
Dictyostelium discoideum: A Model System for Neurological Disorders

 , , and
, , and
Abstract
:1. Introduction
2. Alzheimer’s Disease (AD)
3. Parkinson’s Disease (PD)
3.1. Leucine Rich Repeat Kinase 2 (LRRK2)
3.2. HTRA2 or Omi Protease
3.3. DJ-1
3.4. Alpha-Synuclein
4. Huntington’s Disease
5. Neuronal Ceroid Lipofuscinoses
5.1. CLN1/PPT1
5.2. CLN2/TPP1
5.3. CLN3
5.4. CLN4/DNAJC5
5.5. CLN5
5.6. CLN7/MFSD8
5.7. CLN10/CTSD
5.8. CLN11/GRN
5.9. CLN12/Park9
5.10. CLN13/CTSF
5.11. CLN14/KCTD7
6. Lissencephaly
7. D. discoideum as a Pharmacological Model for Neurological Disorders
8. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Feigin, V.L.; Vos, T. Global Burden of Neurological Disorders: From Global Burden of Disease Estimates to Actions. Neuroepidemiology 2019, 52, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Annesley, S.J.; Chen, S.; Francione, L.M.; Sanislav, O.; Chavan, A.J.; Farah, C.; De Piazza, S.W.; Storey, C.L.; Ilievska, J.; Fernando, S.G.; et al. Dictyostelium, a microbial model for brain disease. BBA-Gen. Subj. 2014, 1840, 20. [Google Scholar] [CrossRef]
- Annesley, S.J.; Fisher, P.R. Lymphoblastoid Cell Lines as Models to Study Mitochondrial Function in Neurological Disorders. Int. J. Mol. Sci. 2021, 22, 4536. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, K.A.; Willicott, C.W.; Caldwell, G.A. Caldwell. Modeling neurodegeneration in Caenorhabditis elegans. Dis. Model. Mech. 2020, 13, dmm046110. [Google Scholar] [CrossRef] [PubMed]
- Cauchi, R.J.; van den Heuvel, M. The fly as a model for neurodegenerative diseases: Is it worth the jump? Neurodegener. Dis. 2006, 3, 338–356. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, J.B.; He, K.J.; Wang, F.; Liu, C.F. Advances of Zebrafish in Neurodegenerative Disease: From Models to Drug Discovery. Front. Pharm. 2021, 12, 713963. [Google Scholar] [CrossRef] [PubMed]
- Surguchov, A. Invertebrate Models Untangle the Mechanism of Neurodegeneration in Parkinson’s Disease. Cells 2021, 10, 407. [Google Scholar] [CrossRef]
- Huber, R.J.; Hughes, S.M.; Liu, W.; Morgan, A.; Tuxworth, R.I.; Russell, C. The contribution of multicellular model organisms to neuronal ceroid lipofuscinosis research. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165614. [Google Scholar] [CrossRef]
- Martin-Gonzalez, J.; Montero-Bullon, J.F.; Lacal, J. Dictyostelium discoideum as a non-mammalian biomedical model. Microb. Biotechnol. 2021, 14, 111–125. [Google Scholar] [CrossRef]
- Ishikawa-Ankerhold, H.C.; Muller-Taubenberger, A. Actin assembly states in Dictyostelium discoideum at different stages of development and during cellular stress. Int. J. Dev. Biol. 2019, 63, 417–427. [Google Scholar] [CrossRef] [Green Version]
- Williams, R.S.; Boeckeler, K.; Graf, R.; Muller-Taubenberger, A.; Li, Z.; Isberg, R.R.; Wessels, D.; Soll, D.R.; Alexander, H.; Alexander, S. Towards a molecular understanding of human diseases using Dictyostelium discoideum. Trends Mol. Med. 2006, 12, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Eichinger, L.; Pachebat, J.A.; Glockner, G.; Rajandream, M.A.; Sucgang, R.; Berriman, M.; Song, J.; Olsen, R.; Szafranski, K.; Xu, Q.; et al. The genome of the social amoeba Dictyostelium discoideum. Nature 2005, 435, 43–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernando, S.; Allan, C.Y.; Mroczek, K.; Pearce, X.; Sanislav, O.; Fisher, P.R.; Annesley, S.J. Cytotoxicity and Mitochondrial Dysregulation Caused by alpha-Synuclein in Dictyostelium discoideum. Cells 2020, 9, 2289. [Google Scholar] [CrossRef] [PubMed]
- Mroczek, K.; Fernando, S.; Fisher, P.R.; Annesley, S.J. Interactions and Cytotoxicity of Human Neurodegeneration-Associated Proteins Tau and α-Synuclein in the Simple Model Dictyostelium discoideum. Front. Cell Dev. Biol. 2021, 9, 2502. [Google Scholar] [CrossRef]
- McMains, V.C.; Myre, M.; Kreppel, L.; Kimmel, A.R. Dictyostelium possesses highly diverged presenilin/gamma-secretase that regulates growth and cell-fate specification and can accurately process human APP: A system for functional studies of the presenilin/gamma-secretase complex. Dis. Model. Mech. 2010, 3, 581–594. [Google Scholar] [CrossRef] [Green Version]
- Pearce, X.G.; Annesley, S.J.; Fisher, P.R. The Dictyostelium model for mitochondrial biology and disease. Int. J. Dev. Biol. 2019, 63, 497–508. [Google Scholar] [CrossRef] [Green Version]
- Smith, P.K.; Sen, M.G.; Fisher, P.R.; Annesley, S.J. Modelling of Neuronal Ceroid Lipofuscinosis Type 2 in Dictyostelium discoideum Suggests That Cytopathological Outcomes Result from Altered TOR Signalling. Cells 2019, 8, 469. [Google Scholar] [CrossRef] [Green Version]
- Annesley, S.J.; Fisher, P.R. Dictyostelium discoideum--a model for many reasons. Mol. Cell. Biochem. 2009, 329, 73–91. [Google Scholar] [CrossRef]
- Kelly, E.; Sharma, D.; Wilkinson, C.J.; Williams, R.S.B. Diacylglycerol kinase (DGKA) regulates the effect of the epilepsy and bipolar disorder treatment valproic acid in Dictyostelium discoideum. Dis. Model. Mech. 2018, 11, dmm035600. [Google Scholar] [CrossRef] [Green Version]
- King, J.S.; Teo, R.; Ryves, J.; Reddy, J.V.; Peters, O.; Orabi, B.; Hoeller, O.; Williams, R.S.; Harwood, A.J. The mood stabiliser lithium suppresses PIP3 signalling in Dictyostelium and human cells. Dis. Model. Mech. 2009, 2, 306–312. [Google Scholar] [CrossRef] [Green Version]
- Chang, P.; Orabi, B.; Deranieh, R.M.; Dham, M.; Hoeller, O.; Shimshoni, J.A.; Yagen, B.; Bialer, M.; Greenberg, M.L.; Walker, M.C.; et al. The antiepileptic drug valproic acid and other medium-chain fatty acids acutely reduce phosphoinositide levels independently of inositol in Dictyostelium. Dis. Model. Mech. 2012, 5, 115–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alzheimer’s Association. Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2020, 16, 391–460. [Google Scholar]
- Matthews, K.A.; Xu, W.; Gaglioti, A.H.; Holt, J.B.; Croft, J.B.; Mack, D.; McGuire, L.C. Racial and ethnic estimates of Alzheimer’s disease and related dementias in the United States (2015-2060) in adults aged >/=65 years. Alzheimer’s Dement. 2019, 15, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Trejo-Lopez, J.A.; Yachnis, A.T.; Prokop, S. Neuropathology of Alzheimer’s Disease. Neurotherapeutics 2021, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Browne, A.; Kim, D.Y.; Tanzi, R.E. Familial Alzheimer’s disease mutations in presenilin 1 do not alter levels of the secreted amyloid-beta protein precursor generated by beta-secretase cleavage. Curr. Alzheimer’s Res. 2010, 7, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Tam, C.; Wong, J.H.; Ng, T.B.; Tsui, S.K.W.; Zuo, T. Drugs for Targeted Therapies of Alzheimer’s Disease. Curr. Med. Chem. 2019, 26, 335–359. [Google Scholar] [CrossRef] [PubMed]
- Ludtmann, M.H.; Otto, G.P.; Schilde, C.; Chen, Z.H.; Allan, C.Y.; Brace, S.; Beesley, P.W.; Kimmel, A.R.; Fisher, P.; Killick, R.; et al. An ancestral non-proteolytic role for presenilin proteins in multicellular development of the social amoeba Dictyostelium discoideum. J. Cell Sci. 2014, 127, 1576–1584. [Google Scholar] [CrossRef] [Green Version]
- Sharma, D.; Otto, G.; Warren, E.C.; Beesley, P.; King, J.S.; Williams, R.S.B. Gamma secretase orthologs are required for lysosomal activity and autophagic degradation in Dictyostelium discoideum, independent of PSEN (presenilin) proteolytic function. Autophagy 2019, 15, 1407–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otto, G.P.; Sharma, D.; Williams, R.S. Non-Catalytic Roles of Presenilin Throughout Evolution. J. Alzheimers Dis. 2016, 52, 1177–1187. [Google Scholar] [CrossRef] [Green Version]
- Walter, J.; Capell, A.; Grunberg, J.; Pesold, B.; Schindzielorz, A.; Prior, R.; Podlisny, M.B.; Fraser, P.; Hyslop, P.S.; Selkoe, D.J.; et al. The Alzheimer’s disease-associated presenilins are differentially phosphorylated proteins located predominantly within the endoplasmic reticulum. Mol. Med. 1996, 2, 673–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Haapasalo, A.; Kim, D.Y.; Ingano, L.A.; Pettingell, W.H.; Kovacs, D.M. Presenilin/gamma-secretase activity regulates protein clearance from the endocytic recycling compartment. FASEB J. 2006, 20, 1176–1178. [Google Scholar] [CrossRef] [PubMed]
- Tamboli, I.Y.; Prager, K.; Thal, D.R.; Thelen, K.M.; Dewachter, I.; Pietrzik, C.U.; St George-Hyslop, P.; Sisodia, S.S.; De Strooper, B.; Heneka, M.T.; et al. Loss of gamma-secretase function impairs endocytosis of lipoprotein particles and membrane cholesterol homeostasis. J. Neurosci. 2008, 28, 12097–12106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 2010, 141, 1146–1158. [Google Scholar] [CrossRef] [Green Version]
- Maiti, P.; Manna, J.; Dunbar, G.L. Current understanding of the molecular mechanisms in Parkinson’s disease: Targets for potential treatments. Transl. Neurodegener. 2017, 6, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Le, W. Profiling Non-motor Symptoms in Monogenic Parkinson’s Disease. Front. Aging Neurosci. 2020, 12, 591183. [Google Scholar] [CrossRef]
- Chartier, S.; Duyckaerts, C. Is Lewy pathology in the human nervous system chiefly an indicator of neuronal protection or of toxicity? Cell Tissue Res. 2018, 373, 149–160. [Google Scholar] [CrossRef]
- Nguyen, M.; Wong, Y.C.; Ysselstein, D.; Severino, A.; Krainc, D. Synaptic, Mitochondrial, and Lysosomal Dysfunction in Parkinson’s Disease. Trends Neurosci. 2019, 42, 140–149. [Google Scholar] [CrossRef]
- Van Egmond, W.N.; van Haastert, P.J. Characterization of the Roco protein family in Dictyostelium discoideum. Eukaryot. Cell 2010, 9, 751–761. [Google Scholar] [CrossRef] [Green Version]
- Kicka, S.; Shen, Z.; Annesley, S.J.; Fisher, P.R.; Lee, S.; Briggs, S.; Firtel, R.A. The LRRK2-related Roco kinase Roco2 is regulated by Rab1A and controls the actin cytoskeleton. Mol. Biol. Cell 2011, 22, 2198–2211. [Google Scholar] [CrossRef]
- Abysalh, J.C.; Kuchnicki, L.L.; Larochelle, D.A. The identification of pats1, a novel gene locus required for cytokinesis in Dictyostelium discoideum. Mol. Biol. Cell 2003, 14, 14–25. [Google Scholar] [CrossRef] [Green Version]
- Abe, T.; Langenick, J.; Williams, J.G. Rapid generation of gene disruption constructs by in vitro transposition and identification of a Dictyostelium protein kinase that regulates its rate of growth and development. Nucleic Acids Res. 2003, 31, e107. [Google Scholar] [CrossRef] [Green Version]
- Gilsbach, B.K.; Ho, F.Y.; Vetter, I.R.; van Haastert, P.J.; Wittinghofer, A.; Kortholt, A. Roco kinase structures give insights into the mechanism of Parkinson disease-related leucine-rich-repeat kinase 2 mutations. Proc. Natl. Acad. Sci. USA 2012, 109, 10322–10327. [Google Scholar] [CrossRef] [Green Version]
- Rosenbusch, K.E.; Oun, A.; Sanislav, O.; Lay, S.T.; Keizer-Gunnink, I.; Annesley, S.J.; Fisher, P.R.; Dolga, A.M.; Kortholt, A. A Conserved Role for LRRK2 and Roco Proteins in the Regulation of Mitochondrial Activity. Front. Cell Dev. Biol. 2021, 9, 734554. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Sanislav, O.; Annesley, S.J.; Fisher, P.R. Mitochondrial HTRA2 Plays a Positive, Protective Role in Dictyostelium discoideum but Is Cytotoxic When Overexpressed. Genes 2018, 9, 355. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Annesley, S.J.; Jasim, R.A.F.; Musco, V.J.; Sanislav, O.; Fisher, P.R. The Parkinson’s disease-associated protein DJ-1 plays a positive nonmitochondrial role in endocytosis in Dictyostelium cells. Dis. Model. Mech. 2017, 10, 1261–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Annesley, S.J.; Jasim, R.A.F.; Fisher, P.R. The Parkinson’s Disease-Associated Protein DJ-1 Protects Dictyostelium Cells from AMPK-Dependent Outcomes of Oxidative Stress. Cells 2021, 10, 1874. [Google Scholar] [CrossRef] [PubMed]
- Blauwendraat, C.; Nalls, M.A.; Singleton, A.B. The genetic architecture of Parkinson’s disease. Lancet Neurol. 2020, 19, 170–178. [Google Scholar] [CrossRef]
- Goldberg, J.M.; Bosgraaf, L.; Van Haastert, P.J.; Smith, J.L. Identification of four candidate cGMP targets in Dictyostelium. Proc. Natl. Acad. Sci. USA 2002, 99, 6749–6754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolosa, E.; Vila, M.; Klein, C.; Rascol, O. LRRK2 in Parkinson disease: Challenges of clinical trials. Nat. Rev. Neurol. 2020, 16, 97–107. [Google Scholar] [CrossRef]
- Annesley, S.J.; Carilla-Latorre, S.; Escalante, R.; Fisher, P.R. Mitochondrial respiratory complex function and the phenotypic consequences of dysfunction. Methods Mol. Biol. 2013, 983, 345–366. [Google Scholar] [CrossRef]
- Bokko, P.B.; Francione, L.; Bandala-Sanchez, E.; Ahmed, A.U.; Annesley, S.J.; Huang, X.; Khurana, T.; Kimmel, A.R.; Fisher, P.R. Diverse cytopathologies in mitochondrial disease are caused by AMP-activated protein kinase signaling. Mol. Biol. Cell 2007, 18, 1874–1886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, K.; Wang, Y.; Guo, D.; Wang, G.; Ren, H. Familial Parkinson’s Disease-Associated L166P Mutant DJ-1 is Cleaved by Mitochondrial Serine Protease Omi/HtrA2. Neurosci. Bull. 2017, 33, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Plun-Favreau, H.; Klupsch, K.; Moisoi, N.; Gandhi, S.; Kjaer, S.; Frith, D.; Harvey, K.; Deas, E.; Harvey, R.J.; McDonald, N.; et al. The mitochondrial protease HtrA2 is regulated by Parkinson’s disease-associated kinase PINK1. Nat. Cell Biol. 2007, 9, 1243–1252. [Google Scholar] [CrossRef]
- Fitzgerald, J.C.; Camprubi, M.D.; Dunn, L.; Wu, H.C.; Ip, N.Y.; Kruger, R.; Martins, L.M.; Wood, N.W.; Plun-Favreau, H. Phosphorylation of HtrA2 by cyclin-dependent kinase-5 is important for mitochondrial function. Cell Death Differ. 2012, 19, 257–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyama, Y.; Harkness, R.W.; Lee, T.Y.T.; Maynes, J.T.; Kay, L.E. Oligomeric assembly regulating mitochondrial HtrA2 function as examined by methyl-TROSY NMR. Proc. Natl. Acad. Sci. USA 2021, 118, e2025022118. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.F.; Zhang, X.W.; Nie, L.L.; Zhang, H.N.; Liao, B.; Li, J.; Wang, L.; Yan, X.X.; Tang, B.S. Mutation analysis of Parkin, PINK1 and DJ-1 genes in Chinese patients with sporadic early onset parkinsonism. J. Neurol. 2010, 257, 1170–1175. [Google Scholar] [CrossRef] [PubMed]
- Repici, M.; Giorgini, F. DJ-1 in Parkinson’s Disease: Clinical Insights and Therapeutic Perspectives. J. Clin. Med. 2019, 8, 1377. [Google Scholar] [CrossRef] [Green Version]
- Mencke, P.; Boussaad, I.; Romano, C.D.; Kitami, T.; Linster, C.L.; Kruger, R. The Role of DJ-1 in Cellular Metabolism and Pathophysiological Implications for Parkinson’s Disease. Cells 2021, 10, 347. [Google Scholar] [CrossRef]
- Van der Merwe, C.; Jalali Sefid Dashti, Z.; Christoffels, A.; Loos, B.; Bardien, S. Evidence for a common biological pathway linking three Parkinson’s disease-causing genes: Parkin, PINK1 and DJ-1. Eur. J. Neurosci. 2015, 41, 1113–1125. [Google Scholar] [CrossRef]
- Kumar, R.; Kumar, S.; Hanpude, P.; Singh, A.K.; Johari, T.; Majumder, S.; Maiti, T.K. Partially oxidized DJ-1 inhibits alpha-synuclein nucleation and remodels mature alpha-synuclein fibrils in vitro. Commun. Biol. 2019, 2, 395. [Google Scholar] [CrossRef]
- Kojima, W.; Kujuro, Y.; Okatsu, K.; Bruno, Q.; Koyano, F.; Kimura, M.; Yamano, K.; Tanaka, K.; Matsuda, N. Unexpected mitochondrial matrix localization of Parkinson’s disease-related DJ-1 mutants but not wild-type DJ-1. Genes Cells 2016, 21, 772–788. [Google Scholar] [CrossRef] [PubMed]
- Kyung, J.W.; Kim, J.M.; Lee, W.; Ha, T.Y.; Cha, S.H.; Chung, K.H.; Choi, D.J.; Jou, I.; Song, W.K.; Joe, E.H.; et al. DJ-1 deficiency impairs synaptic vesicle endocytosis and reavailability at nerve terminals. Proc. Natl. Acad. Sci. USA 2018, 115, 1629–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.S.; Kim, J.S.; Park, J.Y.; Suh, Y.H.; Jou, I.; Joe, E.H.; Park, S.M. DJ-1 associates with lipid rafts by palmitoylation and regulates lipid rafts-dependent endocytosis in astrocytes. Hum. Mol. Genet. 2013, 22, 4805–4817. [Google Scholar] [CrossRef] [Green Version]
- Vines, J.H.; King, J.S. The endocytic pathways of Dictyostelium discoideum. Int. J. Dev. Biol. 2019, 63, 461–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annesley, S.J.; Lay, S.T.; De Piazza, S.W.; Sanislav, O.; Hammersley, E.; Allan, C.Y.; Francione, L.M.; Bui, M.Q.; Chen, Z.P.; Ngoei, K.R.; et al. Immortalized Parkinson’s disease lymphocytes have enhanced mitochondrial respiratory activity. Dis. Model. Mech. 2016, 9, 1295–1305. [Google Scholar] [CrossRef] [Green Version]
- Haylett, W.; Swart, C.; van der Westhuizen, F.; van Dyk, H.; van der Merwe, L.; van der Merwe, C.; Loos, B.; Carr, J.; Kinnear, C.; Bardien, S. Altered Mitochondrial Respiration and Other Features of Mitochondrial Function in Parkin-Mutant Fibroblasts from Parkinson’s Disease Patients. Parkinsons Dis. 2016, 2016, 1819209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ugalde, C.L.; Annesley, S.J.; Gordon, S.E.; Mroczek, K.; Perugini, M.A.; Lawson, V.A.; Fisher, P.R.; Finkelstein, D.I.; Hill, A.F. Misfolded alpha-synuclein causes hyperactive respiration without functional deficit in live neuroblastoma cells. Dis. Model. Mech. 2020, 13, dmm040899. [Google Scholar] [CrossRef] [Green Version]
- Francione, L.; Smith, P.K.; Accari, S.L.; Taylor, P.E.; Bokko, P.B.; Bozzaro, S.; Beech, P.L.; Fisher, P.R. Legionella pneumophila multiplication is enhanced by chronic AMPK signalling in mitochondrially diseased Dictyostelium cells. Dis. Model. Mech. 2009, 2, 479–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.T.; Donzelli, S.; Chiki, A.; Syed, M.M.K.; Lashuel, H.A. A simple, versatile and robust centrifugation-based filtration protocol for the isolation and quantification of alpha-synuclein monomers, oligomers and fibrils: Towards improving experimental reproducibility in alpha-synuclein research. J. NeuroChem. 2020, 153, 103–119. [Google Scholar] [CrossRef]
- Tateno, F.; Sakakibara, R.; Kawai, T.; Kishi, M.; Murano, T. Alpha-synuclein in the cerebrospinal fluid differentiates synucleinopathies (Parkinson Disease, dementia with Lewy bodies, multiple system atrophy) from Alzheimer disease. Alzheimers Dis. Assoc. Disord. 2012, 26, 213–216. [Google Scholar] [CrossRef]
- Sorrentino, Z.A.; Giasson, B.I. The emerging role of alpha-synuclein truncation in aggregation and disease. J. Biol. Chem. 2020, 295, 10224–10244. [Google Scholar] [CrossRef]
- Guardia-Laguarta, C.; Area-Gomez, E.; Schon, E.A.; Przedborski, S. Novel subcellular localization for α-synuclein: Possible functional consequences. Front. Neuroanat. 2015, 9, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardiner, M.; Sandford, A.; Deadman, M.; Poulton, J.; Cookson, W.; Reeders, S.; Jokiaho, I.; Peltonen, L.; Eiberg, H.; Julier, C. Batten disease (Spielmeyer-Vogt disease, juvenile onset neuronal ceroid-lipofuscinosis) gene (CLN3) maps to human chromosome 16. Genomics 1990, 8, 387–390. [Google Scholar] [CrossRef]
- Bartels, T.; Ahlstrom, L.S.; Leftin, A.; Kamp, F.; Haass, C.; Brown, M.F.; Beyer, K. The N-terminus of the intrinsically disordered protein alpha-synuclein triggers membrane binding and helix folding. Biophys. J. 2010, 99, 2116–2124. [Google Scholar] [CrossRef] [Green Version]
- Lindwall, G.; Cole, R.D. Phosphorylation affects the ability of tau protein to promote microtubule assembly. J. Biol. Chem. 1984, 259, 5301–5305. [Google Scholar] [CrossRef]
- Kim, J.; Yang, G.; Kim, Y.; Kim, J.; Ha, J. AMPK activators: Mechanisms of action and physiological activities. Exp. Mol. Med. 2016, 48, e224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blumenstock, S.; Dudanova, I. Cortical and Striatal Circuits in Huntington’s Disease. Front. Neurosci. 2020, 14, 82. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, E.; Zuccato, C.; Tartari, M. Normal huntingtin function: An alternative approach to Huntington’s disease. Nat. Rev. Neurosci. 2005, 6, 919–930. [Google Scholar] [CrossRef]
- Myre, M.A.; Lumsden, A.L.; Thompson, M.N.; Wasco, W.; MacDonald, M.E.; Gusella, J.F. Deficiency of huntingtin has pleiotropic effects in the social amoeba Dictyostelium discoideum. PLoS Genet. 2011, 7, e1002052. [Google Scholar] [CrossRef] [Green Version]
- Santarriaga, S.; Petersen, A.; Ndukwe, K.; Brandt, A.; Gerges, N.; Bruns Scaglione, J.; Scaglione, K.M. The Social Amoeba Dictyostelium discoideum Is Highly Resistant to Polyglutamine Aggregation. J. Biol. Chem. 2015, 290, 25571–25578. [Google Scholar] [CrossRef] [Green Version]
- Bhadoriya, P.; Jain, M.; Kaicker, G.; Saidullah, B.; Saran, S. Deletion of Htt cause alterations in cAMP signaling and spatial patterning in Dictyostelium discoideum. J. Cell. Physiol. 2019, 234, 18858–18871. [Google Scholar] [CrossRef] [PubMed]
- Myre, M.A. Clues to gamma-secretase, huntingtin and Hirano body normal function using the model organism Dictyostelium discoideum. J. Biomed. Sci. 2012, 19, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeitlin, S.; Liu, J.P.; Chapman, D.L.; Papaioannou, V.E.; Efstratiadis, A. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington’s disease gene homologue. Nat. Genet. 1995, 11, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.N.; MacDonald, M.E.; Gusella, J.F.; Myre, M.A. Huntingtin Supplies a csaA-Independent Function Essential for EDTA-Resistant Homotypic Cell Adhesion in Dictyostelium discoideum. J. Huntingt. Dis. 2014, 3, 261–271. [Google Scholar] [CrossRef]
- Wang, Y.; Steimle, P.A.; Ren, Y.; Ross, C.A.; Robinson, D.N.; Egelhoff, T.T.; Sesaki, H.; Iijima, M. Dictyostelium huntingtin controls chemotaxis and cytokinesis through the regulation of myosin II phosphorylation. Mol. Biol. Cell 2011, 22, 2270–2281. [Google Scholar] [CrossRef]
- Johnson, T.B.; Cain, J.T.; White, K.A.; Ramirez-Montealegre, D.; Pearce, D.A.; Weimer, J.M. Therapeutic landscape for Batten disease: Current treatments and future prospects. Nat. Rev. Neurol. 2019, 15, 161–178. [Google Scholar] [CrossRef]
- Mole, S.E.; Schulz, A.; Haltia, M. The neuronal ceroid-lipofuscinoses (batten disease). In Rosenberg’s Molecular and Genetic Basis of Neurological and Psychiatric Disease; Elsevier: Amsterdam, The Netherlands, 2020; pp. 53–71. [Google Scholar]
- Kohlschutter, A.; Schulz, A.; Bartsch, U.; Storch, S. Current and Emerging Treatment Strategies for Neuronal Ceroid Lipofuscinoses. CNS Drugs 2019, 33, 315–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mole, S.E.; Cotman, S.L. Genetics of the neuronal ceroid lipofuscinoses (Batten disease). Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2015, 1852, 2237–2241. [Google Scholar] [CrossRef] [Green Version]
- Carcel-Trullols, J.; Kovacs, A.D.; Pearce, D.A. Cell biology of the NCL proteins: What they do and don’t do. Biochim. Biophys. Acta 2015, 1852, 2242–2255. [Google Scholar] [CrossRef] [Green Version]
- Huber, R.J. Using the social amoeba Dictyostelium to study the functions of proteins linked to neuronal ceroid lipofuscinosis. J. Biomed. Sci. 2016, 23, 83. [Google Scholar] [CrossRef] [Green Version]
- Huber, R.J.; Myre, M.A.; Cotman, S.L. Loss of Cln3 function in the social amoeba Dictyostelium discoideum causes pleiotropic effects that are rescued by human CLN3. PLoS ONE 2014, 9, e110544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakthavatsalam, D.; Gomer, R.H. The secreted proteome profile of developing Dictyostelium discoideum cells. Proteomics 2010, 10, 2556–2559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Journet, A.; Klein, G.; Brugière, S.; Vandenbrouck, Y.; Chapel, A.; Kieffer, S.; Bruley, C.; Masselon, C.; Aubry, L. Investigating the macropinocytic proteome of Dictyostelium amoebae by high-resolution mass spectrometry. Proteomics 2012, 12, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Sillo, A.; Bloomfield, G.; Balest, A.; Balbo, A.; Pergolizzi, B.; Peracino, B.; Skelton, J.; Ivens, A.; Bozzaro, S. Genome-wide transcriptional changes induced by phagocytosis or growth on bacteria in Dictyostelium. BMC Genom. 2008, 9, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, J.E.; Gomer, R.H. Partial genetic suppression of a loss-of-function mutant of the neuronal ceroid lipofuscinosis-associated protease TPP1 in Dictyostelium discoideum. Dis. Model. Mech. 2015, 8, 147–156. [Google Scholar] [CrossRef] [Green Version]
- Huber, R.J. Molecular networking in the neuronal ceroid lipofuscinoses: Insights from mammalian models and the social amoeba Dictyostelium discoideum. J. Biomed. Sci. 2020, 27, 1–16. [Google Scholar]
- Stumpf, M.; Müller, R.; Gaßen, B.; Wehrstedt, R.; Fey, P.; Karow, M.A.; Eichinger, L.; Glöckner, G.; Noegel, A.A. A tripeptidyl peptidase 1 is a binding partner of the Golgi pH regulator (GPHR) in Dictyostelium. Dis. Model. Mech. 2017, 10, 897–907. [Google Scholar] [CrossRef] [Green Version]
- Carilla-Latorre, S.; Calvo-Garrido, J.; Bloomfield, G.; Skelton, J.; Kay, R.R.; Ivens, A.; Martinez, J.L.; Escalante, R. Dictyostelium transcriptional responses to Pseudomonas aeruginosa: Common and specific effects from PAO1 and PA14 strains. BMC Microbiol. 2008, 8, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Mathavarajah, S.; McLaren, M.D.; Huber, R.J. Cln3 function is linked to osmoregulation in a Dictyostelium model of Batten disease. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3559–3573. [Google Scholar] [CrossRef]
- Huber, R.J.; Mathavarajah, S. Cln5 is secreted and functions as a glycoside hydrolase in Dictyostelium. Cell. Signal. 2018, 42, 236–248. [Google Scholar] [CrossRef]
- Huber, R.J.; Mathavarajah, S. Comparative transcriptomics reveals mechanisms underlying cln3-deficiency phenotypes in Dictyostelium. Cell. Signal. 2019, 58, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Huber, R.J. Loss of Cln3 impacts protein secretion in the social amoeba Dictyostelium. Cell. Signal. 2017, 35, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Huber, R.J.; Myre, M.A.; Cotman, S.L. Aberrant adhesion impacts early development in a Dictyostelium model for juvenile neuronal ceroid lipofuscinosis. Cell Adhes. Migr. 2017, 11, 399–418. [Google Scholar] [CrossRef] [Green Version]
- Gotthardt, D.; Blancheteau, V.; Bosserhoff, A.; Ruppert, T.; Delorenzi, M.; Soldati, T. Proteomics Fingerprinting of Phagosome Maturation and Evidence for the Role of a Gα during Uptake* S. Mol. Cell. Proteom. 2006, 5, 2228–2243. [Google Scholar] [CrossRef] [Green Version]
- Reinders, Y.; Schulz, I.; Gräf, R.; Sickmann, A. Identification of Novel Centrosomal Proteins in Dictyostelium d iscoideum by Comparative Proteomic Approaches. J. Proteome Res. 2006, 5, 589–598. [Google Scholar] [CrossRef]
- Huber, R.J.; Mathavarajah, S. Secretion and function of Cln5 during the early stages of Dictyostelium development. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1437–1450. [Google Scholar] [CrossRef]
- Huber, R.J.; Mathavarajah, S.; Yap, S.Q. Mfsd8 localizes to endocytic compartments and influences the secretion of Cln5 and cathepsin D in Dictyostelium. Cell. Signal. 2020, 70, 109572. [Google Scholar] [CrossRef] [PubMed]
- Journet, A.; Chapel, A.; Jehan, S.; Adessi, C.; Freeze, H.; Klein, G.; Garin, J. Characterization of Dictyostelium discoideum cathepsin D. J. Cell Sci. 1999, 112, 3833–3843. [Google Scholar] [CrossRef]
- Hagedorn, M.; Soldati, T. Flotillin and RacH modulate the intracellular immunity of Dictyostelium to Mycobacterium marinum infection. Cell. Microbiol. 2007, 9, 2716–2733. [Google Scholar] [CrossRef]
- Harris, E.; Wang, N.; Wu, W.-l.; Weatherford, A.; De Lozanne, A.; Cardelli, J. Dictyostelium LvsB mutants model the lysosomal defects associated with Chediak-Higashi syndrome. Mol. Biol. Cell 2002, 13, 656–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mir, H.; Rajawat, J.; Begum, R. Staurosporine induced poly (ADP-ribose) polymerase independent cell death in Dictyostelium discoideum. Indian J. Exp. Biol. 2012, 50, 80–86. [Google Scholar] [PubMed]
- Sillo, A.; Matthias, J.; Konertz, R.; Bozzaro, S.; Eichinger, L. Salmonella typhimurium is pathogenic for Dictyostelium cells and subverts the starvation response. Cell. Microbiol. 2011, 13, 1793–1811. [Google Scholar] [CrossRef] [PubMed]
- Lelong, E.; Marchetti, A.; Guého, A.; Lima, W.C.; Sattler, N.; Molmeret, M.; Hagedorn, M.; Soldati, T.; Cosson, P. Role of magnesium and a phagosomal P-type ATPase in intracellular bacterial killing. Cell. Microbiol. 2011, 13, 246–258. [Google Scholar] [CrossRef] [Green Version]
- Le Coadic, M.; Froquet, R.; Lima, W.C.; Dias, M.; Marchetti, A.; Cosson, P. Phg1/TM9 proteins control intracellular killing of bacteria by determining cellular levels of the Kil1 sulfotransferase in Dictyostelium. PLoS ONE 2013, 8, e53259. [Google Scholar] [CrossRef] [PubMed]
- Adessi, C.; Chapel, A.; Vinçon, M.; Rabilloud, T.; Klein, G.; Satre, M.; Garin, J. Identification of major proteins associated with Dictyostelium discoideum endocytic vesicles. J. Cell Sci. 1995, 108, 3331–3337. [Google Scholar] [CrossRef]
- Na, J.; Tunggal, B.; Eichinger, L. STATc is a key regulator of the transcriptional response to hyperosmotic shock. BMC Genom. 2007, 8, 123. [Google Scholar] [CrossRef] [Green Version]
- Bellizzi, J.J., 3rd; Widom, J.; Kemp, C.; Lu, J.Y.; Das, A.K.; Hofmann, S.L.; Clardy, J. The crystal structure of palmitoyl protein thioesterase 1 and the molecular basis of infantile neuronal ceroid lipofuscinosis. Proc. Natl. Acad. Sci. USA 2000, 97, 4573–4578. [Google Scholar] [CrossRef] [Green Version]
- Zeidman, R.; Jackson, C.S.; Magee, A.I. Protein acyl thioesterases (Review). Mol. Membr. Biol. 2009, 26, 32–41. [Google Scholar] [CrossRef]
- Appu, A.P.; Bagh, M.B.; Sadhukhan, T.; Mondal, A.; Casey, S.; Mukherjee, A.B. Cln3-mutations underlying juvenile neuronal ceroid lipofuscinosis cause significantly reduced levels of Palmitoyl-protein thioesterases-1 (Ppt1)-protein and Ppt1-enzyme activity in the lysosome. J. Inherit. Metab. Dis. 2019, 42, 944–954. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Chieng, K.S.; Baheerathan, A.; Hussain, N.; Gosalakkal, J. Novel CLN1 mutation with atypical juvenile neuronal ceroid lipofuscinosis. J. Pediatr. Neurosci. 2013, 8, 49–51. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Lobel, P. Production and characterization of recombinant human CLN2 protein for enzyme-replacement therapy in late infantile neuronal ceroid lipofuscinosis. BioChem. J. 2001, 357, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Prada, A.M.; Schmidt, M.; Morrow, E.M. Generation of pathogenic TPP1 mutations in human stem cells as a model for neuronal ceroid lipofuscinosis type 2 disease. Stem Cell Res. 2021, 53, 102323. [Google Scholar] [CrossRef] [PubMed]
- Ardicli, D.; Haliloglu, G.; Gocmen, R.; Gunbey, C.; Topcu, M. Unraveling neuronal ceroid lipofuscinosis type 2 (CLN2) disease: A tertiary center experience for determinants of diagnostic delay. Eur. J. Paediatr. Neurol. 2021, 33, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Goebel, H.H. The neuronal ceroid-lipofuscinoses. Semin. Pediatr. Neurol. 1996, 3, 270–278. [Google Scholar] [CrossRef]
- Whiting, R.E.H.; Jensen, C.A.; Pearce, J.W.; Gillespie, L.E.; Bristow, D.E.; Katz, M.L. Intracerebroventricular gene therapy that delays neurological disease progression is associated with selective preservation of retinal ganglion cells in a canine model of CLN2 disease. Exp. Eye Res. 2016, 146, 276–282. [Google Scholar] [CrossRef] [Green Version]
- Specchio, N.; Pietrafusa, N.; Trivisano, M. Changing Times for CLN2 Disease: The Era of Enzyme Replacement Therapy. Ther. Clin. Risk Manag. 2020, 16, 213–222. [Google Scholar] [CrossRef] [Green Version]
- Pal, A.; Kraetzner, R.; Gruene, T.; Grapp, M.; Schreiber, K.; Gronborg, M.; Urlaub, H.; Becker, S.; Asif, A.R.; Gartner, J.; et al. Structure of tripeptidyl-peptidase I provides insight into the molecular basis of late infantile neuronal ceroid lipofuscinosis. J. Biol. Chem. 2009, 284, 3976–3984. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Sohar, I.; Lackland, H.; Lobel, P. The human CLN2 protein/tripeptidyl-peptidase I is a serine protease that autoactivates at acidic pH. J. Biol. Chem. 2001, 276, 2249–2255. [Google Scholar] [CrossRef] [Green Version]
- Vines, D.; Warburton, M.J. Purification and characterisation of a tripeptidyl aminopeptidase I from rat spleen. Biochim. Biophys. Acta 1998, 1384, 233–242. [Google Scholar] [CrossRef]
- Chen, Z.R.; Liu, D.T.; Meng, H.; Liu, L.; Bian, W.J.; Liu, X.R.; Zhu, W.W.; He, Y.; Wang, J.; Tang, B.; et al. Homozygous missense TPP1 mutation associated with mild late infantile neuronal ceroid lipofuscinosis and the genotype-phenotype correlation. Seizure 2019, 69, 180–185. [Google Scholar] [CrossRef]
- Palmer, D.N.; Fearnley, I.M.; Walker, J.E.; Hall, N.A.; Lake, B.D.; Wolfe, L.S.; Haltia, M.; Martinus, R.D.; Jolly, R.D. Mitochondrial ATP synthase subunit c storage in the ceroid-lipofuscinoses (Batten disease). Am. J. Med. Genet. 1992, 42, 561–567. [Google Scholar] [CrossRef]
- Golabek, A.A.; Kida, E. Tripeptidyl-peptidase I in health and disease. Biol. Chem. 2006, 387, 1091–1099. [Google Scholar] [CrossRef]
- Seranova, E.; Connolly, K.J.; Zatyka, M.; Rosenstock, T.R.; Barrett, T.; Tuxworth, R.I.; Sarkar, S. Dysregulation of autophagy as a common mechanism in lysosomal storage diseases. Essays BioChem. 2017, 61, 733–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deckstein, J.; van Appeldorn, J.; Tsangarides, M.; Yiannakou, K.; Muller, R.; Stumpf, M.; Sukumaran, S.K.; Eichinger, L.; Noegel, A.A.; Riyahi, T.Y. The Dictyostelium discoideum GPHR ortholog is an endoplasmic reticulum and Golgi protein with roles during development. Eukaryot. Cell 2015, 14, 41–54. [Google Scholar] [CrossRef] [Green Version]
- Järvelä, I.; Autti, T.; Lamminranta, S.; Åberg, L.; Raininko, R.; Santavuori, P. Clinical and magnetic resonance imaging findings in batten disease: Analysis of the major mutation (1.02-Kb Deletion). Ann. Neurol. 1997, 42, 799–802. [Google Scholar] [CrossRef]
- Autti, T.; Hamalainen, J.; Aberg, L.; Lauronen, L.; Tyynela, J.; Van Leemput, K. Thalami and corona radiata in juvenile NCL (CLN3): A voxel-based morphometric study. Eur. J. Neurol. 2007, 14, 447–450. [Google Scholar] [CrossRef]
- Phillips, S.N.; Benedict, J.W.; Weimer, J.M.; Pearce, D.A. CLN3, the protein associated with batten disease: Structure, function and localization. J. Neurosci. Res. 2005, 79, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Chandrachud, U.; Walker, M.W.; Simas, A.M.; Heetveld, S.; Petcherski, A.; Klein, M.; Oh, H.; Wolf, P.; Zhao, W.-N.; Norton, S. Unbiased cell-based screening in a neuronal cell model of Batten disease highlights an interaction between Ca2+ homeostasis, autophagy, and CLN3 protein function. J. Biol. Chem. 2015, 290, 14361–14380. [Google Scholar] [CrossRef] [Green Version]
- Lane, S.C.; Jolly, R.D.; Schmechel, D.E.; Alroy, J.; Boustany, R.M. Apoptosis as the mechanism of neurodegeneration in Batten’s disease. J. NeuroChem. 1996, 67, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Luiro, K.; Yliannala, K.; Ahtiainen, L.; Maunu, H.; Järvelä, I.; Kyttälä, A.; Jalanko, A. Interconnections of CLN3, Hook1 and Rab proteins link Batten disease to defects in the endocytic pathway. Hum. Mol. Genet. 2004, 13, 3017–3027. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Huang, Z.; Chen, Y.; Zhou, J.; Hu, S.; Zhi, Q.; Song, S.; Wang, Y.; Wan, D.; Gu, W. Effect of CLN3 silencing by RNA interference on the proliferation and apoptosis of human colorectal cancer cells. Biomed. Pharmacother. 2014, 68, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Evans, L.P.; Gibson-Corley, K.N.; Mullins, R.F.; Tucker, B.A.; Trent, A.; Stone, E.M.; Jones, K.A. An Unusual Presentation of CLN3-Associated Batten Disease With Classic Histopathologic and Ultrastructural Findings. J. Neuropathol. Exp. Neurol. 2021, 80, 1081–1084. [Google Scholar] [CrossRef] [PubMed]
- Stein, C.S.; Yancey, P.H.; Martins, I.; Sigmund, R.D.; Stokes, J.B.; Davidson, B.L. Osmoregulation of ceroid neuronal lipofuscinosis type 3 in the renal medulla. Am. J. Physiol. Cell Physiol. 2010, 298, C1388–C1400. [Google Scholar] [CrossRef] [PubMed]
- Schmidtke, C.; Tiede, S.; Thelen, M.; Kakela, R.; Jabs, S.; Makrypidi, G.; Sylvester, M.; Schweizer, M.; Braren, I.; Brocke-Ahmadinejad, N.; et al. Lysosomal proteome analysis reveals that CLN3-defective cells have multiple enzyme deficiencies associated with changes in intracellular trafficking. J. Biol. Chem. 2019, 294, 9592–9604. [Google Scholar] [CrossRef] [PubMed]
- Noskova, L.; Stranecky, V.; Hartmannova, H.; Pristoupilova, A.; Baresova, V.; Ivanek, R.; Hulkova, H.; Jahnova, H.; van der Zee, J.; Staropoli, J.F.; et al. Mutations in DNAJC5, encoding cysteine-string protein alpha, cause autosomal-dominant adult-onset neuronal ceroid lipofuscinosis. Am. J. Hum. Genet. 2011, 89, 241–252. [Google Scholar] [CrossRef] [Green Version]
- Cadieux-Dion, M.; Andermann, E.; Lachance-Touchette, P.; Ansorge, O.; Meloche, C.; Barnabe, A.; Kuzniecky, R.I.; Andermann, F.; Faught, E.; Leonberg, S.; et al. Recurrent mutations in DNAJC5 cause autosomal dominant Kufs disease. Clin. Genet. 2013, 83, 571–575. [Google Scholar] [CrossRef]
- Benitez, B.A.; Alvarado, D.; Cai, Y.; Mayo, K.; Chakraverty, S.; Norton, J.; Morris, J.C.; Sands, M.S.; Goate, A.; Cruchaga, C. Exome-sequencing confirms DNAJC5 mutations as cause of adult neuronal ceroid-lipofuscinosis. PLoS ONE 2011, 6, e26741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donnelier, J.; Braun, J.E. CSPalpha-chaperoning presynaptic proteins. Front. Cell. Neurosci. 2014, 8, 116. [Google Scholar] [CrossRef] [Green Version]
- Roosen, D.A.; Blauwendraat, C.; Cookson, M.R.; Lewis, P.A. DNAJC proteins and pathways to parkinsonism. FEBS J. 2019, 286, 3080–3094. [Google Scholar] [CrossRef] [Green Version]
- Savukoski, M.; Klockars, T.; Holmberg, V.; Santavuori, P.; Lander, E.S.; Peltonen, L. CLN5, a novel gene encoding a putative transmembrane protein mutated in Finnish variant late infantile neuronal ceroid lipofuscinosis. Nat. Genet. 1998, 19, 286–288. [Google Scholar] [CrossRef]
- Moharir, A.; Peck, S.H.; Budden, T.; Lee, S.Y. The role of N-glycosylation in folding, trafficking, and functionality of lysosomal protein CLN5. PLoS ONE 2013, 8, e74299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaren, M.D.; Mathavarajah, S.; Kim, W.D.; Yap, S.Q.; Huber, R.J. Aberrant Autophagy Impacts Growth and Multicellular Development in a Dictyostelium Knockout Model of CLN5 Disease. Front. Cell Dev. Biol. 2021, 9, 657406. [Google Scholar] [CrossRef] [PubMed]
- Aiello, C.; Terracciano, A.; Simonati, A.; Discepoli, G.; Cannelli, N.; Claps, D.; Crow, Y.J.; Bianchi, M.; Kitzmuller, C.; Longo, D.; et al. Mutations in MFSD8/CLN7 are a frequent cause of variant-late infantile neuronal ceroid lipofuscinosis. Hum. Mutat. 2009, 30, E530–E540. [Google Scholar] [CrossRef]
- Steenhuis, P.; Herder, S.; Gelis, S.; Braulke, T.; Storch, S. Lysosomal targeting of the CLN7 membrane glycoprotein and transport via the plasma membrane require a dileucine motif. Traffic 2010, 11, 987–1000. [Google Scholar] [CrossRef] [PubMed]
- Siintola, E.; Topcu, M.; Aula, N.; Lohi, H.; Minassian, B.A.; Paterson, A.D.; Liu, X.Q.; Wilson, C.; Lahtinen, U.; Anttonen, A.K.; et al. The novel neuronal ceroid lipofuscinosis gene MFSD8 encodes a putative lysosomal transporter. Am. J. Hum. Genet. 2007, 81, 136–146. [Google Scholar] [CrossRef] [Green Version]
- Capurso, C.; Solfrizzi, V.; D’Introno, A.; Colacicco, A.M.; Capurso, S.A.; Bifaro, L.; Menga, R.; Santamato, A.; Seripa, D.; Pilotto, A.; et al. Short arm of chromosome 11 and sporadic Alzheimer’s disease: Catalase and cathepsin D gene polymorphisms. Neurosci. Lett. 2008, 432, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Schulz, A.; Kohlschutter, A.; Mink, J.; Simonati, A.; Williams, R. NCL diseases—Clinical perspectives. Biochim. Biophys. Acta 2013, 1832, 1801–1806. [Google Scholar] [CrossRef] [Green Version]
- Fritchie, K.; Siintola, E.; Armao, D.; Lehesjoki, A.E.; Marino, T.; Powell, C.; Tennison, M.; Booker, J.M.; Koch, S.; Partanen, S.; et al. Novel mutation and the first prenatal screening of cathepsin D deficiency (CLN10). Acta Neuropathol. 2009, 117, 201–208. [Google Scholar] [CrossRef]
- Siintola, E.; Partanen, S.; Stromme, P.; Haapanen, A.; Haltia, M.; Maehlen, J.; Lehesjoki, A.E.; Tyynela, J. Cathepsin D deficiency underlies congenital human neuronal ceroid-lipofuscinosis. Brain 2006, 129, 1438–1445. [Google Scholar] [CrossRef] [Green Version]
- Tayebi, N.; Lopez, G.; Do, J.; Sidransky, E. Pro-cathepsin D, Prosaposin, and Progranulin: Lysosomal Networks in Parkinsonism. Trends Mol. Med. 2020, 26, 913–923. [Google Scholar] [CrossRef] [PubMed]
- Neuray, C.; Sultan, T.; Alvi, J.R.; Franca, M.C.; Assmann, B.; Wagner, M.; Canafoglia, L.; Franceschetti, S.; Rossi, G.; Santana, I.; et al. Early-onset phenotype of bi-allelic GRN mutations. Brain 2021, 144, e22. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, X.; Li, Y.; Chen, D.; Liu, Z.; Zhao, Y.; Tu, L.; Wang, S. Regulation of progranulin expression and location by sortilin in oxygen-glucose deprivation/reoxygenation injury. Neurosci. Lett. 2020, 738, 135394. [Google Scholar] [CrossRef] [PubMed]
- Ban, R.; Pu, C.; Fang, F.; Shi, Q. A novel homozygous mutation in ATP13A2 gene causing pure hereditary spastic paraplegia. Parkinsonism Relat. Disord. 2021, 86, 58–60. [Google Scholar] [CrossRef] [PubMed]
- Gowda, V.K.; Srinivasan, V.M.; Shivappa, S.K. Kufor-Rakeb Syndrome/Parkinson Disease Type 9. Indian J. Pediatr. 2020, 87, 231–232. [Google Scholar] [CrossRef] [PubMed]
- Covy, J.P.; Waxman, E.A.; Giasson, B.I. Characterization of cellular protective effects of ATP13A2/PARK9 expression and alterations resulting from pathogenic mutants. J. Neurosci. Res. 2012, 90, 2306–2316. [Google Scholar] [CrossRef] [Green Version]
- Smith, K.R.; Dahl, H.H.; Canafoglia, L.; Andermann, E.; Damiano, J.; Morbin, M.; Bruni, A.C.; Giaccone, G.; Cossette, P.; Saftig, P.; et al. Cathepsin F mutations cause Type B Kufs disease, an adult-onset neuronal ceroid lipofuscinosis. Hum. Mol. Genet. 2013, 22, 1417–1423. [Google Scholar] [CrossRef]
- Staropoli, J.F.; Karaa, A.; Lim, E.T.; Kirby, A.; Elbalalesy, N.; Romansky, S.G.; Leydiker, K.B.; Coppel, S.H.; Barone, R.; Xin, W.; et al. A homozygous mutation in KCTD7 links neuronal ceroid lipofuscinosis to the ubiquitin-proteasome system. Am. J. Hum. Genet. 2012, 91, 202–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azizieh, R.; Orduz, D.; Van Bogaert, P.; Bouschet, T.; Rodriguez, W.; Schiffmann, S.N.; Pirson, I.; Abramowicz, M.J. Progressive myoclonic epilepsy-associated gene KCTD7 is a regulator of potassium conductance in neurons. Mol. Neurobiol. 2011, 44, 111–121. [Google Scholar] [CrossRef]
- Guerrini, R.; Dobyns, W.B.; Barkovich, A.J. Abnormal development of the human cerebral cortex: Genetics, functional consequences and treatment options. Trends Neurosci. 2008, 31, 154–162. [Google Scholar] [CrossRef]
- De Rijk-van Andel, J.; Arts, W.; Hofman, A.; Staal, A.; Niermeijer, M. Epidemiology of lissencephaly type I. Neuroepidemiology 1991, 10, 200–204. [Google Scholar] [CrossRef]
- Leventer, R.J. Genotype-phenotype correlation in lissencephaly and subcortical band heterotopia: The key questions answered. J. Child. Neurol. 2005, 20, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Verloes, A.; Elmaleh, M.; Gonzales, M.; Laquerrière, A.; Gressens, P. Genetic and clinical aspects of lissencephaly. Rev. Neurol. 2007, 163, 533–547. [Google Scholar] [CrossRef]
- Leventer, R.J.; Pilz, D.T.; Matsumoto, N.; Ledbetter, D.H.; Dobyns, W.B. Lissencephaly and subcortical band heterotopia: Molecular basis and diagnosis. Mol. Med. Today 2000, 6, 277–284. [Google Scholar] [CrossRef]
- Elshenawy, M.M.; Kusakci, E.; Volz, S.; Baumbach, J.; Bullock, S.L.; Yildiz, A. Lis1 activates dynein motility by modulating its pairing with dynactin. Nat. Cell Biol. 2020, 22, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Guerrini, R.; Parrini, E. Neuronal migration disorders. Neurobiol. Dis. 2010, 38, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Meyer, I.; Kuhnert, O.; Graf, R. Functional analyses of lissencephaly-related proteins in Dictyostelium. Semin. Cell Dev. Biol. 2011, 22, 89–96. [Google Scholar] [CrossRef]
- Rehberg, M.; Kleylein-Sohn, J.; Faix, J.; Ho, T.H.; Schulz, I.; Graf, R. Dictyostelium LIS1 is a centrosomal protein required for microtubule/cell cortex interactions, nucleus/centrosome linkage, and actin dynamics. Mol. Biol. Cell 2005, 16, 2759–2771. [Google Scholar] [CrossRef] [Green Version]
- Schaf, J.; Damstra-Oddy, J.; Williams, R.S.B. Dictyostelium discoideum as a pharmacological model system to study the mechanisms of medicinal drugs and natural products. Int. J. Dev. Biol. 2019, 63, 541–550. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, K.; Iriki, H.; Kamimura, Y.; Muramoto, T. CRISPR Toolbox for Genome Editing in Dictyostelium. Front. Cell Dev. Biol. 2021, 9, 721630. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Kuspa, A.; Loomis, W.F. Tagging developmental genes in Dictyostelium by restriction enzyme-mediated integration of plasmid DNA. Proc. Natl. Acad. Sci. USA 1992, 89, 8803–8807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruenheit, N.; Baldwin, A.; Stewart, B.; Jaques, S.; Keller, T.; Parkinson, K.; Salvidge, W.; Baines, R.; Brimson, C.; Wolf, J.B. Mutant resources for functional genomics in Dictyostelium discoideum using REMI-seq technology. BMC Biol. 2021, 19, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Loomis, W.F. A better way to discover gene function in the social amoeba Dictyostelium discoideum. Genome Res. 2016, 26, 1161–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, C.J.; Finch, P.; Müller-Taubenberger, A.; Leung, K.Y.; Warren, E.C.; Damstra-Oddy, J.; Sharma, D.; Patra, P.H.; Glyn, S.; Boberska, J. A new mechanism for cannabidiol in regulating the one-carbon cycle and methionine levels in Dictyostelium and in mammalian epilepsy models. Br. J. Pharmacol. 2020, 177, 912–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damstra-Oddy, J.L.; Warren, E.C.; Perry, C.J.; Desfougeres, Y.; Fitzpatrick, J.K.; Schaf, J.; Costelloe, L.; Hind, W.; Downer, E.J.; Saiardi, A.; et al. Phytocannabinoid-dependent mTORC1 regulation is dependent upon inositol polyphosphate multikinase activity. Br. J. Pharm. 2021, 178, 1149–1163. [Google Scholar] [CrossRef] [PubMed]
- Waheed, A.; Ludtmann, M.H.; Pakes, N.; Robery, S.; Kuspa, A.; Dinh, C.; Baines, D.; Williams, R.S.; Carew, M.A. Naringenin inhibits the growth of Dictyostelium and MDCK-derived cysts in a TRPP2 (polycystin-2)-dependent manner. Br. J. Pharm. 2014, 171, 2659–2670. [Google Scholar] [CrossRef]
- Cocorocchio, M.; Baldwin, A.J.; Stewart, B.; Kim, L.; Harwood, A.J.; Thompson, C.R.L.; Andrews, P.L.R.; Williams, R.S.B. Curcumin and derivatives function through protein phosphatase 2A and presenilin orthologues in Dictyostelium discoideum. Dis. Model. Mech. 2018, 11, dmm032375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warren, E.C.; Dooves, S.; Lugara, E.; Damstra-Oddy, J.; Schaf, J.; Heine, V.M.; Walker, M.C.; Williams, R.S.B. Decanoic acid inhibits mTORC1 activity independent of glucose and insulin signaling. Proc. Natl. Acad. Sci. USA 2020, 117, 23617–23625. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.S.; Eames, M.; Ryves, W.J.; Viggars, J.; Harwood, A.J. Loss of a prolyl oligopeptidase confers resistance to lithium by elevation of inositol (1,4,5) trisphosphate. EMBO J. 1999, 18, 2734–2745. [Google Scholar] [CrossRef] [Green Version]
- Williams, R.S.; Cheng, L.; Mudge, A.W.; Harwood, A.J. A common mechanism of action for three mood-stabilizing drugs. Nature 2002, 417, 292–295. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.; Walker, M.C.; Williams, R.S. Seizure-induced reduction in PIP3 levels contributes to seizure-activity and is rescued by valproic acid. Neurobiol. Dis. 2014, 62, 296–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, P.; Zuckermann, A.M.; Williams, S.; Close, A.J.; Cano-Jaimez, M.; McEvoy, J.P.; Spencer, J.; Walker, M.C.; Williams, R.S. Seizure control by derivatives of medium chain fatty acids associated with the ketogenic diet show novel branching-point structure for enhanced potency. J. Pharm. Exp. 2015, 352, 43–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, P.; Terbach, N.; Plant, N.; Chen, P.E.; Walker, M.C.; Williams, R.S. Seizure control by ketogenic diet-associated medium chain fatty acids. Neuropharmacology 2013, 69, 105–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoeler, N.E.; Orford, M.; Vivekananda, U.; Simpson, Z.; Van de Bor, B.; Smith, H.; Balestrini, S.; Rutherford, T.; Brennan, E.; McKenna, J.; et al. Vita: A feasibility study of a blend of medium chain triglycerides to manage drug-resistant epilepsy. Brain Commun. 2021, 3, fcab160. [Google Scholar] [CrossRef] [PubMed]
- Warren, E.C.; Walker, M.C.; Williams, R.S.B. All You Need Is Fats-for Seizure Control: Using Amoeba to Advance Epilepsy Research. Front. Cell. Neurosci. 2018, 12, 199. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Human Gene | D. discoideum Homologue | Mutant Strain | Multicellular Phenotype | Mitochondrial Respiration | Main Findings | Ref. |
|---|---|---|---|---|---|---|
| LRRK2 | Roco genes (gbpC, qkgA, pats1, roco4-11) | gbpC- | Decreased mound size, aberrant fruiting body morphology and decreased chemotaxis | Not tested | The Roco kinases have diverse roles in D. discoideum and it is yet to be determined which of the Roco proteins are true functional homologues of LRRK2. GbpC and Pats1 play a role in chemotaxis. Pats1 null mutants show defects in cytokinesis. | [38,39,40,41,42] |
| qkgA- | Decreased chemotaxis | Not tested | QkgA plays a role in cell proliferation where null mutants grow quicker in shaking culture and overexpression of QkgA results in slower growth. | [38,39,40,41] | ||
| roco4- | Aberrant fruiting body morphology and slug migratory defect | Elevated parameters | Deletion of Roco4 results in aberrant multicellular development and mutants display elevated mitochondrial respiratory parameters. | [38,42,43] | ||
| roco11- | Aberrant fruiting body morphology | Not tested | Roco11 results in aberrant multicellular development | [38,43] | ||
| HTRA2 htrA | htra2 knockdown | Aberrant fruiting body morphology | No defect | Hyperactivity of HTRA protease activity in D. discoideum is lethal. HTRA localises to the mitochondria. HTRA knock-down and protease-dead HTRA strains display phenotypic defects reminiscent of mitochondrial dysfunction including altered fruiting body morphology and decreased growth rates with no endocytic defect and implicates the protease domain in these functions. Seahorse respiratory measurements indicate no defect in mitochondrial respiration in either the knock down or protease dead strains. | [44] | |
| htra2 protease dead | Aberrant fruiting body morphology | No defect | HTRA knock-down and protease-dead HTRA strains display phenotypic defects reminiscent of mitochondrial dysfunction including altered fruiting body morphology and decreased growth rates with no endocytic defect and implicates the protease domain in these functions. Seahorse respiratory measurements indicate no defect in mitochondrial respiration in either the knock down or protease dead strains | [44] | ||
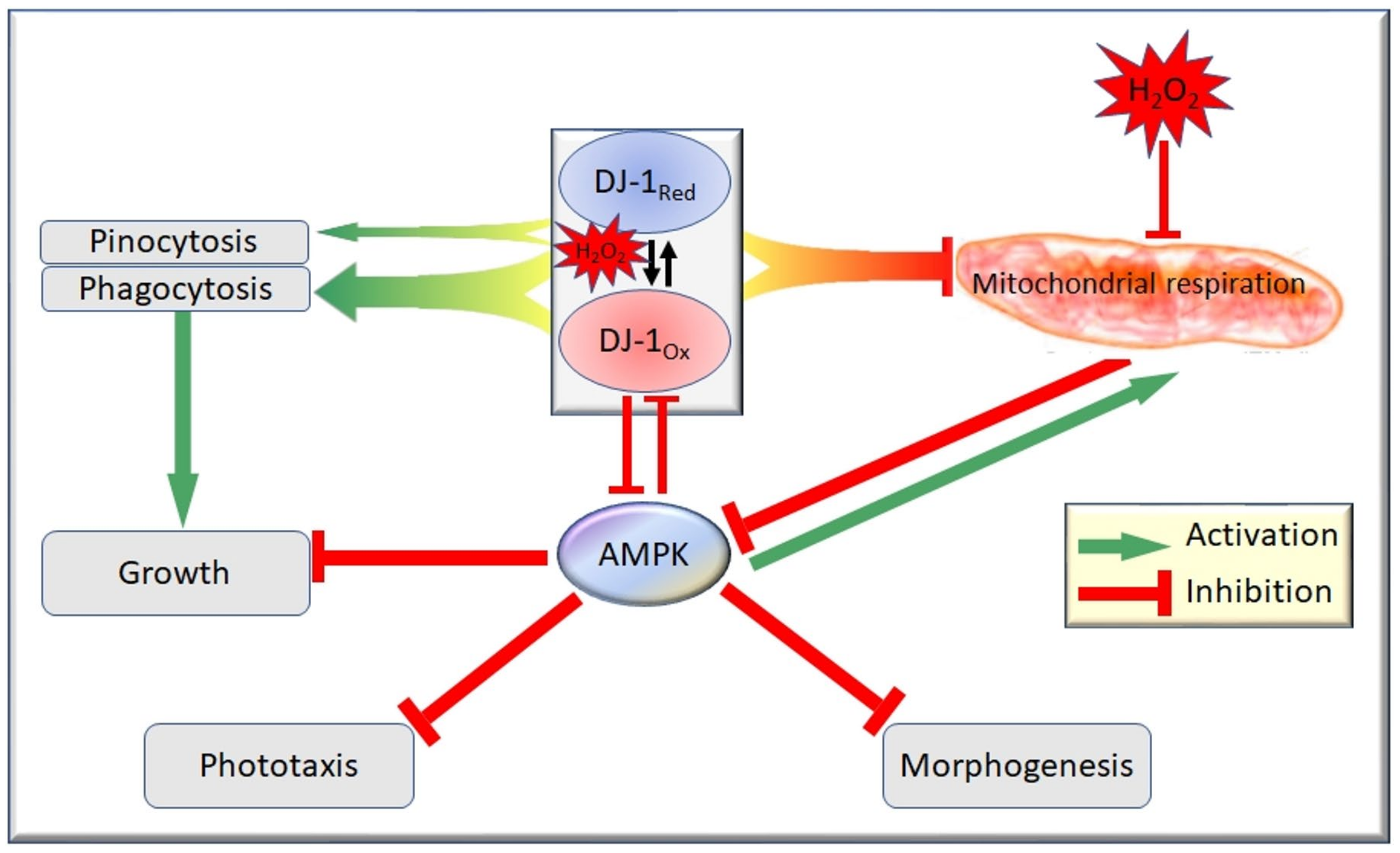
| DJ-1 (Park7) deeJ | deeJ knockdown | Aberrant fruiting body morphology | Elevated parameters | DJ-1 antisense inhibition elevates mitochondrial respiration in D. discoideum. | [45] | |
| deeJ knockdown exposed to H2O2 | Aberrant fruiting body morphology and phototaxis (rescued by antisense AMPK) | Elevated parameters (but less elevated compared to deeJ knockdown alone) | Knockdown of AMPK rescues phenotypic defects observed in DJ-1 knock-down strains, suggesting that DJ-1 may play a role downstream of mitochondria by mitigating the consequences of AMPK activation. | [46] | ||
| deeJ Overex-pression | No defects observed | Decreased parameters | DJ-1 overexpression inhibits mitochondrial respiration in D. discoideum. DJ-1 localises to the cytoplasm, and has roles in multicellular development, growth and endocytosis. | [45] | ||
| SNCA | No homologue. Expression of human α-synuclein in D. discoideum | Full length (WT) human SNCA C-terminally truncated human SNCA A53T human SNCA | Mild phototactic defect (not significant) Impaired phototaxis and aberrant fruiting body morphology Impaired phototaxis and thermotaxis | Elevated parameters Elevated parameters No defect | In D. discoideum α-synuclein was observed at the cortex and localisation was attributed to the 20 most C-terminal residues. Expression of α-synuclein caused cytotoxic phagocytosis defects. Mitochondrial respirometry parameters were elevated in strains expressing WT and truncated α-synuclein. | [13] |
| Tau | No homologue. Expression of human Tau in Dictyostelium | Human Tau (longest isoform) Co-expression of human Tau and full length SNCA | Impaired phototaxis and thermotaxis in addition to aberrant fruiting body morphology Exacerbated defects in phototaxis and fruiting body morphology when co-expressed compared to when independently expressed | Defect in complex V Normal parameters observed when co-expressed | Tau causes AMPK-dependent phototactic defects exacerbated by the co-expression of α-synuclein. Tau expression reduced axenic growth, increased Legionella susceptibility and impaired ATP synthesis. Tau and α-synuclein interact directly as evident by close proximity experiments. Tau and α-synuclein interact functionally in a complex relationship and when coexpressed can rescue, exacerbate or rescue the phenotypic defects evident when expressed singly. Proteomic analysis of Tau, α-synuclein and Tau/α-synuclein co-transformants revealed a distinct set of dysregulated proteins which supported functional and localisation studies. | [14] |
| Human Gene | D. discoideum Homologue | Localisation | Impaired Phenotypes | Interactions with Other NCL Proteins | Main Findings |
|---|---|---|---|---|---|
| CLN1/PPT | ppt1 | Extracellular space [93] Macropinosome [94] | Unknown | Unknown | ppt1 mRNA is downregulated during phagocytosis [95]. |
| CLN2/TPP-1 | tpp1A tpp1B tpp1C tpp1D tpp1E tpp1F | Late endosome/lysosome [96] Predicted Golgi [97]/extracellular space [93] Unknown Macropinosome [94] Unknown Endocytic compartments, Golgi complex, ER, extracellular space [97,98] | tpp1A null:precocious development, abnormal spore formation, reduced autophagy [96]. Tpp1 antisense inhibited: reduced growth, increased endocytosis, small fruiting bodies [17] Unknown Transcriptional response to Pseudomonas aeruginosa [99] Unknown Unknown No obvious defects in growth or development [98] | Cln3 deficiency increases tpp1A expression during osmotic stress [100] Interacts with Cln5 [101] Unknown Cln3 deficiency decreases tpp1D expression during starvation [102] Unknown Cln3 deficiency increases tpp1F expression and secretion during starvation [103] | Tpp1A regulates autophagy. Tpp1A mediates its effects via mTOR pathway as tpp1A antisense inhibited strains the defective phenotypes were rescued by overexpression of Rheb and mimicked by exposure to rapamycin. A suppressor of Tpp1A (stpA) was identified via REMI [96]. Tpp1B and Tpp1F were discovered to interact with a Golgi membrane channel (GPHR) ubiquitous to eukaryotes. This was the first time a TPP1 homologue was found to interact with GPHR [98]. |
| CLN3 | cln3 | CV, endocytic compartments, Golgi complex [92] | CLN3 loss impairs aggregation, chemotaxis, multicellular development, cell adhesion, protein secretion, osmoregulation and pinocytosis. Increases proliferation [92,100,101,102,103,104] | Cln3 deficiency increases expression of CLN5, tpp1A and tpp1F but decreases tpp1D expression under certain conditions [100,102,103] Loss of Cln3 also reduces grn expression during starvation and reduces the secretion of CprA and CprB [103] | Loss of Cln3 in Dictyostelium results in reduced resistance to osmotic stress and reduced spore viability [100] Loss of Cln3 accelerated each stage of the multicellular developmental and this was rescued by calcium chelation [92] |
| CLN4/DNAJC5 | DDB_ G0290017 ddj1 | Macropinosome [94] Phagosome [105], centrosome [106] and Macropinosome [94] | Unknown Phagocytosis | Unknown Unknown | A true functional homologue of Cln4 in Dictyostelium is yet to be elucidated. |
| CLN5 | cln5 | CV [107], extracellular space [107], ER [101], cortex [107], macropinosome [94] | CLN5 null has reduced chemotaxis to folic acid, cell adhesion and autophagy [107] | Colocalises with CLN3 in CV [107]. Interacts with CtsD, Tpp1B [101], and is reduced in mfsd8 null [108] | Both D. discoideum Cln5 and human CLN5 function as a glycoside hydrolase [101]. |
| CLN7/MFSD8 | mfsd8 | Macropinosome [94] | Protein secretion [108] | Loss of mfsd8 results in reduced secretion of Cln5 and CtsD during starvation [108] | The roles of Mfsd8 may be conserved in D. discoideum. In both humans and D. discoideum it regulates protein secretion and interacts with other NCL proteins [108]. |
| CLN10/CTSD | ctsD | Extracellular space [103], macropinosome [94], lysosome, phagosome [109,110,111] | ctsD null causes development delay, bacterial degradation and cell death [109,112,113] | Interacts with Cln5 and reduced in mfsd8 null [108]. | Both Dictyostelium and human CTSD contain secretion peptides. Dictyostelium CtsD is highly expressed during growth and aggregation and plays similar roles in humans [108]. |
| CLN11/PGRN | grn | Unknown | Unknown | Reduced by the loss of cln3 during starvation [102] | |
| CLN12/Park9 | kil2 | Phagosome [114] and macropinosome [94] | Growth, defense against bacteria, fungi and metal toxicity [114,115] | Unknown | Kil2 acts as a magnesium pump and protects the cell against cation stress [114]. |
| CLN13/CTSF | cprA cprB DDB0252831 | Extracellular space [103] Extracellular space [103] Extracellular space [93,103] and endocytic vesicles [116] | Osmoregulation [117] Phagocytosis [95] Transcriptional response to Pseudomonas aeruginosa [99] | cln3 deficiency reduces secretion of CprA and CprB [103] | CprA identified as the likely homologue of human CTSF [91]. |
| CLN14/KCTD7 | kctd9 DDB0238663, DDB0346929 DDB0347398 | Unknown Unknown Macropinosome [94] Unknown | Unknown Unknown Unknown Unknown | No known interactions in Dictyostelium for any of the proposed homologues | Four potential homologues [8]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Storey, C.L.; Williams, R.S.B.; Fisher, P.R.; Annesley, S.J. Dictyostelium discoideum: A Model System for Neurological Disorders. Cells 2022, 11, 463. https://doi.org/10.3390/cells11030463
Storey CL, Williams RSB, Fisher PR, Annesley SJ. Dictyostelium discoideum: A Model System for Neurological Disorders. Cells. 2022; 11(3):463. https://doi.org/10.3390/cells11030463
Chicago/Turabian StyleStorey, Claire Louise, Robin Simon Brooke Williams, Paul Robert Fisher, and Sarah Jane Annesley. 2022. "Dictyostelium discoideum: A Model System for Neurological Disorders" Cells 11, no. 3: 463. https://doi.org/10.3390/cells11030463