
Extracellular Vesicles Derived from Primed Mesenchymal Stromal Cells Loaded on Biphasic Calcium Phosphate Biomaterial Exhibit Enhanced Macrophage Polarization


Abstract
:1. Introduction
2. Materials and Methods
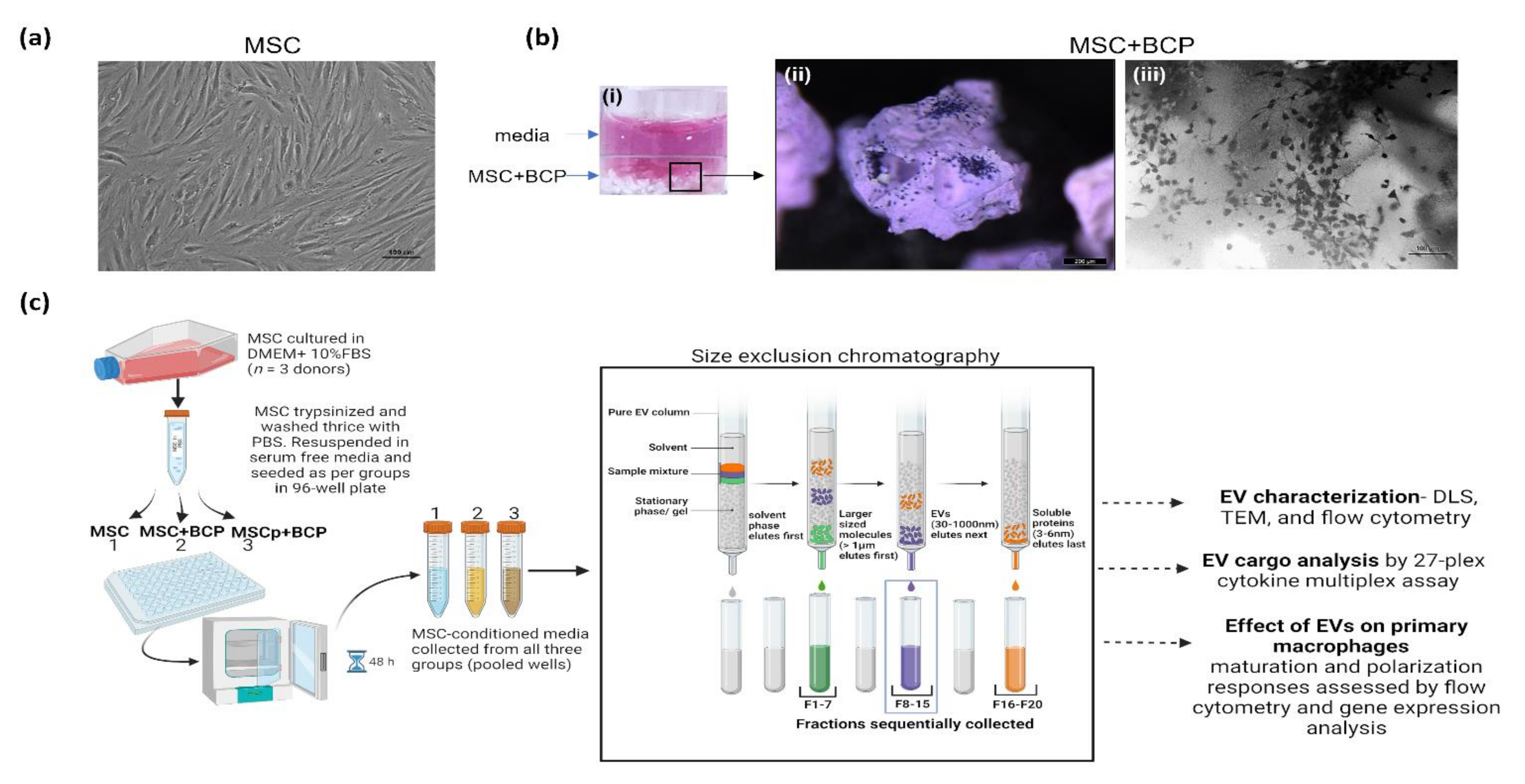
2.1. EV Isolation from MSC Conditioned Medium
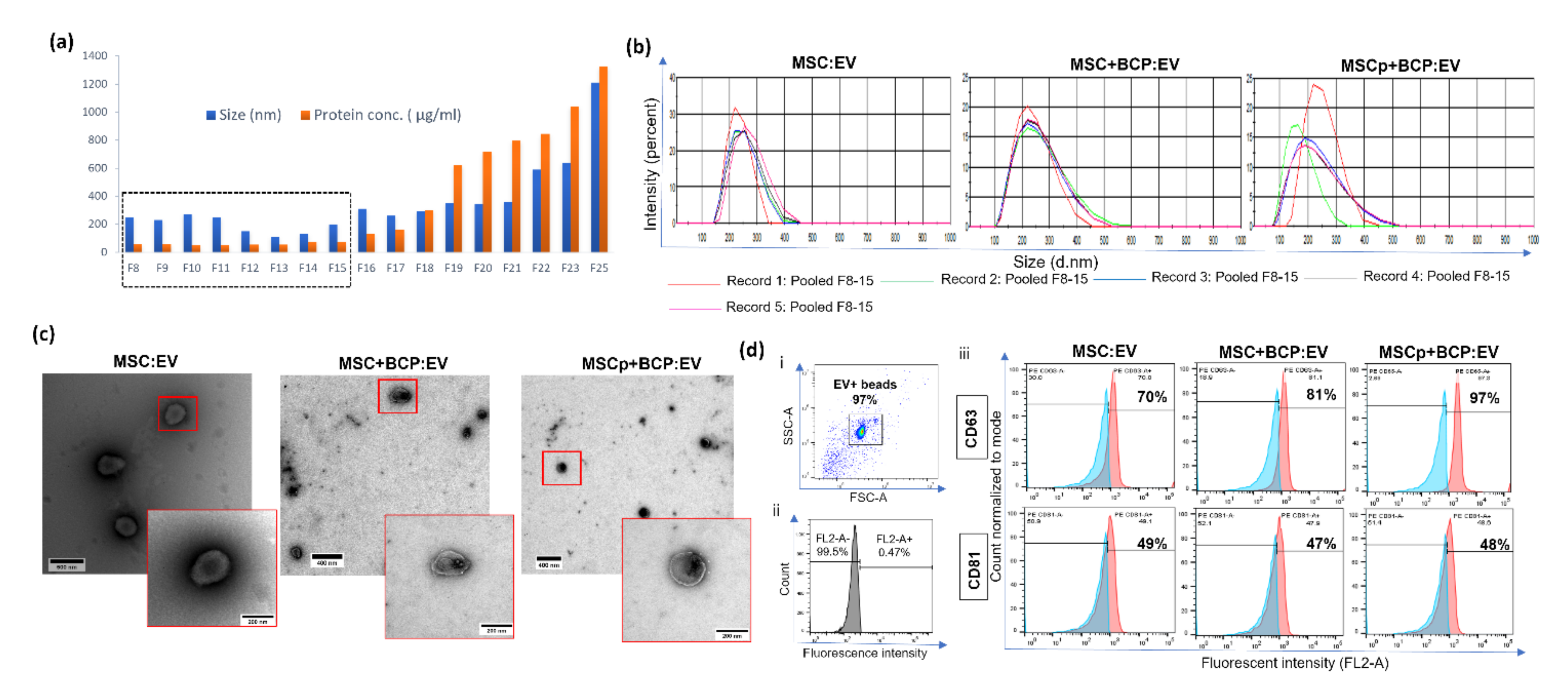
2.2. Size Exclusion Chromatography
2.3. Transmission Electron Microscopy
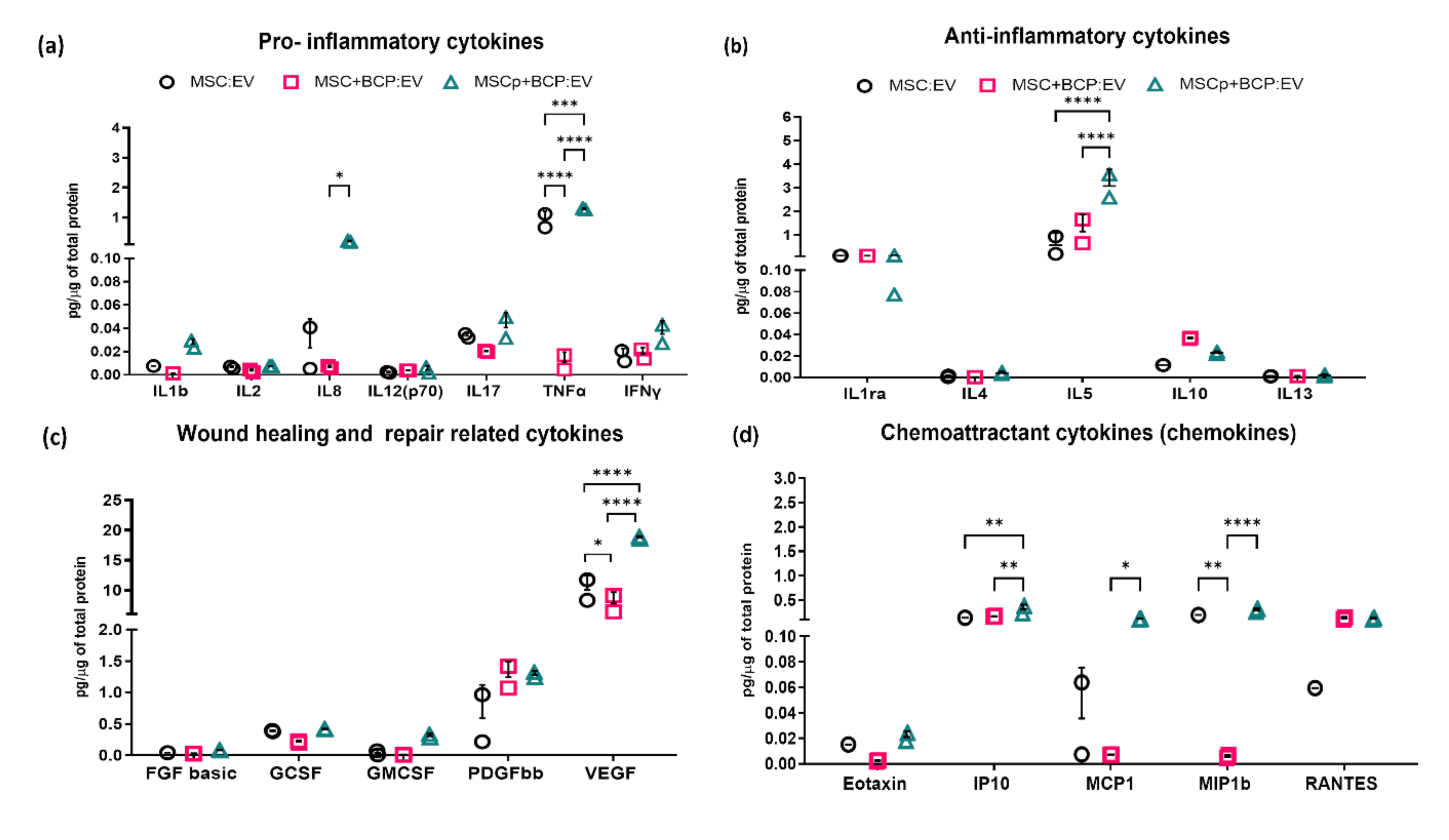
2.4. Cytokine Bioplex Immunoassay
2.5. CD14+CD16- Human Monocyte Isolation from Peripheral Blood
2.6. Generation of M0/M1/M2 Differentiated Macrophages and EV Treatment
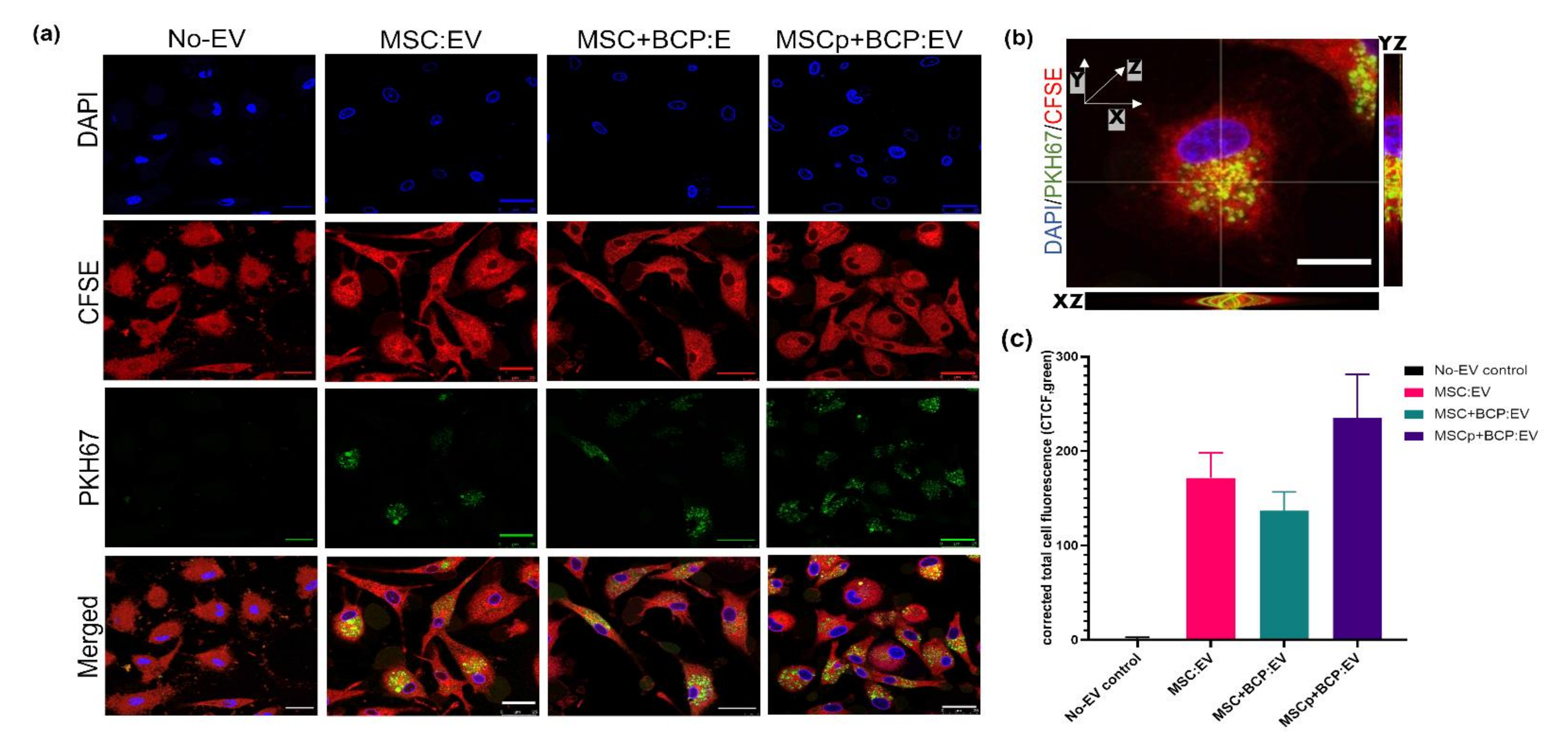
2.7. Uptake of EVs by Macrophages
2.8. Flow Cytometry
2.9. Gene Expression Analysis
2.10. Statistical Analysis
3. Results
3.1. Isolation and Characterization of MSC and MSC + BCP Derived EVs
3.2. EV Cytokine Bioplex Immunoassay
3.3. Uptake of EVs Derived from Different MSC Groups by Macrophages in Culture
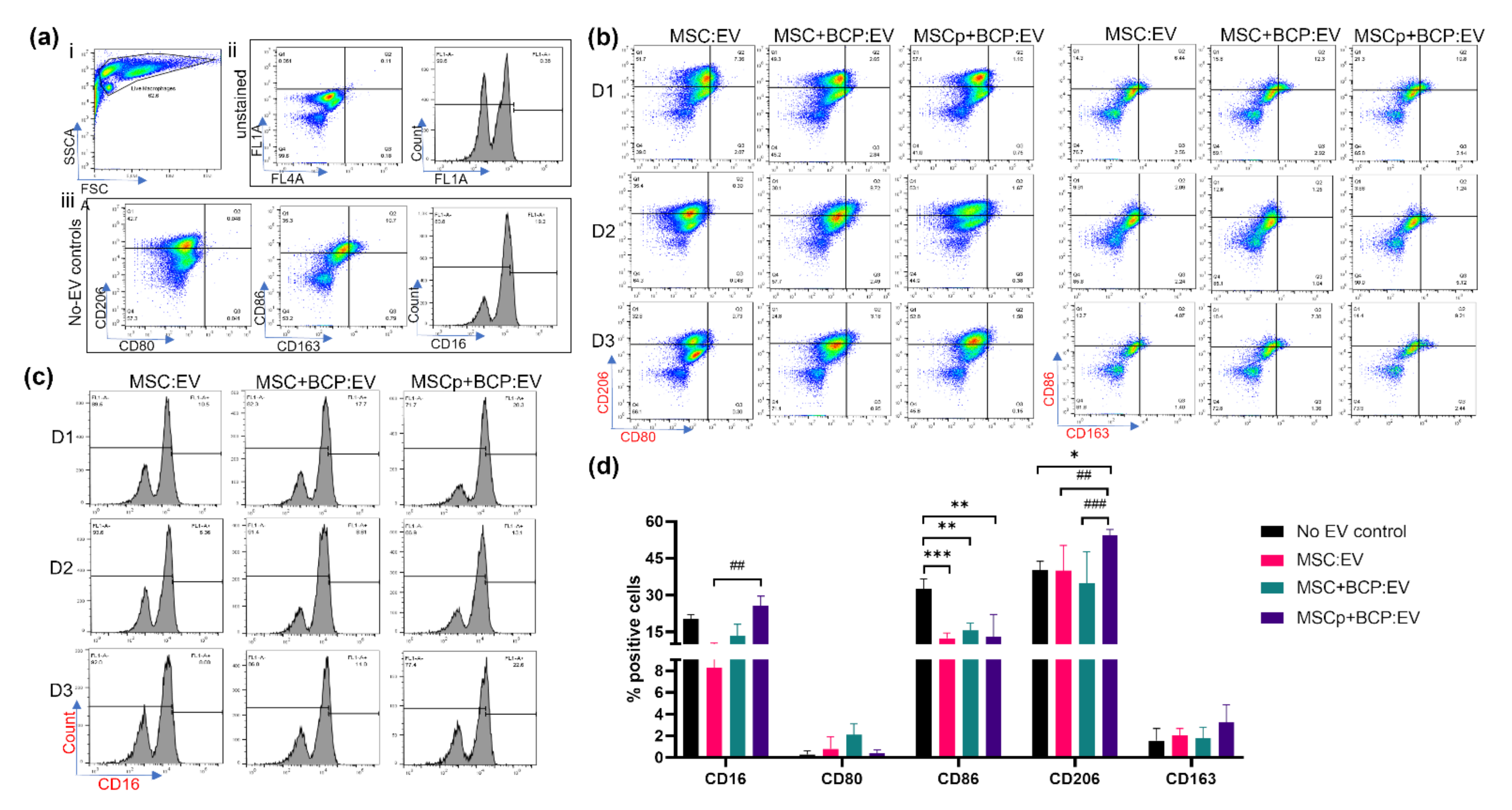
3.4. Effect of MSC-Derived EVs on Macrophage (M0) Maturation
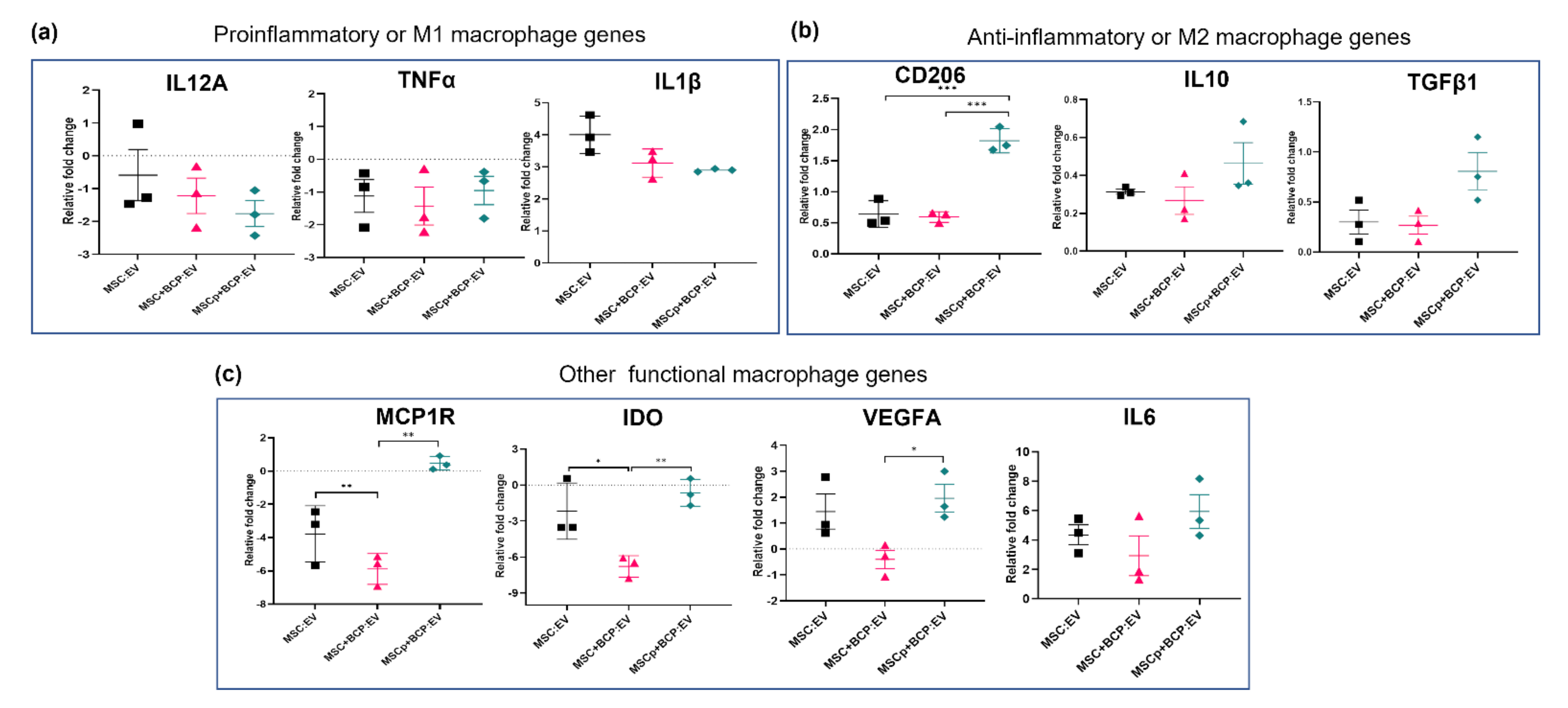
3.5. Effect of MSC-Derived EVs in Regulating Macrophage Gene Expression
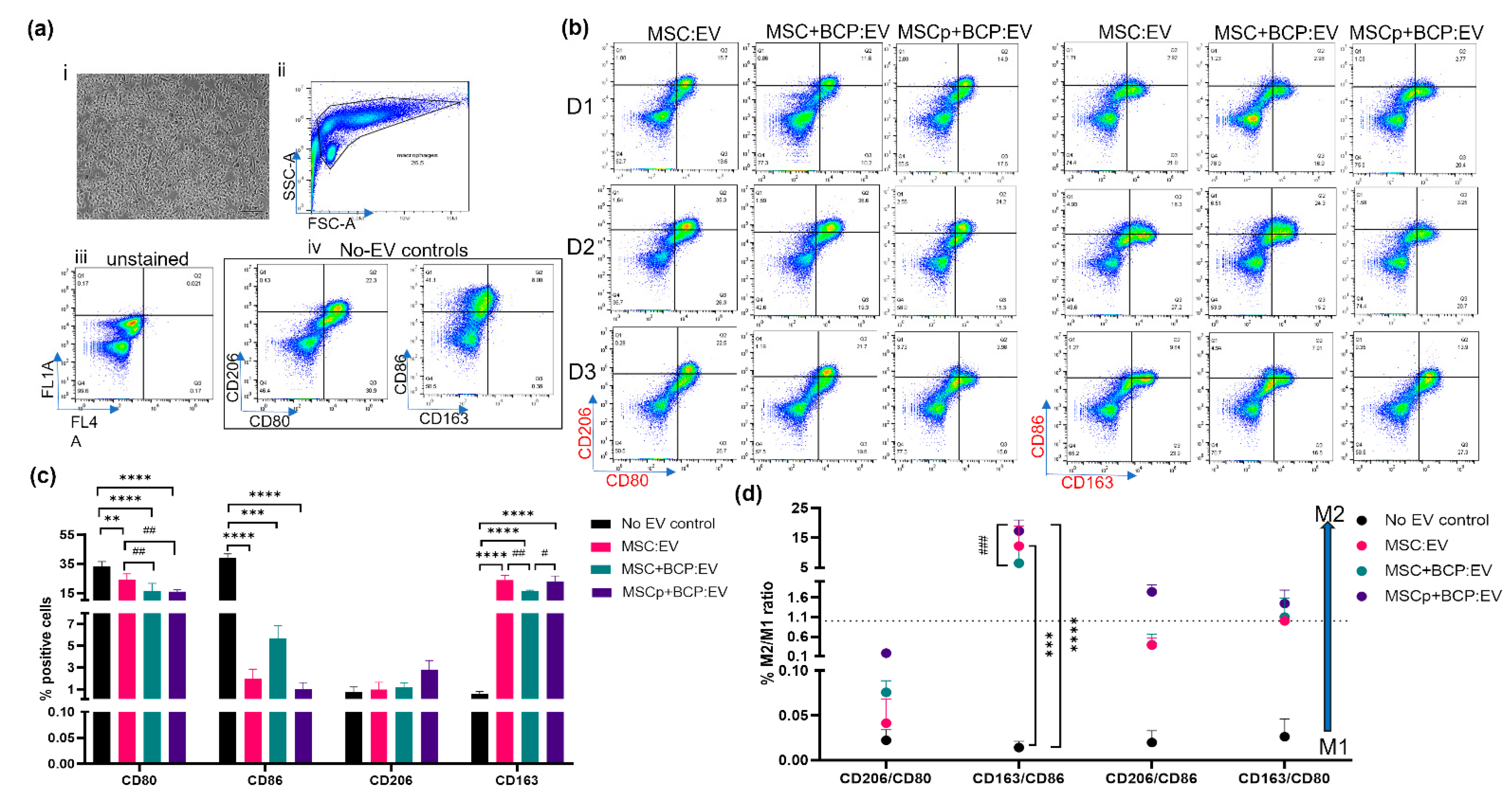
3.6. Effect of MSC-Derived EVs on M1 Polarized Macrophages
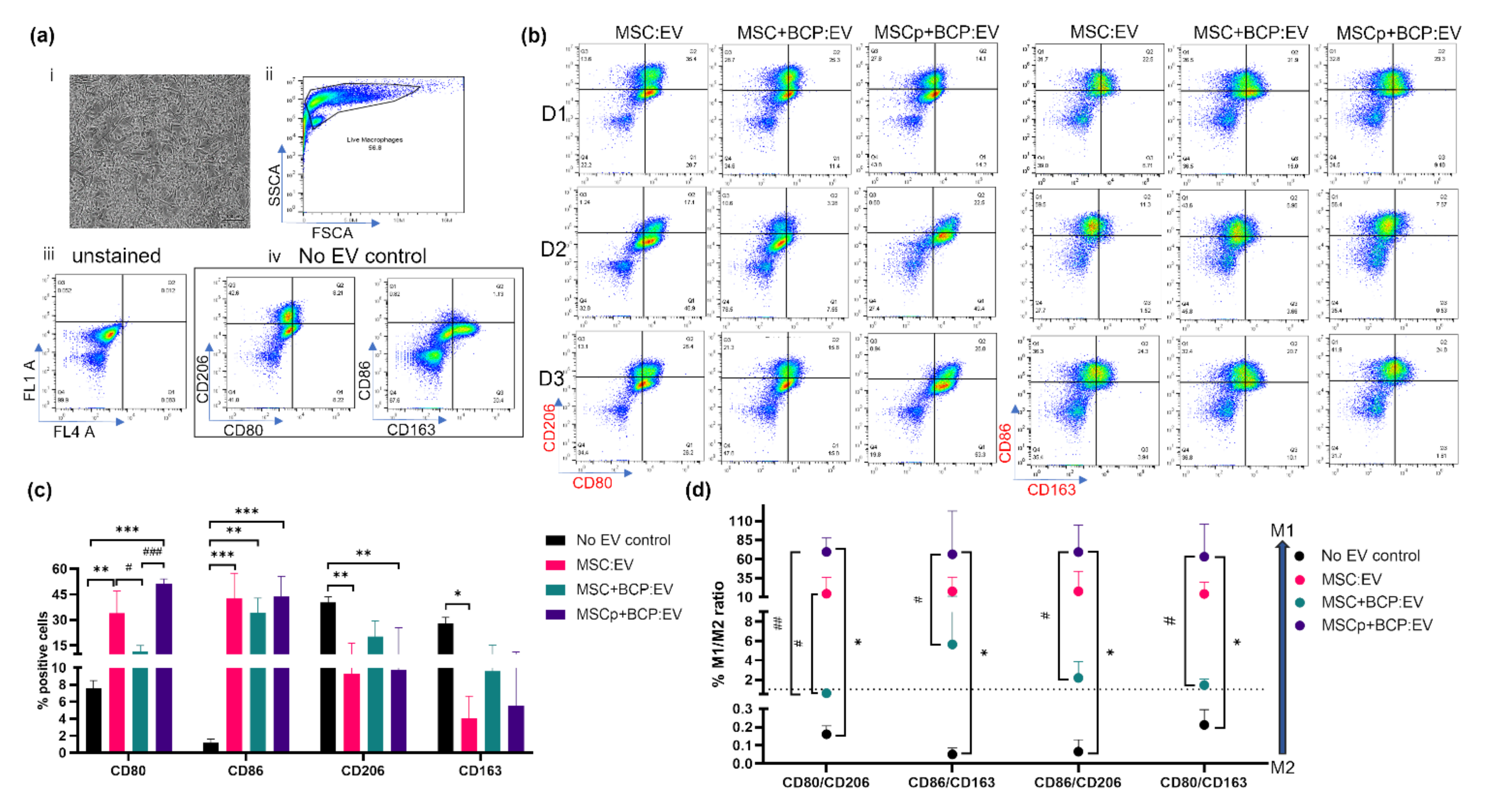
3.7. Effect of MSC-Derived EVs on M2 Polarized Macrophages
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Presen, D.M.; Traweger, A.; Gimona, M.; Redl, H. Mesenchymal Stromal Cell-Based Bone Regeneration Therapies: From Cell Transplantation and Tissue Engineering to Therapeutic Secretomes and Extracellular Vesicles. Front. Bioeng. Biotechnol. 2019, 7, 352. [Google Scholar] [CrossRef]
- Frohlich, M.; Grayson, W.; Wan, L.; Marolt, D.; Drobnic, M.; Vunjak-Novakovic, G. Tissue Engineered Bone Grafts: Biological Requirements, Tissue Culture and Clinical Relevance. Curr. Stem Cell Res. Ther. 2008, 3, 254–264. [Google Scholar] [CrossRef] [Green Version]
- Brennan, M.Á.; Renaud, A.; Amiaud, J.; Rojewski, M.T.; Schrezenmeier, H.; Heymann, D.; Trichet, V.; Layrolle, P. Pre-clinical studies of bone regeneration with human bone marrow stromal cells and biphasic calcium phosphate. Stem Cell Res. Ther. 2014, 5, 114. [Google Scholar] [CrossRef] [Green Version]
- Gjerde, C.; Mustafa, K.; Hellem, S.; Rojewski, M.; Gjengedal, H.; Yassin, M.A.; Feng, X.; Skaale, S.; Berge, T.; Rosen, A.; et al. Cell therapy induced regeneration of severely atrophied mandibular bone in a clinical trial. Stem Cell Res. Ther. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Meirelles, L.D.S.; Fontes, A.M.; Covas, D.T.; Caplan, A.I. Mechanisms involved in the therapeutic properties of mesenchymal stem cells. Cytokine Growth Factor Rev. 2009, 20, 419–427. [Google Scholar] [CrossRef]
- Medhat, D.; Rodríguez, C.I.; Infante, A. Immunomodulatory Effects of MSCs in Bone Healing. Int. J. Mol. Sci. 2019, 20, 5467. [Google Scholar] [CrossRef] [Green Version]
- Luque-Campos, N.; Bustamante-Barrientos, F.A.; Pradenas, C.; García, C.; Araya, M.J.; Bohaud, C.; Contreras-López, R.; Elizondo-Vega, R.; Djouad, F.; Luz-Crawford, P.; et al. The Macrophage Response Is Driven by Mesenchymal Stem Cell-Mediated Metabolic Reprogramming. Front. Immunol. 2021, 12, 624746. [Google Scholar] [CrossRef]
- Chen, Z.; Wu, C.; Gu, W.; Klein, T.; Crawford, R.; Xiao, Y. Osteogenic differentiation of bone marrow MSCs by β-tricalcium phosphate stimulating macrophages via BMP2 signalling pathway. Biomaterials 2014, 35, 1507–1518. [Google Scholar] [CrossRef]
- Maggini, J.; Mirkin, G.; Geffner, J.R.; Bognanni, I.; Holmberg, J.; Piazzón, I.M.; Nepomnaschy, I.; Costa, H.; Cañones, C.; Raiden, S.; et al. Mouse Bone Marrow-Derived Mesenchymal Stromal Cells Turn Activated Macrophages into a Regulatory-Like Profile. PLoS ONE 2010, 5, e9252. [Google Scholar] [CrossRef]
- Wise, A.F.; Williams, T.M.; Kiewiet, M.B.G.; Payne, N.L.; Siatskas, C.; Samuel, C.S.; Ricardo, S.D. Human mesenchymal stem cells alter macrophage phenotype and promote regeneration via homing to the kidney following ischemia-reperfusion injury. Am. J. Physiol. Physiol. 2014, 306, F1222–F1235. [Google Scholar] [CrossRef] [Green Version]
- Tylek, T.; Schilling, T.; Schlegelmilch, K.; Ries, M.; Rudert, M.; Jakob, F.; Groll, J. Platelet lysate outperforms FCS and human serum for co-culture of primary human macrophages and hMSCs. Sci. Rep. 2019, 9, 3533. [Google Scholar] [CrossRef] [Green Version]
- Vasandan, A.B.; Jahnavi, S.; Shashank, C.; Prasad, P.; Kumar, A.; Prasanna, S.J. Human Mesenchymal stem cells program macrophage plasticity by altering their metabolic status via a PGE2-dependent mechanism. Sci. Rep. 2016, 6, 38308. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Deng, Z.; Zhang, J.; Yang, C.; Liu, J.; Han, W.; Ye, P.; Si, Y.; Chen, G. Mesenchymal stem cells promote type 2 macrophage polarization to ameliorate the myocardial injury caused by diabetic cardiomyopathy. J. Transl. Med. 2019, 17, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, W.; Freeman, M.L.; Lederman, M.M.; Vasilieva, E.; Romero, R.; Margolis, L. A System of Cytokines Encapsulated in ExtraCellular Vesicles. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [Green Version]
- Liu, A.; Jin, S.; Fu, C.; Cui, S.; Zhang, T.; Zhu, L.; Wang, Y.; Shen, S.G.F.; Jiang, N.; Liu, Y. Macrophage-derived small extracellular vesicles promote biomimetic mineralized collagen-mediated endogenous bone regeneration. Int. J. Oral Sci. 2020, 12, 1–10. [Google Scholar] [CrossRef]
- Zhang, D.; Lee, H.; Wang, X.; Rai, A.; Groot, M.; Jin, Y. Exosome-Mediated Small RNA Delivery: A Novel Therapeutic Approach for Inflammatory Lung Responses. Mol. Ther. 2018, 26, 2119–2130. [Google Scholar] [CrossRef] [Green Version]
- Fang, S.-B.; Zhang, H.-Y.; Meng, X.-C.; Wang, C.; He, B.-X.; Peng, Y.-Q.; Xu, Z.-B.; Fan, X.-L.; Wu, Z.-J.; Wu, Z.-C.; et al. Small extracellular vesicles derived from human MSCs prevent allergic airway inflammation via immunomodulation on pulmonary macrophages. Cell Death Dis. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Lo Sicco, C.; Reverberi, D.; Balbi, C.; Ulivi, V.; Principi, E.; Pascucci, L.; Becherini, P.; Bosco, M.C.; Varesio, L.; Franzin, C.; et al. Mesenchymal Stem Cell-Derived Extracellular Vesicles as Mediators of Anti-Inflammatory Effects: Endorsement of Macrophage Polarization. Stem Cells Transl. Med. 2017, 6, 1018–1028. [Google Scholar] [CrossRef]
- Harting, M.T.; Srivastava, A.K.; Zhaorigetu, S.; Bair, H.; Prabhakara, K.S.; Furman, N.E.T.; Vykoukal, J.V.; Ruppert, K.A.; Cox, C.S., Jr.; Olson, S.D. Inflammation-Stimulated Mesenchymal Stromal Cell-Derived Extracellular Vesicles Attenuate Inflammation. Stem Cells 2018, 36, 79–90. [Google Scholar] [CrossRef] [Green Version]
- Ragni, E.; Orfei, C.P.; De Luca, P.; Mondadori, C.; Viganò, M.; Colombini, A.; de Girolamo, L. Inflammatory priming enhances mesenchymal stromal cell secretome potential as a clinical product for regenerative medicine approaches through secreted factors and EV-miRNAs: The example of joint disease. Stem Cell Res. Ther. 2020, 11, 1–19. [Google Scholar] [CrossRef] [PubMed]
- da Silva, L.H.A.; Antunes, M.A.; Dos Santos, C.C.; Weiss, D.J.; Cruz, F.F.; Rocco, P.R.M. Strategies to improve the therapeutic effects of mesenchymal stromal cells in respiratory diseases. Stem Cell Res. Ther. 2018, 9, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sisakhtnezhad, S.; Alimoradi, E.; Akrami, H. External factors influencing mesenchymal stem cell fate in vitro. Eur. J. Cell Biol. 2017, 96, 13–33. [Google Scholar] [CrossRef] [PubMed]
- Leuning, D.G.; Beijer, N.; du Fossé, N.; Vermeulen, S.; Lievers, E.; Van Kooten, C.; Rabelink, T.; De Boer, J. The cytokine secretion profile of mesenchymal stromal cells is determined by surface structure of the microenvironment. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Brennan, M.Á.; Layrolle, P.; Mooney, D.J. Biomaterials Functionalized with MSC Secreted Extracellular Vesicles and Soluble Factors for Tissue Regeneration. Adv. Funct. Mater. 2020, 30, 1909125. [Google Scholar] [CrossRef] [PubMed]
- Barbeck, M.; Schröder, M.-L.; Alkildani, S.; Jung, O.; Unger, R. Exploring the Biomaterial-Induced Secretome: Physical Bone Substitute Characteristics Influence the Cytokine Expression of Macrophages. Int. J. Mol. Sci. 2021, 22, 4442. [Google Scholar] [CrossRef]
- Shanbhag, S.; Mohamed-Ahmed, S.; Lunde, T.H.F.; Suliman, S.; Bolstad, A.I.; Hervig, T.; Mustafa, K. Influence of platelet storage time on human platelet lysates and platelet lysate-expanded mesenchymal stromal cells for bone tissue engineering. Stem Cell Res. Ther. 2020, 11, 1–18. [Google Scholar] [CrossRef]
- Mohamed-Ahmed, S.; Fristad, I.; Lie, S.A.; Suliman, S.; Mustafa, K.; Vindenes, H.; Idris, S.B. Adipose-derived and bone marrow mesenchymal stem cells: A donor-matched comparison. Stem Cell Res. Ther. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Mathew, B.; Torres, L.; Acha, L.G.; Tran, S.; Liu, A.; Patel, R.; Chennakesavalu, M.; Aneesh, A.; Huang, C.-C.; Feinstein, D.; et al. Uptake and Distribution of Administered Bone Marrow Mesenchymal Stem Cell Extracellular Vesicles in Retina. Cells 2021, 10, 730. [Google Scholar] [CrossRef]
- Brylka, L.J.; Schinke, T. Chemokines in Physiological and Pathological Bone Remodeling. Front. Immunol. 2019, 10, 2182. [Google Scholar] [CrossRef] [Green Version]
- McWhorter, F.Y.; Wang, T.; Nguyen, P.; Chung, T.; Liu, W.F. Modulation of macrophage phenotype by cell shape. Proc. Natl. Acad. Sci. USA 2013, 110, 17253–17258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Jiao, G.; Ren, S.; Zhang, X.; Li, C.; Wu, W.; Wang, H.; Liu, H.; Zhou, H.; Chen, Y. Exosomes from bone marrow mesenchymal stem cells enhance fracture healing through the promotion of osteogenesis and angiogenesis in a rat model of nonunion. Stem Cell Res. Ther. 2020, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Barrena, E.; Padilla-Eguiluz, N.; Rosset, P.; Gebhard, F.; Hernigou, P.; Baldini, N.; Rouard, H.; Sensebé, L.; Gonzalo-Daganzo, R.-M.; Giordano, R.; et al. Early efficacy evaluation of mesenchymal stromal cells (MSC) combined to biomaterials to treat long bone non-unions. Injury 2020, 51, S63–S73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dabrowska, S.; Andrzejewska, A.; Janowski, M.; Lukomska, B. Immunomodulatory and Regenerative Effects of Mesenchymal Stem Cells and Extracellular Vesicles: Therapeutic Outlook for Inflammatory and Degenerative Diseases. Front. Immunol. 2021, 11, 591065. [Google Scholar] [CrossRef]
- Chen, Y.; Shu, Z.; Qian, K.; Wang, J.; Zhu, H. Harnessing the properties of biomaterial to enhance the immunomodulation of mesenchymal stem cells. Tissue Eng. Part. B Rev. 2019, 25, 492–499. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Zhang, K.; Zhao, R.; Ye, X.; Chen, X.; Xiao, Z.; Yang, X.; Zhu, X.; Zhang, K.; Fan, Y.; et al. Bone regeneration with micro/nano hybrid-structured biphasic calcium phosphate bioceramics at segmental bone defect and the induced immunoregulation of MSCs. Biomaterials 2017, 147, 133–144. [Google Scholar] [CrossRef]
- Yano, S.; Mentaverri, R.; Kanuparthi, D.; Bandyopadhyay, S.; Rivera, A.; Brown, E.M.; Chattopadhyay, N. Functional Expression of β-Chemokine Receptors in Osteoblasts: Role of Regulated upon Activation, Normal T Cell Expressed and Secreted (RANTES) in Osteoblasts and Regulation of Its Secretion by Osteoblasts and Osteoclasts. Endocrinology 2005, 146, 2324–2335. [Google Scholar] [CrossRef] [Green Version]
- Xie, H.; Wang, Z.; Zhang, L.; Lei, Q.; Zhao, A.; Wang, H.; Li, Q.; Cao, Y.; Zhang, W.J.; Chen, Z. Extracellular Vesicle-functionalized Decalcified Bone Matrix Scaffolds with Enhanced Pro-angiogenic and Pro-bone Regeneration Activities. Sci. Rep. 2017, 7, srep45622. [Google Scholar] [CrossRef]
- Murali, V.P.; Holmes, C.A. Mesenchymal stromal cell-derived extracellular vesicles for bone regeneration therapy. Bone Rep. 2021, 14, 101093. [Google Scholar] [CrossRef]
- Cheng, A.; Choi, D.; Lora, M.; Shum-Tim, D.; Rak, J.; Colmegna, I. Human multipotent mesenchymal stromal cells cytokine priming promotes RAB27B-regulated secretion of small extracellular vesicles with immunomodulatory cargo. Stem Cell Res. Ther. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Zhang, J.; Hu, X.; Wang, Z.; Wu, S.; Yi, Y. Extracellular vesicles derived from human adipose-derived stem cells promote the exogenous angiogenesis of fat grafts via the let-7/AGO1/VEGF signalling pathway. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-García, L.; Castro-Manrreza, M.E. TNF-α and IFN-γ Participate in Improving the Immunoregulatory Capacity of Mesenchymal Stem/Stromal Cells: Importance of Cell–Cell Contact and Extracellular Vesicles. Int. J. Mol. Sci. 2021, 22, 9531. [Google Scholar] [CrossRef] [PubMed]
- Kou, P.M.; Babensee, J.E. Macrophage and dendritic cell phenotypic diversity in the context of biomaterials. J. Biomed. Mater. Res. Part. A 2010, 96, 239–260. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Böse, T.; Unger, R.E.; Jansen, J.A.; Kirkpatrick, C.J.; Beucken, J.J.J.P.V.D. Macrophage type modulates osteogenic differentiation of adipose tissue MSCs. Cell Tissue Res. 2017, 369, 273–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, B.N.; Londono, R.; Tottey, S.; Zhang, L.; Kukla, K.A.; Wolf, M.T.; Daly, K.A.; Reing, J.E.; Badylak, S.F. Macrophage phenotype as a predictor of constructive remodeling following the implantation of biologically derived surgical mesh materials. Acta Biomater. 2012, 8, 978–987. [Google Scholar] [CrossRef] [Green Version]
- Cho, D.I.; Kim, M.R.; Jeong, H.Y.; Jeong, H.C.; Jeong, M.H.; Yoon, S.H.; Kim, Y.S.; Ahn, Y. Mesenchymal stem cells reciprocally regulate the M1/M2 balance in mouse bone marrow-derived macrophage. Exp. Mol. Med. 2014, 46, e70. [Google Scholar] [CrossRef]
- Wiklander, O.P.B.; Brennan, M.Á.; Lötvall, J.; Breakefield, X.O.; El Andaloussi, S. Advances in therapeutic applications of extracellular vesicles. Sci. Transl. Med. 2019, 11, 492. [Google Scholar] [CrossRef]
- Zhang, B.; Tian, X.; Hao, J.; Xu, G.; Zhang, W. Mesenchymal Stem Cell-Derived Extracellular Vesicles in Tissue Regeneration. Cell Transplant. 2020, 29, 963689720908500. [Google Scholar] [CrossRef] [Green Version]
- Tour, G.; Wendel, M.; Tcacencu, I. Bone marrow stromal cells enhance the osteogenic properties of hydroxyapatite scaffolds by modulating the foreign body reaction. J. Tissue Eng. Regen. Med. 2014, 8, 841–849. [Google Scholar] [CrossRef]
- Gamblin, A.-L.; Brennan, M.Á.; Renaud, A.; Yagita, H.; Lézot, F.; Heymann, D.; Trichet, V.; Layrolle, P. Bone tissue formation with human mesenchymal stem cells and biphasic calcium phosphate ceramics: The local implication of osteoclasts and macrophages. Biomaterials 2014, 35, 9660–9667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuta, T.; Miyaki, S.; Ishitobi, H.; Ogura, T.; Kato, Y.; Kamei, N.; Miyado, K.; Higashi, Y.; Ochi, M. Mesenchymal Stem Cell-Derived Exosomes Promote Fracture Healing in a Mouse Model. STEM CELLS Transl. Med. 2016, 5, 1620–1630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drela, K.; Stanaszek, L.; Nowakowski, A.; Kuczynska, Z.; Lukomska, B. Experimental Strategies of Mesenchymal Stem Cell Propagation: Adverse Events and Potential Risk of Functional Changes. Stem Cells Int. 2019, 2019, 7012692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukomska, B.; Stanaszek, L.; Zuba-Surma, E.; Legosz, P.; Sarzynska, S.; Drela, K. Challenges and Controversies in Human Mesenchymal Stem Cell Therapy. Stem Cells Int. 2019, 2019, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordin, J.Z.; Lee, Y.; Vader, P.; Mäger, I.; Johansson, H.J.; Heusermann, W.; Wiklander, O.P.; Hällbrink, M.; Seow, Y.; Bultema, J.J.; et al. Ultrafiltration with size-exclusion liquid chromatography for high yield isolation of extracellular vesicles preserving intact biophysical and functional properties. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 879–883. [Google Scholar] [CrossRef] [Green Version]
- Mol, E.A.; Goumans, M.-J.; Doevendans, P.A.; Sluijter, J.P.G.; Vader, P. Higher functionality of extracellular vesicles isolated using size-exclusion chromatography compared to ultracentrifugation. Nanomedicine 2017, 13, 2061–2065. [Google Scholar] [CrossRef] [PubMed]
- Tkach, M.; Kowal, J.; Théry, C. Why the need and how to approach the functional diversity of extracellular vesicles. Philos. Trans. R. Soc. B: Biol. Sci. 2018, 373, 20160479. [Google Scholar] [CrossRef]
- Gardiner, C.; Di Vizio, D.; Sahoo, S.; Théry, C.; Witwer, K.W.; Wauben, M.; Hill, A.F. Techniques used for the isolation and characterization of extracellular vesicles: Results of a worldwide survey. J. Extracell. Vesicles 2016, 5, 32945. [Google Scholar] [CrossRef] [PubMed]
- Corso, G.; Mäger, I.; Lee, Y.; Görgens, A.; Bultema, J.; Giebel, B.; Wood, M.J.A.; Nordin, J.Z.; El Andaloussi, S. Reproducible and scalable purification of extracellular vesicles using combined bind-elute and size exclusion chromatography. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Xue, D.; Chen, E.; Zhong, H.; Zhang, W.; Wang, S.; Joomun, M.U.; Yao, T.; Tan, Y.; Lin, S.; Zheng, Q.; et al. Immunomodulatory properties of graphene oxide for osteogenesis and angiogenesis. Int. J. Nanomed. 2018, ume 13, 5799–5810. [Google Scholar] [CrossRef] [Green Version]
- Deng, M.; Tan, J.; Dai, Q.; Luo, F.; Xu, J. Macrophage-Mediated Bone Formation in Scaffolds Modified With MSC-Derived Extracellular Matrix Is Dependent on the Migration Inhibitory Factor Signaling Pathway. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Riddy, D.M.; Goy, E.; Delerive, P.; Summers, R.; Sexton, P.M.; Langmead, C.J. Comparative genotypic and phenotypic analysis of human peripheral blood monocytes and surrogate monocyte-like cell lines commonly used in metabolic disease research. PLoS ONE 2018, 13, e0197177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadtler, K.; Estrellas, K.; Allen, B.W.; Wolf, M.T.; Fan, H.; Tam, A.J.; Patel, C.H.; Luber, B.S.; Wang, H.; Wagner, K.R.; et al. Developing a pro-regenerative biomaterial scaffold microenvironment requires T helper 2 cells. Science 2016, 352, 366–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, M.C.; Gantzel, R.H.; Clària, J.; Trebicka, J.; Møller, H.J.; Grønbæk, H. Macrophage Activation Markers, CD163 and CD206, in Acute-on-Chronic Liver Failure. Cells 2020, 9, 1175. [Google Scholar] [CrossRef] [PubMed]
- Ono, T.; Okamoto, K.; Nakashima, T.; Nitta, T.; Hori, S.; Iwakura, Y.; Takayanagi, H. IL-17-producing γδ T cells enhance bone regeneration. Nat. Commun. 2016, 7, 10928. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abbreviation | Cytokine |
|---|---|
| bFGF | Basic fibroblast growth factor |
| Eotaxin/CCL11 | C-C chemokine 11 |
| GCSF | Granulocyte colony stimulating factor |
| GMCSF | Granulocyte-macrophage colony-stimulating factor |
| IFNγ | Interferon-γ |
| IL1b | Interleukin-1β |
| IL1ra | Interleukin 1 receptor antagonist |
| IL2 | Interleukin-2 |
| IL4 | Interleukin-4 |
| IL5 | Interleukin-5 |
| IL6 | Interleukin-6 |
| IL7 | Interleukin-7 |
| IL8 | Interleukin-8 |
| IL9 | Interleukin-9 |
| IL10 | Interleukin-10 |
| IL12 (p70) | Interleukin-12 |
| IL13 | Interleukin-13 |
| IL15 | Interleukin-15 |
| IL17A | Interleukin-17 |
| IP10/CXCL10 | Interferon gamma-induced protein 10/CXC chemokine 10 |
| MCP1/CCL2 | Monocyte Chemoattractant Protein-1 |
| MIP1α/CCL3 | Macrophage inflammatory protein |
| MIP1β/CCL4 | Macrophage inflammatory protein |
| PDGFBB | Platelet-derived growth factor-BB |
| RANTES/CCL5 | Regulated on activation, normal T cell expressed and secreted |
| TNFα | Tumour Necrosis Factor-α |
| VEGF | Vascular endothelial growth factor |
| Gene ID | Gene Name | TaqMan® Assay ID | Amplicon Length |
|---|---|---|---|
| GAPDH | Glyceraldehyde 3-Phosphate Dehydrogenase | Hs99999905_m1 | 122 |
| IL12A | Interleukin 12A | Hs00168405_m1 | 67 |
| TNFα | Tumor Necrosis Factor alpha | Hs00174128_m1 | 80 |
| IL1β | Interleukin 1 beta | Hs01555410_m1 | 91 |
| CD206/MRC1 | Mannose Receptor, C type 1 | Hs00168405_m1 | 82 |
| IL10 | Interleukin 10 | Hs00961622_m1 | 74 |
| TGFβ1 | Transforming Growth Factor, beta 1 | Hs00998133_m1 | 57 |
| MCP1R/CCR2 | Monocyte Chemoattractant Protein 1 Receptor | Hs00704702_s1 | 61 |
| IDO | Indoleamine 2,3-Dioxygenase 1 | Hs00984148_m1 | 66 |
| VEGFA | Vascular Endothelial Growth Factor A | Hs00900055_m1 | 59 |
| IL6 | Interleukin 6 | Hs00985641_m1 | 89 |
| Cytokine Concentrations: (pg/µg) | MSC:EV | MSC + BCP:EV | MSCp + BCP:EV | |||
|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | Mean | SD | |
| Pro-inflammatory cytokines | ||||||
| IL1b | 0.00752 | - | 0.00121 | - | 0.02624 | 0.00439 |
| IL2 | 0.06248 | 0.00126 | 0.00335 | 0.00157 | 0.00743 | - |
| IL8 | 0.02300 | 0.02501 | 0.00666 | 0.00142 | 0.19358 | 0.05257 |
| IL12(p70) | 0.00208 | 0.00060 | 0.00365 | 0.00012 | 0.00425 | 0.00338 |
| IL17 | 0.00226 | 0.00226 | 0.02007 | 0.00172 | 0.04048 | 0.0126 |
| TNFα | 0.88730 | 0.32121 | 0.01065 | 0.00877 | 1.28098 | 0.03592 |
| IFNγ | 0.01602 | 0.00651 | 0.01782 | 0.00559 | 0.03505 | 0.01117 |
| Anti-inflammatory cytokines | ||||||
| IL1ra | 0.11891 | 0.00594 | 0.11318 | 0.00410 | 0.10508 | 0.03924 |
| IL4 | 0.00101 | 0.00102 | 0.00027 | 0.00013 | 0.00399 | 0.00134 |
| IL5 | 0.56833 | 0.52276 | 1.14836 | 0.70858 | 3.07281 | 0.69376 |
| 1L10 | 0.01150 | 0.00031 | 0.03547 | 0.00055 | 0.01774 | 0.00871 |
| 1L13 | 0.00112 | 0.00046 | 0.00132 | 0.00031 | 0.00183 | 0.00117 |
| Wound healing and repair cytokines | ||||||
| FGF basic | 0.04181 | - | 0.02819 | 0.00560 | 0.07910 | 0.00835 |
| GCSF | 0.38614 | 0.01427 | 0.21457 | 0.02511 | 0.41665 | 0.01024 |
| GMCSF | 0.03895 | 0.04553 | 0.00755 | 0.00433 | 0.30745 | 0.03843 |
| PDGFbb | 0.59094 | 0.53008 | 1.24396 | 0.24791 | 1.28193 | 0.05983 |
| VEGF | 10.0391 | 2.40937 | 7.77386 | 1.8794 | 18.7309 | 0.30316 |
| Chemokines | ||||||
| Eotaxin | 0.01515 | - | 0.00225 | 0.00054 | 0.02109 | 0.00445 |
| IP10 | 0.13681 | - | 0.16551 | 0.00610 | 0.30435 | 0.10212 |
| MCP1 | 0.03561 | 0.03995 | 0.00704 | 0.00024 | 0.10736 | 0.00844 |
| MIP1b | 0.19382 | - | 0.00562 | 0.00124 | 0.29157 | 0.04188 |
| RANTES | 0.05952 | - | 0.12446 | 0.02725 | 0.11663 | 0.02002 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rana, N.; Suliman, S.; Al-Sharabi, N.; Mustafa, K. Extracellular Vesicles Derived from Primed Mesenchymal Stromal Cells Loaded on Biphasic Calcium Phosphate Biomaterial Exhibit Enhanced Macrophage Polarization. Cells 2022, 11, 470. https://doi.org/10.3390/cells11030470
Rana N, Suliman S, Al-Sharabi N, Mustafa K. Extracellular Vesicles Derived from Primed Mesenchymal Stromal Cells Loaded on Biphasic Calcium Phosphate Biomaterial Exhibit Enhanced Macrophage Polarization. Cells. 2022; 11(3):470. https://doi.org/10.3390/cells11030470
Chicago/Turabian StyleRana, Neha, Salwa Suliman, Niyaz Al-Sharabi, and Kamal Mustafa. 2022. "Extracellular Vesicles Derived from Primed Mesenchymal Stromal Cells Loaded on Biphasic Calcium Phosphate Biomaterial Exhibit Enhanced Macrophage Polarization" Cells 11, no. 3: 470. https://doi.org/10.3390/cells11030470