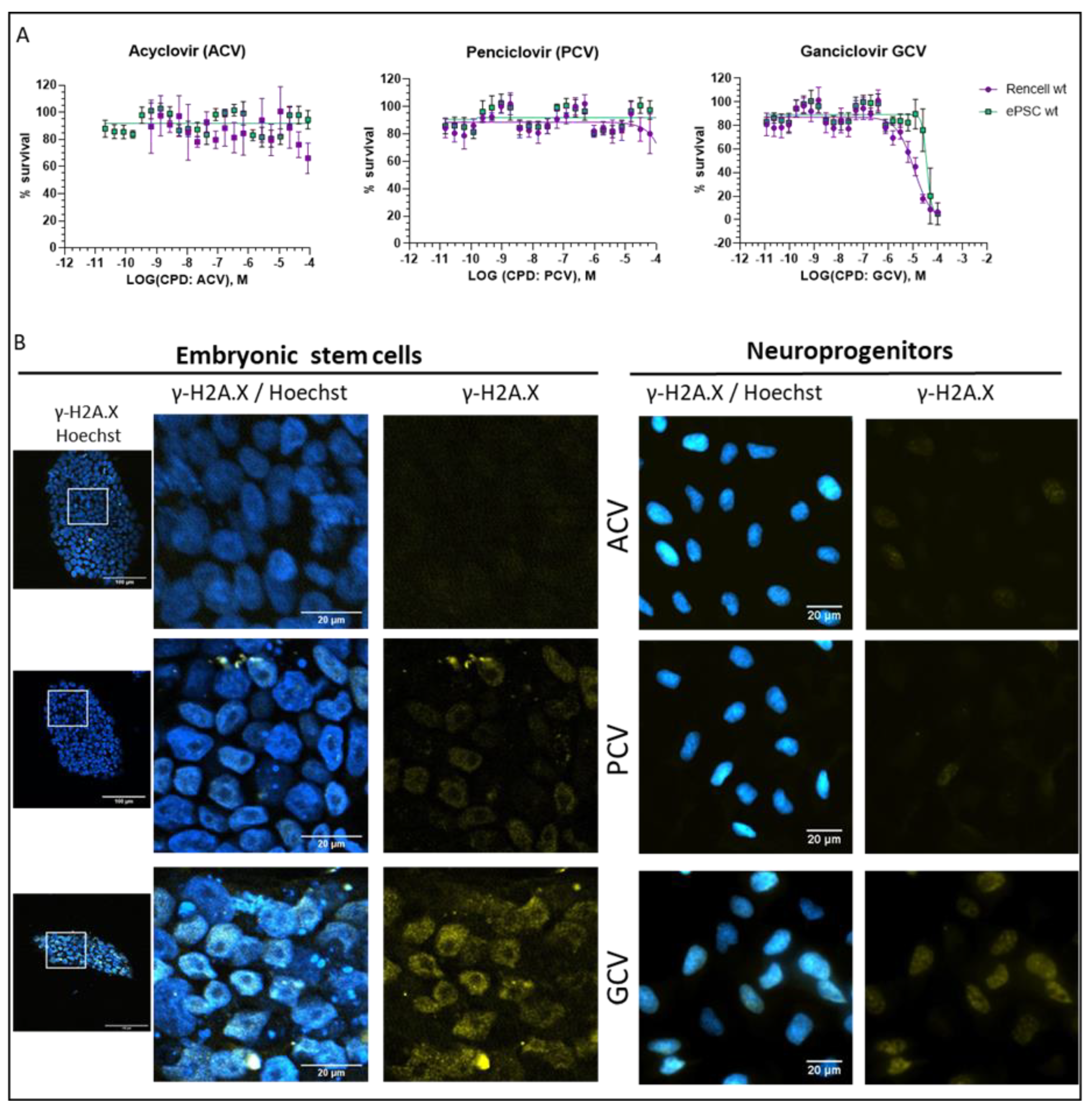
2.5. Optimization of Suicide-Inducing Nucleoside Analogs
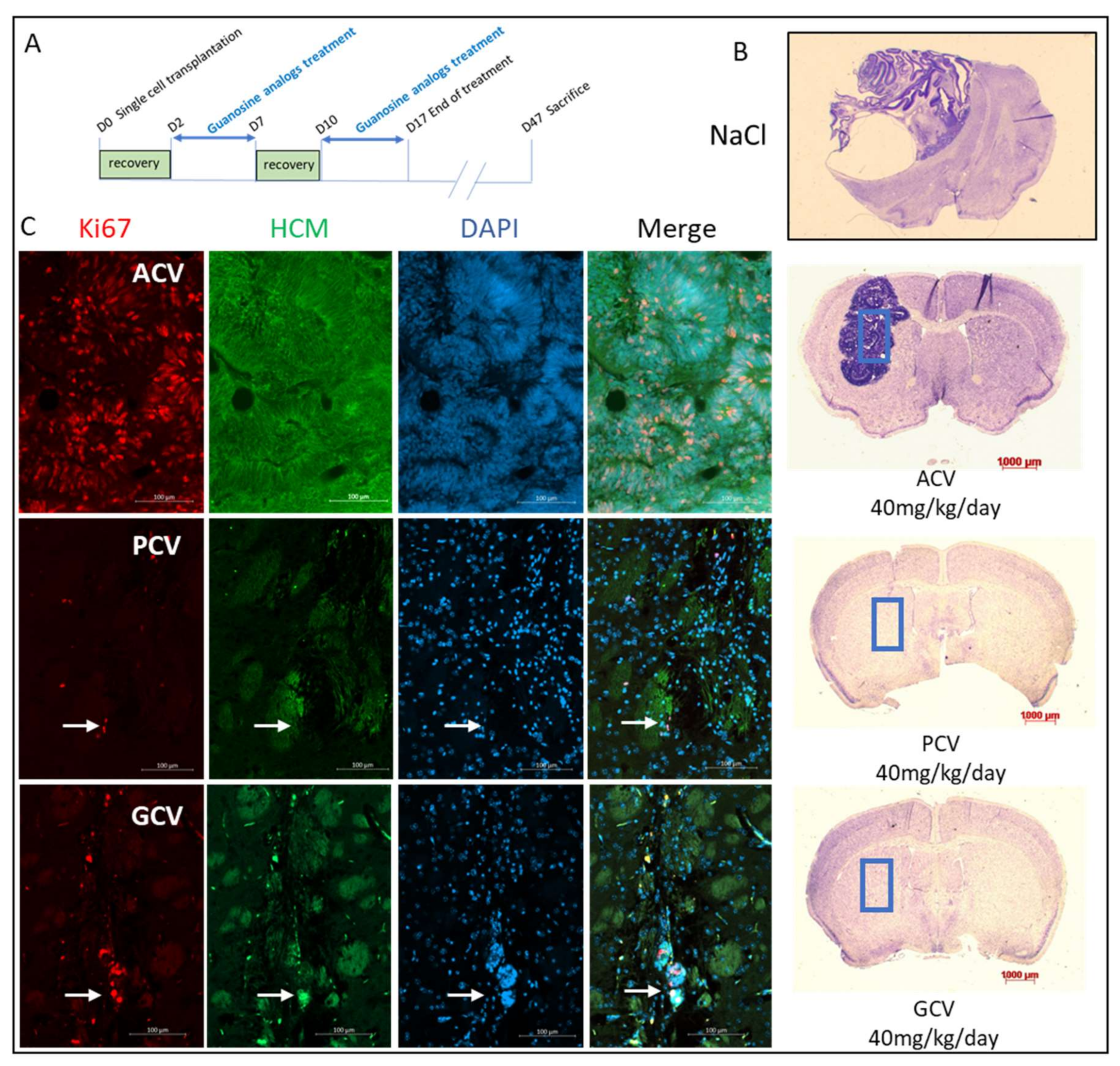
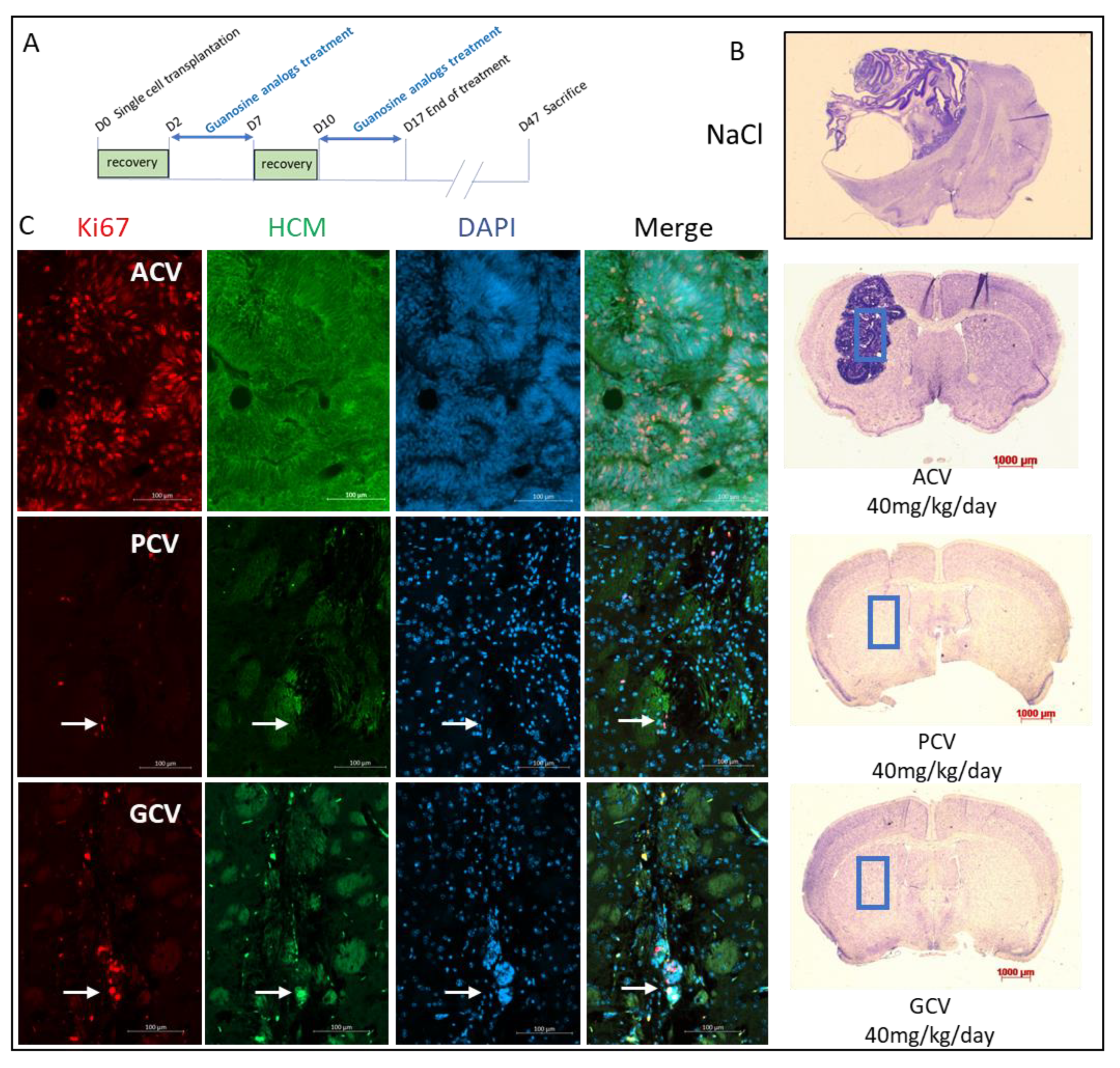
2.6. In Vivo Validation
3. Discussion
- (i).
- Differences in mode of action: ACV is thought to stop DNA synthesis through chain termination, but there is no wide-spread activation of cell death mechanism. More specifically, no fragmentation of host cell DNA in response to ACV was observed [31]. In contrast, PCV and GCV, which allow continuation of elongation despite integration of “wrong nucleosides” have the potential to lead to multiple double strand breaks within the nascent DNA. Accordingly, both compounds have been shown to induce fragmentation of host cell DNA [31]. These differences in mode of action are explained by the presence in PCV and GCV of a 3′-hydroxyl group, which is absent in ACV. The 3′-hydroxyl group, which is also present in naturally occurring nucleosides, is necessary for the continuation of DNA elongation. Thus, elongation with “wrong nucleotides” rather than chain termination might underlie the induction of cell death.
- (ii).
- Differences in TK activity on different nucleosides: Wild type TK has indeed a much lower affinity for ACV than for GCV; however, this is not the case for SR39 [12]. Thus, the low capacity of ACV to induced cell death in SR39-expressing cells is not due to a decrease TK phosphorylation of ACV.
- (iii).
- Differences in selectivity for viral vs. mammalian DNA polymerases: A lower affinity for the mammalian DNA polymerase could account for the fact that ACV does not induce cell death. From a theoretical point of view, this is a pertinent explanation, but there are no systematic studies addressing this question. One article compares affinities of nucleosides for viral and mammalian DNA polymerases, based on numbers from different publications [32]. In this context, it is important to point out that humans have 16 different DNA polymerases for replication and repair of nuclear DNA, as well as one additional DNA polymerase for mitochondrial DNA replication [33]. At this point, it is not clear which of these human DNA polymerases are able to use GCV triphosphate and PCV triphosphate as substrate for DNA synthesis. Given the rapid integration of GCV into the DNA dividing cells, it is tempting to speculate that DNA polymerase ε and δ might be involved. However, given the cell death induction by GCV in post-mitotic SR39h-expressing neurons, it appears that there is also a role for other polymerases, either those involved in DNA repair in post-mitotic cells [34] or the mitochondrial DNA polymerase. However, the markedly higher GCV concentrations were necessary to kill SR39h-expressing post-mitotic neurons. There are several explanations for this difference in terms of EC50, for example a different affinity of different polymerases for GCV triphosphate, or the fact that DNA repair in post-mitotic cells requires much less polymerase activity than DNA synthesis during replication in dividing cells. However, we cannot exclude a different mode of action of GCV in post-mitotic cells.
4. Materials and Methods
4.1. Human Embryonic Stem Cells (hESChESC): Maintenance Culture and Neural Differentiation
4.2. Culture of ReNcells
4.3. Humanization of TK DNA Sequence
4.4. Lentiviral Vector Construction
4.5. Cell Transduction and Selection
4.6. ATP-Based Cytotoxicity and Kinetic of Cell Death Assay
4.7. RNA Extraction and Quantitative Real Time Polymerase Chain Reaction
4.8. Immunocytochemistry
4.9. Flow Cytometry
4.10. Stereotaxic Engraftment and Treatment with Antiviral Nucleoside Analogs
4.11. Mouse Sacrifice and Brain Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Prim. 2017, 3, 17013. [Google Scholar] [CrossRef]
- Kefalopoulou, Z.; Politis, M.; Piccini, P.; Mencacci, N.; Bhatia, K.; Jahanshahi, M.; Widner, H.; Rehncrona, S.; Brundin, P.; Björklund, A.; et al. Long-term clinical outcome of fetal cell transplantation for parkinson disease: Two case reports. JAMA Neurol. 2014, 71, 83–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pera, M.F.; Trounson, A.O. Human embryonic stem cells: Prospects for development. Development 2004, 131, 5515–5525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tieng, V.; Cherpin, O.; Gutzwiller, E.; Zambon, A.C.; Delgado, C.; Salmon, P.; Dubois-Dauphin, M.; Krause, K.-H. Elimination of proliferating cells from CNS grafts using a Ki67 promoter-driven thymidine kinase. Mol. Ther.-Methods Clin. Dev. 2016, 3, 16069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dey, D.; Evans, G.R.D. Suicide Gene Therapy by Herpes Simplex Virus-1 Thymidine Kinase (HSV-TK). In Targets in Gene Therapy; InTech: London, UK, 2011. [Google Scholar]
- Wild, K.; Bohner, T.; Folkers, G.; Schulz, G.E. The Structures of Thymidine Kinase from Herpes Simplex Virus Type 1 in Complex with Substrates and a Substrate Analogue; Cambridge University Press: Cambridge, UK, 1997; Volume 6. [Google Scholar]
- Miller, W.H.; Miller, R.L. Phosphorylation of acyclovir (acycloguanosine) monophosphate by GMP kinase. J. Biol. Chem. 1980, 255, 7204–7207. [Google Scholar] [CrossRef]
- Reardon, J.E. Herpes simplex virus type 1 and human DNA polymerase interactions with 2’-deoxyguanosine 5’-triphosphate analogues. Kinetics of incorporation into DNA and induction of inhibition. J. Biol. Chem. 1989, 264, 19039–19044. [Google Scholar] [CrossRef]
- Foti, M.; Marshalko, S.; Schurter, E.; Kumar, S.; Beardsley, G.P.; Schweitzer, B.I. Solution Structure of a DNA Decamer Containing the Antiviral Drug Ganciclovir: Combined Use of NMR, Restrained Molecular Dynamics, and Full Relaxation Matrix Refinement. Biochemistry 1997, 36, 5336–5345. [Google Scholar] [CrossRef] [PubMed]
- Laberge, R.M.; Adler, D.; DeMaria, M.; Mechtouf, N.; Teachenor, R.; Cardin, G.B.; Desprez, P.Y.; Campisi, J.; Rodier, F. Mitochondrial DNA damage induces apoptosis in senescent cells. Cell Death Dis. 2013, 4, e727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Black, M.E.; Kokoris, M.S.; Sabo, P. Herpes Simplex Virus-1 Thymidine Kinase Mutants Created by Semi-Random Sequence Mutagenesis Improve Prodrug-mediated Tumor Cell Killing 1. Cancer Res. 2001, 61, 3022–3026. [Google Scholar] [PubMed]
- Kokoris, M.S.; Black, M.E. Characterization of herpes simplex virus type 1 thymidine kinase mutants engineered for improved ganciclovir or acyclovir activity. Protein Sci. 2002, 11, 2267–2272. [Google Scholar] [CrossRef] [Green Version]
- Donato, R.; Miljan, E.A.; Hines, S.J.; Aouabdi, S.; Pollock, K.; Patel, S.; Edwards, F.A.; Sinden, J.D. Differential development of neuronal physiological responsiveness in two human neural stem cell lines. BMC Neurosci. 2007, 8, 36. [Google Scholar] [CrossRef] [Green Version]
- Bennett, R.P.; Cox, C.A.; Hoeffler, J.P. Fusion of green fluorescent protein with the Zeocin(TM)-resistance marker allows visual screening and drug selection of transfected eukaryotic cells. Biotechniques 1998, 24, 478–482. [Google Scholar] [CrossRef] [PubMed]
- Ardiani, A.; Sanchez-Bonilla, M.; Black, M.E. Fusion Enzymes Containing HSV-1 Thymidine Kinase Mutants and Guanylate Kinase Enhance Prodrug Sensitivity In Vitro and In Vivo HHS Public Access. Cancer Gene Ther. 2010, 17, 86–96. [Google Scholar] [CrossRef] [Green Version]
- Ijichi, O.; Michel, D.; Mertens, T.; Miyata, K.; Eizuru, Y. GCV resistance due to the mutation A594P in the cytomegalovirus protein UL97 is partially reconstituted by a second mutation at D605E. Antiviral Res. 2002, 53, 135–142. [Google Scholar] [CrossRef]
- Wiegand, C.; Hipler, U.-C. Methods for the measurement of cell and tissue compatibility including tissue regeneration processes. GMS Krankenhhyg. Interdiszip. 2008, 3, Doc12. [Google Scholar]
- Tieng, V.; Stoppini, L.; Villy, S.; Fathi, M.; Dubois-Dauphin, M.; Krause, K.-H. Engineering of midbrain organoids containing long-lived dopaminergic neurons. Stem Cells Dev. 2014, 23, 1535–1547. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhao, X.; Fang, Y.; Jiang, X.; Duong, T.; Fan, C.; Huang, C.C.; Kain, S.R. Generation of destabilized green fluorescent protein as a transcription reporter. J. Biol. Chem. 1998, 273, 34970–34975. [Google Scholar] [CrossRef] [Green Version]
- Greco, O.; Dachs, G.U. Gene directed enzyme/prodrug therapy of cancer: Historical appraisal and future prospectives. J. Cell. Physiol. 2001, 187, 22–36. [Google Scholar] [CrossRef]
- Jacobson, J.G.; Renau, T.E.; Nassiri, M.R.; Sweier, D.G.; Breitenbach, J.M.; Townsend, L.B.; Drach, J.C. Nonnucleoside pyrrolopyrimidines with a unique mechanism of action against human cytomegalovirus. Antimicrob. Agents Chemother. 1999, 43, 1888–1894. [Google Scholar] [CrossRef] [Green Version]
- De Clercq, E. Discovery and development of BVDU (brivudin) as a therapeutic for the treatment of herpes zoster. Biochem. Pharmacol. 2004, 68, 2301–2315. [Google Scholar] [CrossRef]
- Poole, C.L.; James, S.H. Antiviral Therapies for Herpesviruses: Current Agents and New Directions. Clin. Ther. 2018, 40, 1282–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podhorecka, M.; Skladanowski, A.; Bozko, P. H2AX phosphorylation: Its role in DNA damage response and cancer therapy. J. Nucleic Acids 2010, 2010, 9. [Google Scholar] [CrossRef] [Green Version]
- Thust, R.; Tomicic, M.; Klöcking, R.; Wutzler, P.; Kaina, B. Cytogenetic genotoxicity of anti-herpes purine nucleoside analogues in CHO cells expressing the thymidine kinase gene of herpes simplex virus type 1: Comparison of ganciclovir, penciclovir and aciclovir. Mutagenesis 2000, 15, 177–184. [Google Scholar] [CrossRef] [Green Version]
- Brand, K.; Arnold, W.; Bartels, T.; Lieber, A.; Kay, M.A.; Strauss, M.; Dörken, B. Liver-associated toxicity of the HSV-tk/GCV approach and adenoviral vectors. Cancer Gene Ther. 1997, 4, 9–16. [Google Scholar] [PubMed]
- Thust, R.; Tomicic, M.; Klöcking, R.; Voutilainen, N.; Wutzler, P.; Kaina, B. Comparison of the genotoxic and apoptosis-inducing properties of ganciclovir and penciclovir in Chinese hamster ovary cells transfected with the thymidine kinase gene of herpes simplex virus-1: Implications for gene therapeutic approaches. Cancer Gene Ther. 2000, 7, 107–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wassilew, S.W.; Wutzler, P. Oral brivudin in comparison with acyclovir for improved therapy of herpes zoster in immunocompetent patients: Results of a randomized, double-blind, multicentered study. Antiviral Res. 2003, 59, 49–56. [Google Scholar] [CrossRef]
- Kłysik, K.; Pietraszek, A.; Karewicz, A.; Nowakowska, M. Acyclovir in the Treatment of Herpes Viruses—A Review. Curr. Med. Chem. 2018, 27, 4118–4137. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.E.; Harrell, A.W.; Chenery, R.J. Role of aldehyde oxidase in the in vitro conversion of famciclovir to penciclovir in human liver. Drug Metab. Dispos. 1995, 23, 251–254. [Google Scholar]
- Shaw, M.; Gurr, W.; Watts, P.; Littler, E.; Field, H. Ganciclovir and Penciclovir, but Not Acyclovir, Induce Apoptosis in Herpes Simplex Virus Thymidine Kinasetransformed Baby Hamster Kidney Cells. Antivir. Chem. Chemother. 2001, 12, 175–186. [Google Scholar] [CrossRef]
- Topalis, D.; Gillemot, S.; Snoeck, R.; Andrei, G. Distribution and effects of amino acid changes in drug-resistant α and β herpesviruses DNA polymerase. Nucleic Acids Res. 2016, 44, 9530–9554. [Google Scholar] [CrossRef] [Green Version]
- García-Gómez, S.; Reyes, A.; Martínez-Jiménez, M.I.; Chocrón, E.S.; Mourón, S.; Terrados, G.; Powell, C.; Salido, E.; Méndez, J.; Holt, I.J.; et al. PrimPol, an Archaic Primase/Polymerase Operating in Human Cells. Mol. Cell 2013, 52, 541–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simandi, Z. The role of DNA polymerase β in neural genome stability. J. Neurosci. 2017, 37, 11069–11071. [Google Scholar] [CrossRef] [PubMed]
- Iwasawa, C.; Tamura, R.; Sugiura, Y.; Suzuki, S.; Kuzumaki, N.; Narita, M.; Suematsu, M.; Nakamura, M.; Yoshida, K.; Toda, M.; et al. Increased Cytotoxicity of Herpes Simplex Virus Thymidine Kinase Expression in Human Induced Pluripotent Stem Cells. Int. J. Mol. Sci. 2019, 20, 810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caldés, A.; Gil-Vernet, S.; Armendariz, Y.; Colom, H.; Pou, L.; Niubó, J.; Lladó, L.; Torras, J.; Manito, N.; Rufí, G.; et al. Sequential treatment of cytomegalovirus infection or disease with a short course of intravenous ganciclovir followed by oral valganciclovir: Efficacy, safety, and pharmacokinetics. Transpl. Infect. Dis. 2010, 12, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Lazarus, H.M.; Belanger, R.; Candoni, A.; Aoun, M.; Jurewicz, R.; Marks, L. Intravenous penciclovir for treatment of herpes simplex infections in immunocompromised patients: Results of a multicenter, acyclovir-controlled trial. Antimicrob. Agents Chemother. 1999, 43, 1192–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Forward | Reverse | |
|---|---|---|
| GAPDH | CAAGATCATCAGCAATGCCT | CTTCCACGATACCAAAGTTGTC |
| SR39h | CAGCGAGACAATCGCCAAC | CCAGCACAGCATCTGTCAC |
| OCT4, | CTTGCTGCAGAAGTGGGTGGAGGAA | CTGCAGTGTGGGTTTCGGGCA |
| NANOG | CAAAGGCAAACAACCCACTT | TCTGCTGGAGGCTGAGGTAT |
| SOX2 | GCCGAGTGGAAACTTTTGTCG | GGCAGCGTGTACTTATCCTTCT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Locatelli, M.; Delhaes, F.; Cherpin, O.; Black, M.E.; Carnesecchi, S.; Preynat-Seauve, O.; Hibaoui, Y.; Krause, K.-H. Optimization of Thymidine Kinase-Based Safety Switch for Neural Cell Therapy. Cells 2022, 11, 502. https://doi.org/10.3390/cells11030502
Locatelli M, Delhaes F, Cherpin O, Black ME, Carnesecchi S, Preynat-Seauve O, Hibaoui Y, Krause K-H. Optimization of Thymidine Kinase-Based Safety Switch for Neural Cell Therapy. Cells. 2022; 11(3):502. https://doi.org/10.3390/cells11030502
Chicago/Turabian StyleLocatelli, Manon, Flavien Delhaes, Ophélie Cherpin, Margaret E. Black, Stéphanie Carnesecchi, Olivier Preynat-Seauve, Youssef Hibaoui, and Karl-Heinz Krause. 2022. "Optimization of Thymidine Kinase-Based Safety Switch for Neural Cell Therapy" Cells 11, no. 3: 502. https://doi.org/10.3390/cells11030502