
Disruption of Multiple Overlapping Functions Following Stepwise Inactivation of the Extended Myc Network
 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice, MEFs, and Documentation of Target Gene Excision Efficiency
2.2. MEF Immortalization
2.3. Puromycin Labeling, Immunoblotting, and Staining
2.4. Live Cell and Immunohistochemical Staining, Imaging, and Flow Cytometry
2.5. Lactate Measurements
2.6. Respirometry Studies
2.7. Transcriptional Profiling and Bioinformatics Analyses
3. Results
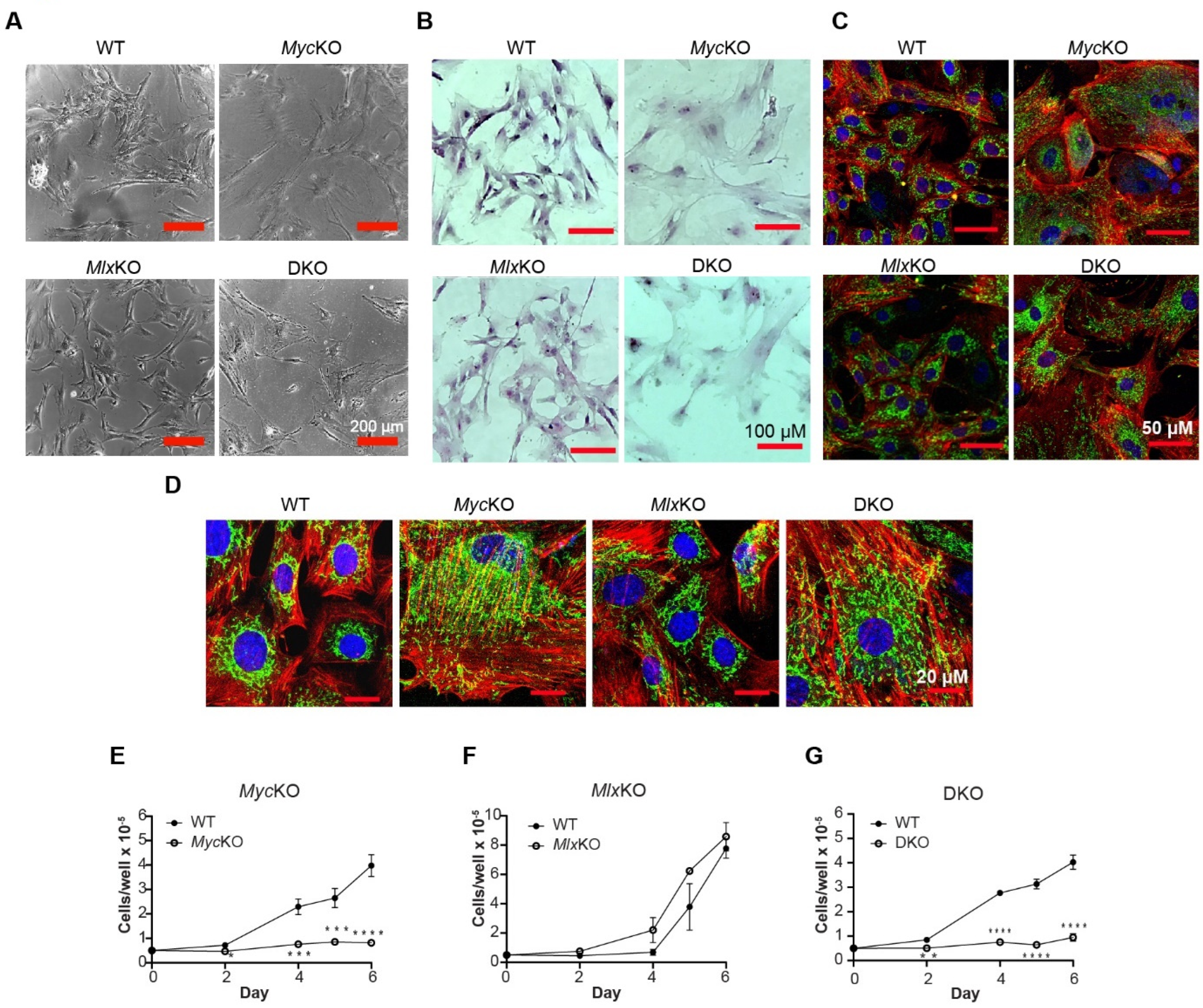
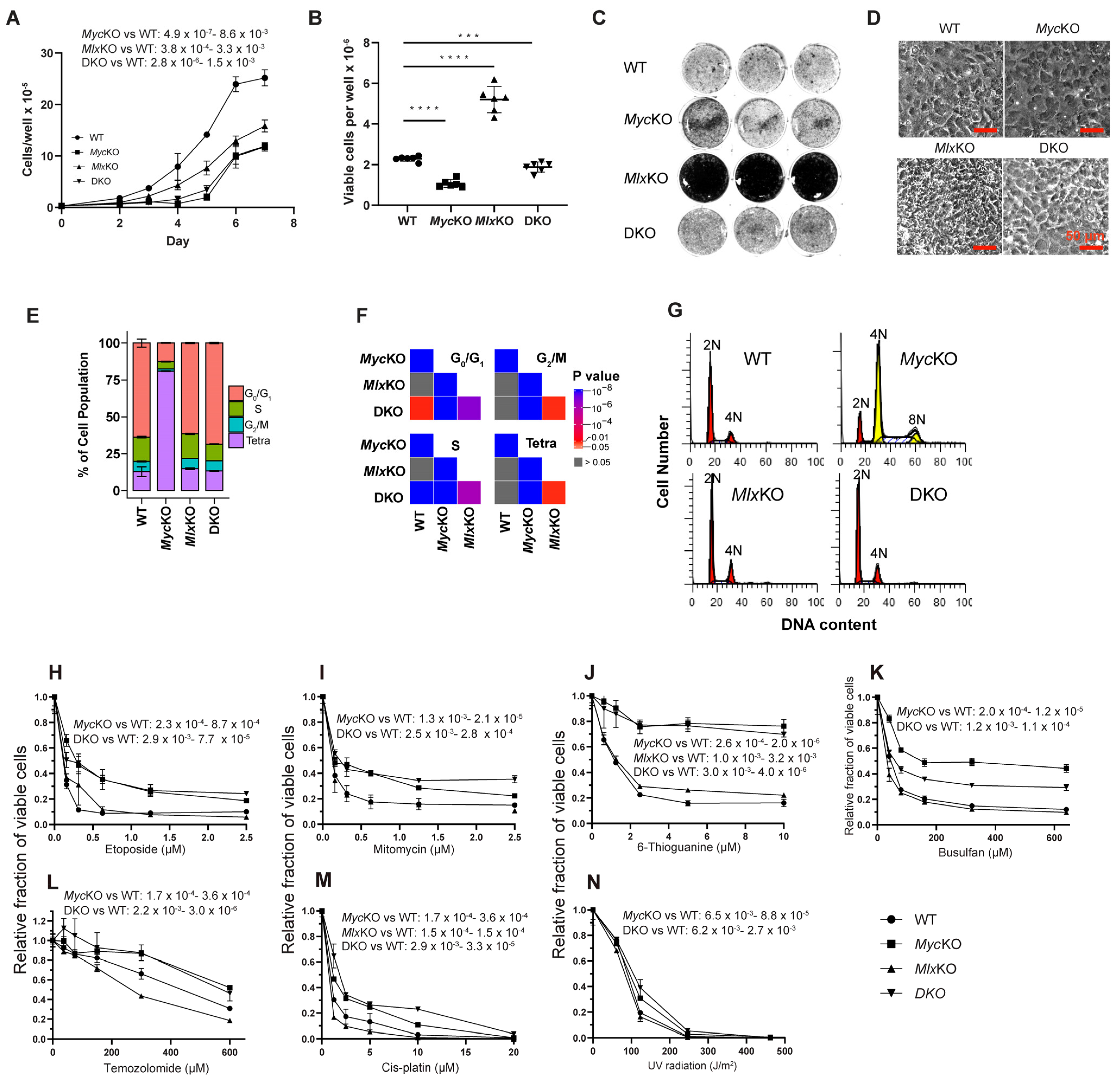
3.1. The Morphology and Proliferation of Primary MEFs Are More Dependent on Myc Than on Mlx
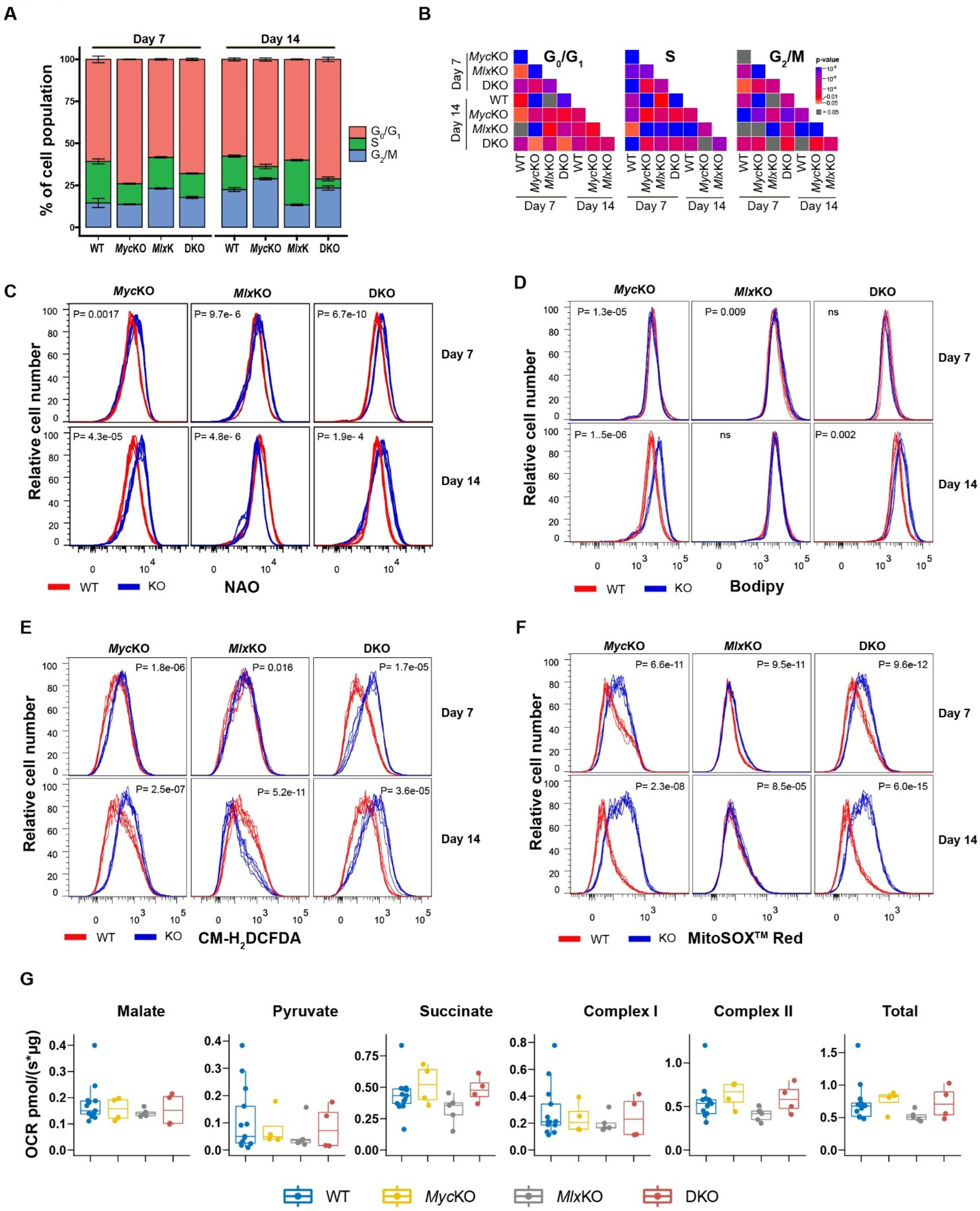
3.2. Loss of Myc and/or Mlx Differentially Affects Cell-Cycle Progression and Mitochondrial Structure and Function
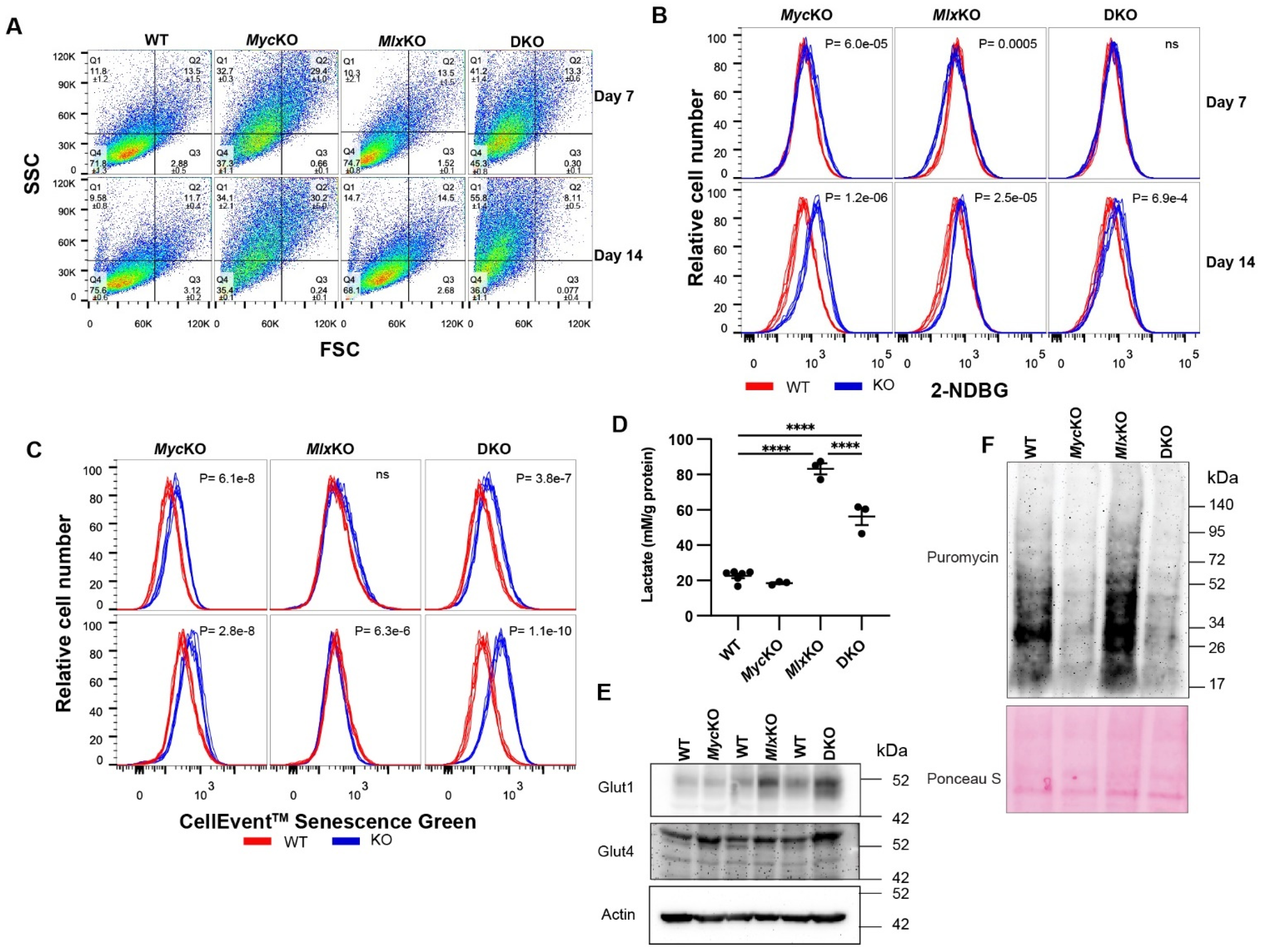
3.3. MycKO and DKO MEFs Display Features of Premature Aging and Senescence
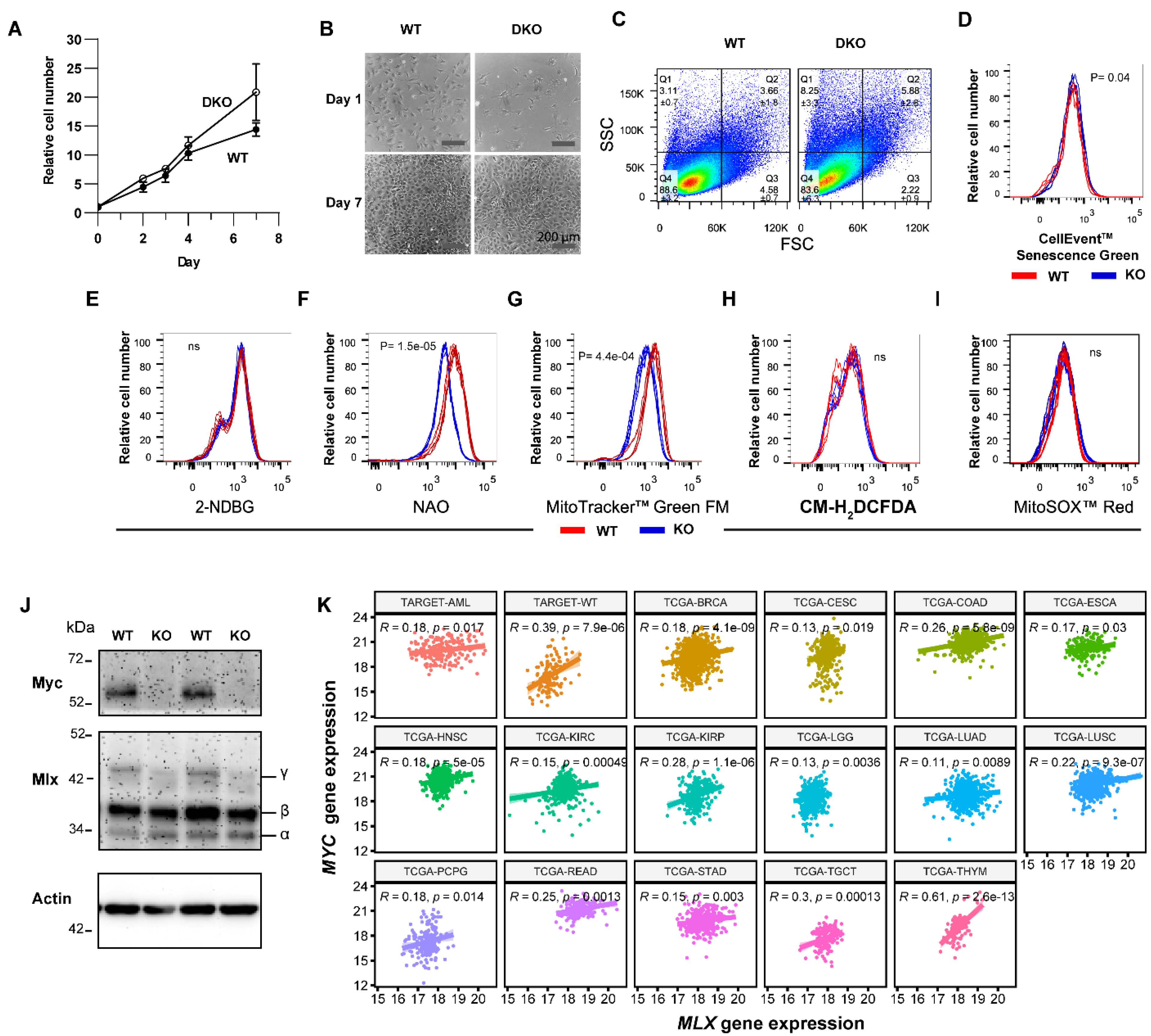
3.4. The Senescence-like State of DKO MEFs Is Spontaneously Reversible
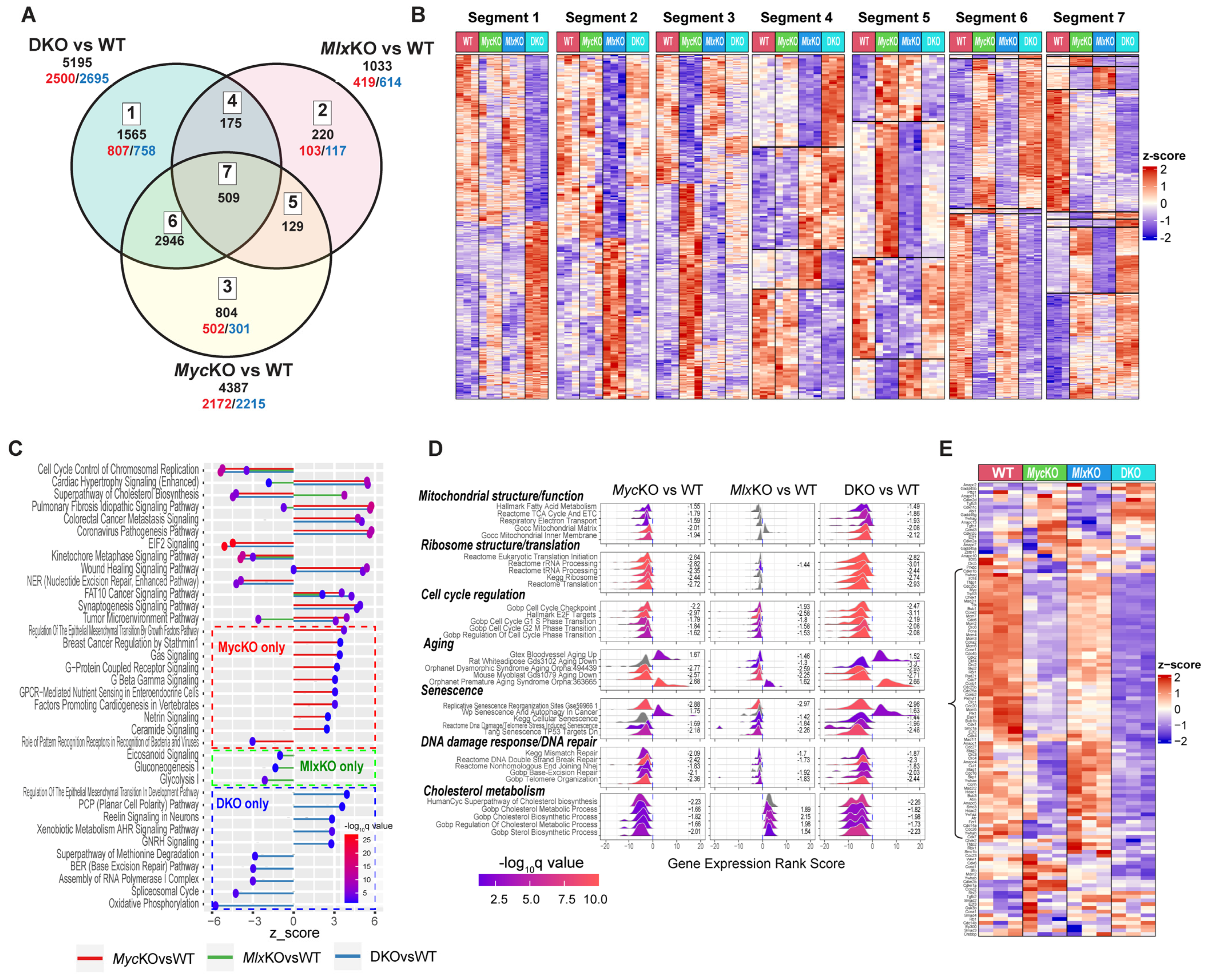
3.5. Co-Regulated Myc and Mlx Target Genes Impact Similar Pathways and Functions
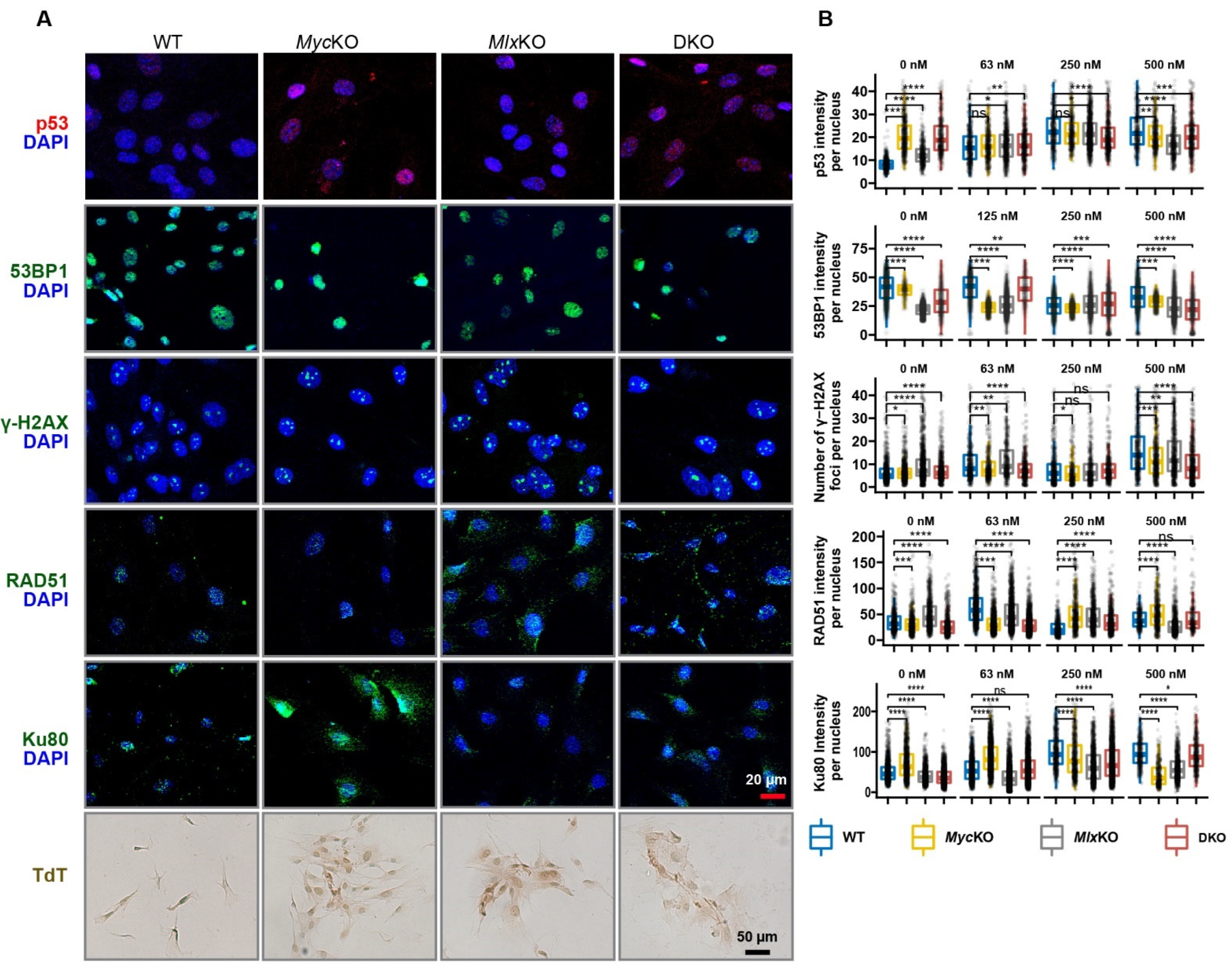
3.6. All KO MEFs Have Multiple but Distinct Defects in DNA Damage Recognition and Repair Pathways
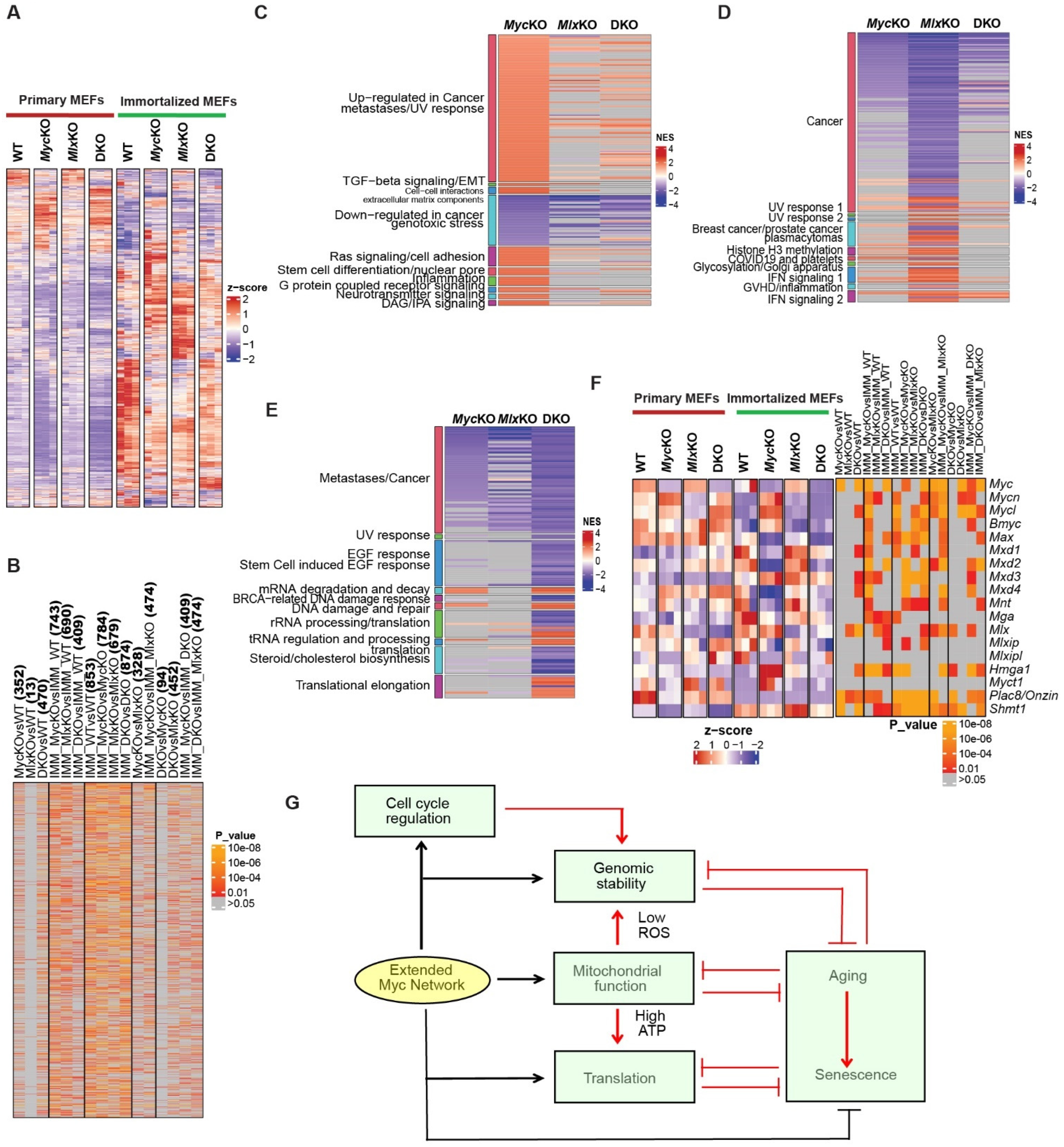
3.7. Immortalized MEFs Proliferate despite the Absence of Myc and Show Differential Chromosomal Instability and Resistance to DNA Damage
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Carroll, P.A.; Freie, B.W.; Cheng, P.F.; Kasinathan, S.; Gu, H.; Hedrich, T.; Dowdle, J.A.; Venkataramani, V.; Ramani, V.; Wu, X.; et al. The glucose-sensing transcription factor MLX balances metabolism and stress to suppress apoptosis and maintain spermatogenesis. PLoS Biol. 2021, 19, e3001085. [Google Scholar] [CrossRef]
- Prochownik, E.V.; Wang, H. Normal and Neoplastic Growth Suppression by the Extended Myc Network. Cells 2022, 11, 747. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lu, J.; Alencastro, F.; Roberts, A.; Fiedor, J.; Carroll, P.; Eisenman, R.N.; Ranganathan, S.; Torbenson, M.; Duncan, A.W.; et al. Coordinated Cross-Talk Between the Myc and Mlx Networks in Liver Regeneration and Neoplasia. Cell Mol. Gastroenterol. Hepatol. 2022, 13, 1785–1804. [Google Scholar] [CrossRef] [PubMed]
- Lourenco, C.; Resetca, D.; Redel, C.; Lin, P.; MacDonald, A.S.; Ciaccio, R.; Kenney, T.M.G.; Wei, Y.; Andrews, D.W.; Sunnerhagen, M.; et al. MYC protein interactors in gene transcription and cancer. Nat. Rev. Cancer 2021, 21, 579–591. [Google Scholar] [CrossRef]
- Gartel, A.L.; Shchors, K. Mechanisms of c-myc-mediated transcriptional repression of growth arrest genes. Exp. Cell Res. 2003, 283, 17–21. [Google Scholar] [CrossRef]
- Wiese, K.E.; Walz, S.; von Eyss, B.; Wolf, E.; Athineos, D.; Sansom, O.; Eilers, M. The role of MIZ-1 in MYC-dependent tumorigenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a014290. [Google Scholar] [CrossRef]
- Kalkat, M.; De Melo, J.; Hickman, K.A.; Lourenco, C.; Redel, C.; Resetca, D.; Tamachi, A.; Tu, W.B.; Penn, L.Z. MYC Deregulation in Primary Human Cancers. Genes 2017, 8, 151. [Google Scholar] [CrossRef] [Green Version]
- Dolezal, J.M.; Wang, H.; Kulkarni, S.; Jackson, L.; Lu, J.; Ranganathan, S.; Goetzman, E.S.; Bharathi, S.S.; Beezhold, K.; Byersdorfer, C.A.; et al. Sequential adaptive changes in a c-Myc-driven model of hepatocellular carcinoma. J. Biol. Chem. 2017, 292, 10068–10086. [Google Scholar] [CrossRef] [Green Version]
- Goetzman, E.S.; Prochownik, E.V. The Role for Myc in Coordinating Glycolysis, Oxidative Phosphorylation, Glutaminolysis, and Fatty Acid Metabolism in Normal and Neoplastic Tissues. Front. Endocrinol. 2018, 9, 129. [Google Scholar] [CrossRef] [Green Version]
- Graves, J.A.; Wang, Y.; Sims-Lucas, S.; Cherok, E.; Rothermund, K.; Branca, M.F.; Elster, J.; Beer-Stolz, D.; Van Houten, B.; Vockley, J.; et al. Mitochondrial structure, function and dynamics are temporally controlled by c-Myc. PLoS ONE 2012, 7, e37699. [Google Scholar] [CrossRef]
- Wang, H.; Dolezal, J.M.; Kulkarni, S.; Lu, J.; Mandel, J.; Jackson, L.E.; Alencastro, F.; Duncan, A.W.; Prochownik, E.V. Myc and ChREBP transcription factors cooperatively regulate normal and neoplastic hepatocyte proliferation in mice. J. Biol. Chem. 2018, 293, 14740–14757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billin, A.N.; Ayer, D.E. The Mlx network: Evidence for a parallel Max-like transcriptional network that regulates energy metabolism. Curr. Top Microbiol. Immunol. 2006, 302, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.C.; Wims, M.; Spotts, G.D.; Hann, S.R.; Bradley, A. A null c-myc mutation causes lethality before 10.5 days of gestation in homozygotes and reduced fertility in heterozygous female mice. Genes Dev. 1993, 7, 671–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edmunds, L.R.; Otero, P.A.; Sharma, L.; D’Souza, S.; Dolezal, J.M.; David, S.; Lu, J.; Lamm, L.; Basantani, M.; Zhang, P.; et al. Abnormal lipid processing but normal long-term repopulation potential of myc−/− hepatocytes. Oncotarget 2016, 7, 30379–30395. [Google Scholar] [CrossRef] [Green Version]
- Mateyak, M.K.; Obaya, A.J.; Adachi, S.; Sedivy, J.M. Phenotypes of c-Myc-deficient rat fibroblasts isolated by targeted homologous recombination. Cell Growth Differ. 1997, 8, 1039–1048. [Google Scholar]
- Trumpp, A.; Refaeli, Y.; Oskarsson, T.; Gasser, S.; Murphy, M.; Martin, G.R.; Bishop, J.M. c-Myc regulates mammalian body size by controlling cell number but not cell size. Nature 2001, 414, 768–773. [Google Scholar] [CrossRef]
- Rosselot, C.; Kumar, A.; Lakshmipathi, J.; Zhang, P.; Lu, G.; Katz, L.S.; Prochownik, E.V.; Stewart, A.F.; Lambertini, L.; Scott, D.K.; et al. Myc Is Required for Adaptive beta-Cell Replication in Young Mice but Is Not Sufficient in One-Year-Old Mice Fed With a High-Fat Diet. Diabetes 2019, 68, 1934–1949. [Google Scholar] [CrossRef]
- Yin, X.Y.; Grove, L.; Datta, N.S.; Katula, K.; Long, M.W.; Prochownik, E.V. Inverse regulation of cyclin B1 by c-Myc and p53 and induction of tetraploidy by cyclin B1 overexpression. Cancer Res. 2001, 61, 6487–6493. [Google Scholar]
- Wang, H.; Lu, J.; Kulkarni, S.; Zhang, W.; Gorka, J.E.; Mandel, J.A.; Goetzman, E.S.; Prochownik, E.V. Metabolic and oncogenic adaptations to pyruvate dehydrogenase inactivation in fibroblasts. J. Biol. Chem. 2019, 294, 5466–5486. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Lu, J.; Mandel, J.A.; Zhang, W.; Schwalbe, M.; Gorka, J.; Liu, Y.; Marburger, B.; Wang, J.; Ranganathan, S.; et al. Patient-Derived Mutant Forms of NFE2L2/NRF2 Drive Aggressive Murine Hepatoblastomas. Cell Mol. Gastroenterol. Hepatol. 2021, 12, 199–228. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.J.; Gazin, C.; Lawson, N.D.; Pages, H.; Lin, S.M.; Lapointe, D.S.; Green, M.R. ChIPpeakAnno: A Bioconductor package to annotate ChIP-seq and ChIP-chip data. BMC Bioinform. 2010, 11, 237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Sharma, L.; Lu, J.; Finch, P.; Fletcher, S.; Prochownik, E.V. Structurally diverse c-Myc inhibitors share a common mechanism of action involving ATP depletion. Oncotarget 2015, 6, 15857–15870. [Google Scholar] [CrossRef] [Green Version]
- Korolchuk, V.I.; Miwa, S.; Carroll, B.; von Zglinicki, T. Mitochondria in Cell Senescence: Is Mitophagy the Weakest Link? EBioMedicine 2017, 21, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Popay, T.M.; Wang, J.; Adams, C.M.; Howard, G.C.; Codreanu, S.G.; Sherrod, S.D.; McLean, J.A.; Thomas, L.R.; Lorey, S.L.; Machida, Y.J.; et al. MYC regulates ribosome biogenesis and mitochondrial gene expression programs through its interaction with host cell factor-1. Elife 2021, 10, e60191. [Google Scholar] [CrossRef]
- Zhuang, D.; Mannava, S.; Grachtchouk, V.; Tang, W.H.; Patil, S.; Wawrzyniak, J.A.; Berman, A.E.; Giordano, T.J.; Prochownik, E.V.; Soengas, M.S.; et al. C-MYC overexpression is required for continuous suppression of oncogene-induced senescence in melanoma cells. Oncogene 2008, 27, 6623–6634. [Google Scholar] [CrossRef] [Green Version]
- Hydbring, P.; Bahram, F.; Su, Y.; Tronnersjo, S.; Hogstrand, K.; von der Lehr, N.; Sharifi, H.R.; Lilischkis, R.; Hein, N.; Wu, S.; et al. Phosphorylation by Cdk2 is required for Myc to repress Ras-induced senescence in cotransformation. Proc. Natl. Acad. Sci. USA 2010, 107, 58–63. [Google Scholar] [CrossRef] [Green Version]
- Cerni, C. Telomeres, telomerase, and myc. An update. Mutat. Res. 2000, 462, 31–47. [Google Scholar] [CrossRef]
- Bittles, A.H.; Harper, N. Increased glycolysis in ageing cultured human diploid fibroblasts. Biosci. Rep. 1984, 4, 751–756. [Google Scholar] [CrossRef] [PubMed]
- Payea, M.J.; Anerillas, C.; Tharakan, R.; Gorospe, M. Translational Control during Cellular Senescence. Mol. Cell Biol. 2021, 41, e00512-20. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.C.; Prescott, J.E.; Campbell, S.E.; Shim, H.; Orlowski, R.Z.; Dang, C.V. Tumor induction by the c-Myc target genes rcl and lactate dehydrogenase A. Cancer Res. 2000, 60, 6178–6183. [Google Scholar]
- Goldstein, S.; Ballantyne, S.R.; Robson, A.L.; Moerman, E.J. Energy metabolism in cultured human fibroblasts during aging in vitro. J. Cell Physiol. 1982, 112, 419–424. [Google Scholar] [CrossRef]
- Osthus, R.C.; Shim, H.; Kim, S.; Li, Q.; Reddy, R.; Mukherjee, M.; Xu, Y.; Wonsey, D.; Lee, L.A.; Dang, C.V. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J. Biol. Chem. 2000, 275, 21797–21800. [Google Scholar] [CrossRef] [Green Version]
- Nikiforov, M.A.; Chandriani, S.; O’Connell, B.; Petrenko, O.; Kotenko, I.; Beavis, A.; Sedivy, J.M.; Cole, M.D. A functional screen for Myc-responsive genes reveals serine hydroxymethyltransferase, a major source of the one-carbon unit for cell metabolism. Mol. Cell Biol. 2002, 22, 5793–5800. [Google Scholar] [CrossRef] [Green Version]
- Rothermund, K.; Rogulski, K.; Fernandes, E.; Whiting, A.; Sedivy, J.; Pu, L.; Prochownik, E.V. C-Myc-independent restoration of multiple phenotypes by two C-Myc target genes with overlapping functions. Cancer Res. 2005, 65, 2097–2107. [Google Scholar] [CrossRef] [Green Version]
- Meroni, G.; Cairo, S.; Merla, G.; Messali, S.; Brent, R.; Ballabio, A.; Reymond, A. Mlx, a new Max-like bHLHZip family member: The center stage of a novel transcription factors regulatory pathway? Oncogene 2000, 19, 3266–3277. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Brouwer, C. Pathview: An R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics 2013, 29, 1830–1831. [Google Scholar] [CrossRef] [Green Version]
- Boddu, P.C.; Kadia, T.M. Molecular pathogenesis of acquired aplastic anemia. Eur. J. Haematol. 2019, 102, 103–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedelnikova, O.A.; Horikawa, I.; Redon, C.; Nakamura, A.; Zimonjic, D.B.; Popescu, N.C.; Bonner, W.M. Delayed kinetics of DNA double-strand break processing in normal and pathological aging. Aging Cell 2008, 7, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.W. Telomeres and aging. Curr. Opin. Cell Biol. 2018, 52, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ness, K.K.; Kirkland, J.L.; Gramatges, M.M.; Wang, Z.; Kundu, M.; McCastlain, K.; Li-Harms, X.; Zhang, J.; Tchkonia, T.; Pluijm, S.M.F.; et al. Premature Physiologic Aging as a Paradigm for Understanding Increased Risk of Adverse Health Across the Lifespan of Survivors of Childhood Cancer. J. Clin. Oncol. 2018, 36, 2206–2215. [Google Scholar] [CrossRef] [PubMed]
- Richardson, R.B. Ionizing radiation and aging: Rejuvenating an old idea. Aging 2009, 1, 887–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, R.R.; Milholland, B.; de Bruin, A.; Curran, S.; Laberge, R.M.; van Steeg, H.; Campisi, J.; Maslov, A.Y.; Vijg, J. Controlled induction of DNA double-strand breaks in the mouse liver induces features of tissue ageing. Nat. Commun. 2015, 6, 6790. [Google Scholar] [CrossRef] [Green Version]
- Vafa, O.; Wade, M.; Kern, S.; Beeche, M.; Pandita, T.K.; Hampton, G.M.; Wahl, G.M. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: A mechanism for oncogene-induced genetic instability. Mol. Cell 2002, 9, 1031–1044. [Google Scholar] [CrossRef]
- Prochownik, E.V. c-Myc: Linking transformation and genomic instability. Curr. Mol. Med. 2008, 8, 446–458. [Google Scholar] [CrossRef]
- Wang, Y.H.; Ho, T.L.F.; Hariharan, A.; Goh, H.C.; Wong, Y.L.; Verkaik, N.S.; Lee, M.Y.; Tam, W.L.; van Gent, D.C.; Venkitaraman, A.R.; et al. Rapid recruitment of p53 to DNA damage sites directs DNA repair choice and integrity. Proc. Natl. Acad. Sci. USA 2022, 119, e2113233119. [Google Scholar] [CrossRef]
- Williams, A.B.; Schumacher, B. p53 in the DNA-Damage-Repair Process. Cold Spring Harb Perspect Med. 2016, 6, a026070. [Google Scholar] [CrossRef] [Green Version]
- Gregoire, M.C.; Massonneau, J.; Leduc, F.; Arguin, M.; Brazeau, M.A.; Boissonneault, G. Quantification and genome-wide mapping of DNA double-strand breaks. DNA Repair 2016, 48, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, J.W.; Zhao, X.; De Cecco, M.; Peterson, A.L.; Pagliaroli, L.; Manivannan, J.; Hubbard, G.B.; Ikeno, Y.; Zhang, Y.; Feng, B.; et al. Reduced expression of MYC increases longevity and enhances healthspan. Cell 2015, 160, 477–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef] [PubMed]
- Hydbring, P.; Larsson, L.G. Cdk2: A key regulator of the senescence control function of Myc. Aging 2010, 2, 244–250. [Google Scholar] [CrossRef] [Green Version]
- Xu, J. Preparation, culture, and immortalization of mouse embryonic fibroblasts. Curr. Protoc. Mol. Biol. 2005, 70. [Google Scholar] [CrossRef]
- Adimoolam, S.; Ford, J.M. p53 and regulation of DNA damage recognition during nucleotide excision repair. DNA Repair 2003, 2, 947–954. [Google Scholar] [CrossRef]
- Cook, R.; Zoumpoulidou, G.; Luczynski, M.T.; Rieger, S.; Moquet, J.; Spanswick, V.J.; Hartley, J.A.; Rothkamm, K.; Huang, P.H.; Mittnacht, S. Direct involvement of retinoblastoma family proteins in DNA repair by non-homologous end-joining. Cell Rep. 2015, 10, 2006–2018. [Google Scholar] [CrossRef] [Green Version]
- Velez-Cruz, R.; Manickavinayaham, S.; Biswas, A.K.; Clary, R.W.; Premkumar, T.; Cole, F.; Johnson, D.G. RB localizes to DNA double-strand breaks and promotes DNA end resection and homologous recombination through the recruitment of BRG1. Genes Dev. 2016, 30, 2500–2512. [Google Scholar] [CrossRef] [Green Version]
- Rogulski, K.; Li, Y.; Rothermund, K.; Pu, L.; Watkins, S.; Yi, F.; Prochownik, E.V. Onzin, a c-Myc-repressed target, promotes survival and transformation by modulating the Akt-Mdm2-p53 pathway. Oncogene 2005, 24, 7524–7541. [Google Scholar] [CrossRef] [Green Version]
- Yin, X.; Grove, L.; Rogulski, K.; Prochownik, E.V. Myc target in myeloid cells-1, a novel c-Myc target, recapitulates multiple c-Myc phenotypes. J. Biol. Chem. 2002, 277, 19998–20010. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.M. Replicative senescence and oxidant-induced premature senescence. Beyond the control of cell cycle checkpoints. Ann. N. Y. Acad. Sci. 2000, 908, 111–125. [Google Scholar] [CrossRef] [PubMed]
- Jongmans, W.; Vuillaume, M.; Chrzanowska, K.; Smeets, D.; Sperling, K.; Hall, J. Nijmegen breakage syndrome cells fail to induce the p53-mediated DNA damage response following exposure to ionizing radiation. Mol. Cell Biol. 1997, 17, 5016–5022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rego, M.A.; Kolling, F.W.t.; Howlett, N.G. The Fanconi anemia protein interaction network: Casting a wide net. Mutat. Res. 2009, 668, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Ceraline, J.; Deplanque, G.; Duclos, B.; Limacher, J.M.; Hajri, A.; Noel, F.; Orvain, C.; Frebourg, T.; Klein-Soyer, C.; Bergerat, J.P. Inactivation of p53 in normal human cells increases G2/M arrest and sensitivity to DNA-damaging agents. Int. J. Cancer 1998, 75, 432–438. [Google Scholar] [CrossRef]
- Galofre, C.; Gonul Geyik, O.; Asensio, E.; Wangsa, D.; Hirsch, D.; Parra, C.; Saez, J.; Molla, M.; Yuce, Z.; Castells, A.; et al. Tetraploidy-Associated Genetic Heterogeneity Confers Chemo-Radiotherapy Resistance to Colorectal Cancer Cells. Cancers 2020, 12, 1118. [Google Scholar] [CrossRef] [PubMed]
- Beausejour, C.M.; Krtolica, A.; Galimi, F.; Narita, M.; Lowe, S.W.; Yaswen, P.; Campisi, J. Reversal of human cellular senescence: Roles of the p53 and p16 pathways. EMBO J. 2003, 22, 4212–4222. [Google Scholar] [CrossRef] [PubMed]
- Bratic, A.; Larsson, N.G. The role of mitochondria in aging. J. Clin. Investig. 2013, 123, 951–957. [Google Scholar] [CrossRef] [Green Version]
- Gerashchenko, M.V.; Peterfi, Z.; Yim, S.H.; Gladyshev, V.N. Translation elongation rate varies among organs and decreases with age. Nucleic Acids Res. 2021, 49, e9. [Google Scholar] [CrossRef]
- Srivastava, S. The Mitochondrial Basis of Aging and Age-Related Disorders. Genes 2017, 8, 398. [Google Scholar] [CrossRef] [Green Version]
- Turi, Z.; Lacey, M.; Mistrik, M.; Moudry, P. Impaired ribosome biogenesis: Mechanisms and relevance to cancer and aging. Aging 2019, 11, 2512–2540. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Stevens, T.; Lu, J.; Airik, M.; Airik, R.; Prochownik, E.V. Disruption of Multiple Overlapping Functions Following Stepwise Inactivation of the Extended Myc Network. Cells 2022, 11, 4087. https://doi.org/10.3390/cells11244087
Wang H, Stevens T, Lu J, Airik M, Airik R, Prochownik EV. Disruption of Multiple Overlapping Functions Following Stepwise Inactivation of the Extended Myc Network. Cells. 2022; 11(24):4087. https://doi.org/10.3390/cells11244087
Chicago/Turabian StyleWang, Huabo, Taylor Stevens, Jie Lu, Merlin Airik, Rannar Airik, and Edward V. Prochownik. 2022. "Disruption of Multiple Overlapping Functions Following Stepwise Inactivation of the Extended Myc Network" Cells 11, no. 24: 4087. https://doi.org/10.3390/cells11244087