


Pre-Transplant Prediction of Acute Graft-versus-Host Disease Using the Gut Microbiome
 , , ,
, , ,
Abstract
:1. Introduction
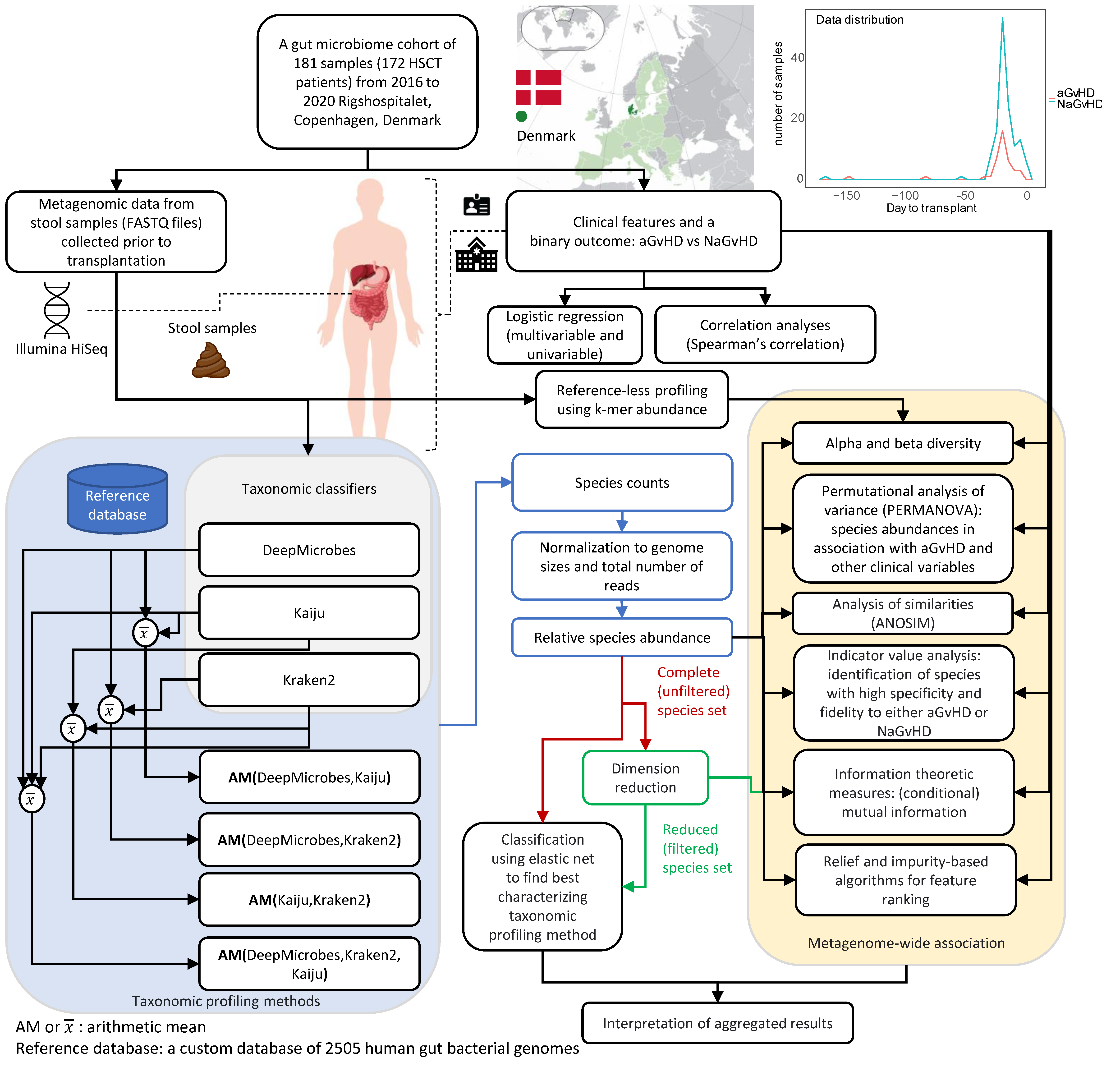
2. Materials and Methods
2.1. Data Collection and Categorization
2.2. Data Analysis
2.3. Clinical Variables
2.4. Taxonomic Classification
2.5. Benchmarking the Taxonomy Profiling Configurations
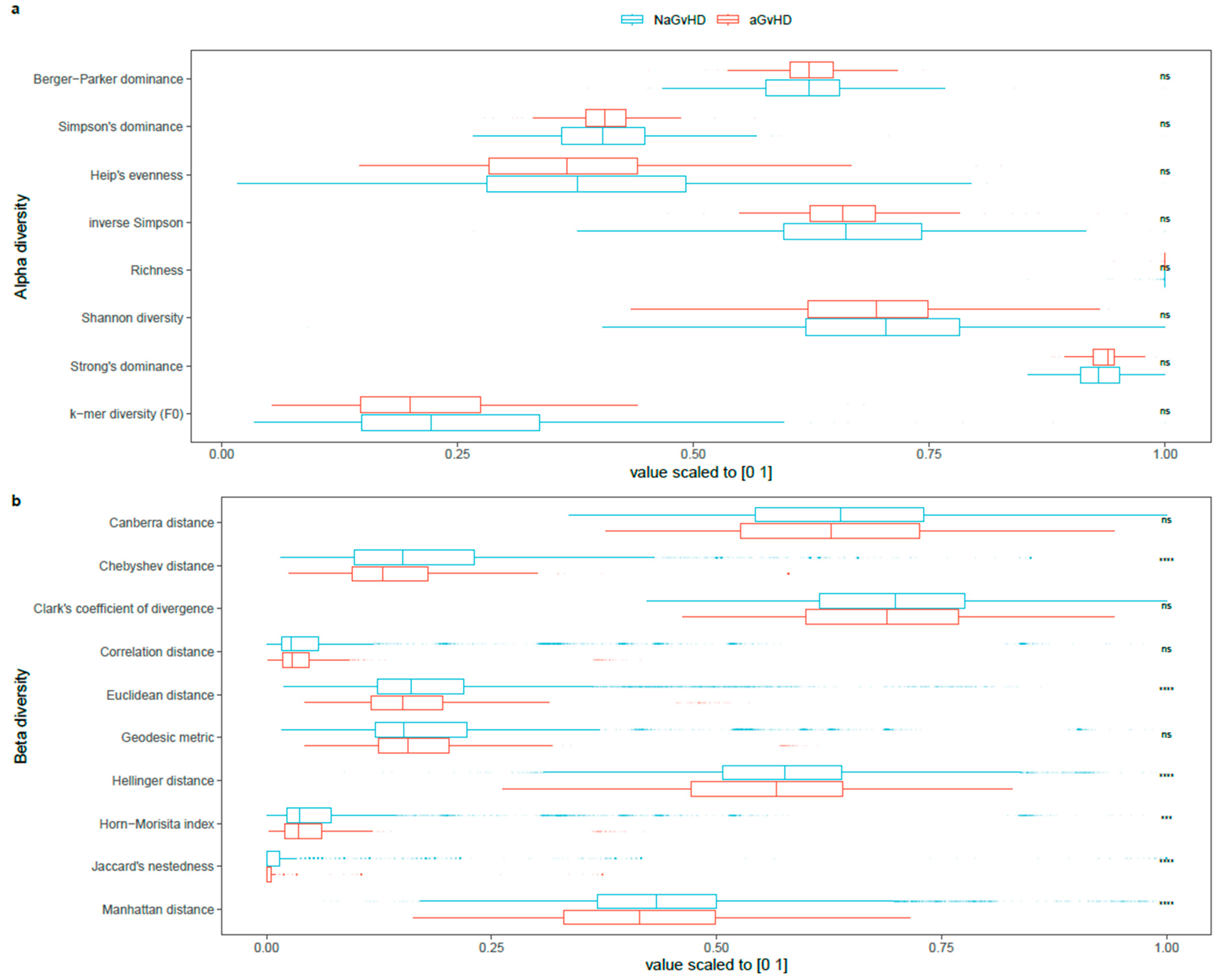
2.6. Diversity Measures
2.7. Dissimilarity Measures
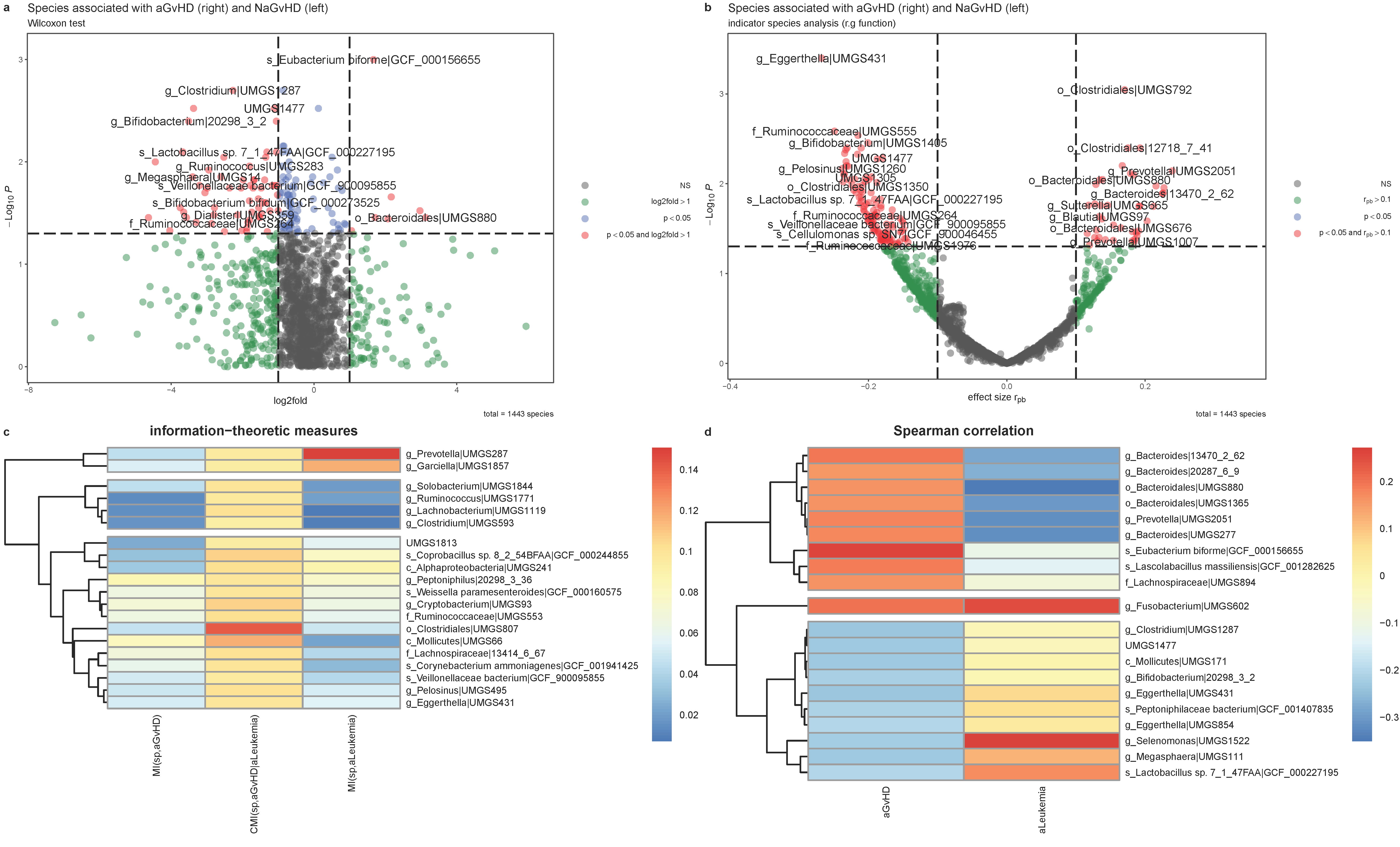
2.8. Information-Theoretic Measures
2.9. Relief and Impurity-Based Algorithms for Metagenomic Feature Ranking
3. Results
3.1. Cohort Characteristics: Clinical and Metagenomic Data
3.2. Clinical Variables in Association with aGvHD
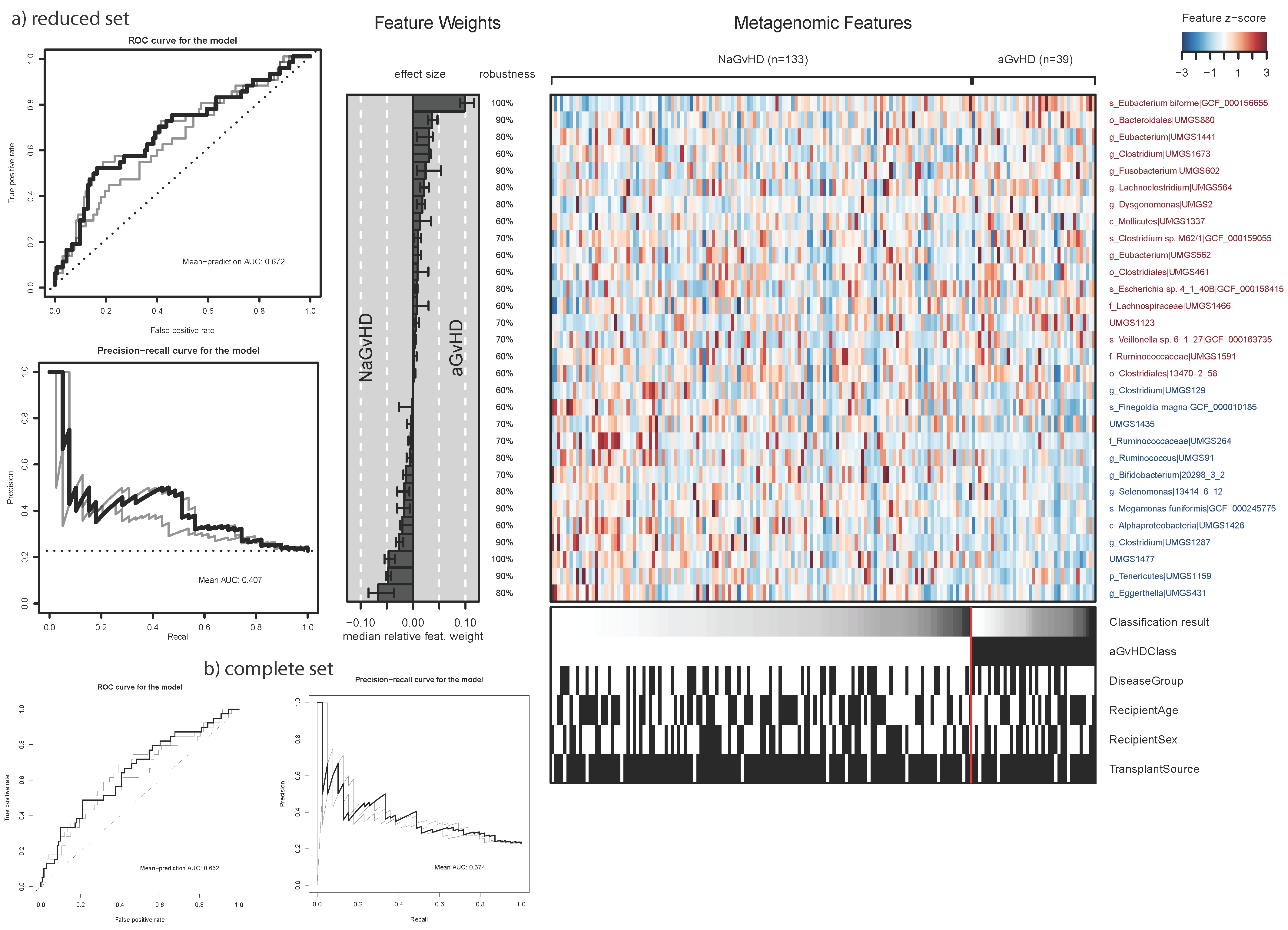
3.3. Benchmarking the Taxonomy Profiling Configurations
3.4. Species Diversity in Association with aGvHD
3.5. Dissimilarity Measures
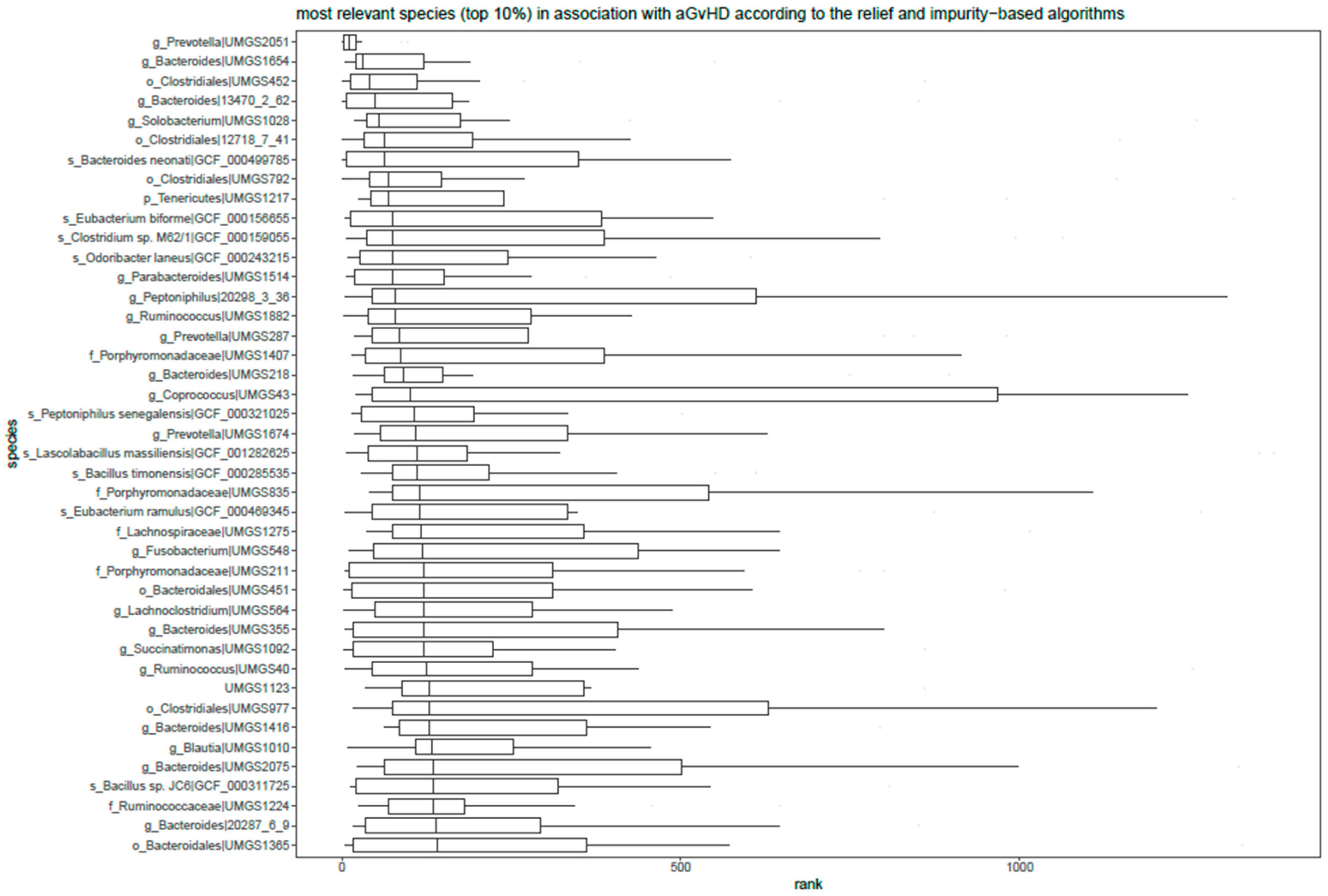
3.6. Relief and Impurity-Based Algorithms for Feature Ranking
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jacobsohn, D.A.; Vogelsang, G.B. Acute Graft versus Host Disease. Orphanet J. Rare Dis. 2007, 2, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.-S.; Huang, X.-J. Seeking Biomarkers for Acute Graft-versus-Host Disease: Where We Are and Where We Are Heading? Biomark. Res. 2019, 7, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, E.M.; Moate, P.J.; Marett, L.C.; Cocks, B.G.; Hayes, B.J. Metagenomic Predictions: From Microbiome to Complex Health and Environmental Phenotypes in Humans and Cattle. PLoS ONE 2013, 8, e73056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.; Haneuse, S.; Wang, H.-L.; Rose, S.; Spellman, S.R.; Verneris, M.; Hsu, K.C.; Fleischhauer, K.; Lee, S.J.; Abdi, R. Prediction of Absolute Risk of Acute Graft-versus-Host Disease Following Hematopoietic Cell Transplantation. PLoS ONE 2018, 13, e0190610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingham, A.C.; Kielsen, K.; Mordhorst, H.; Ifversen, M.; Aarestrup, F.M.; Müller, K.G.; Pamp, S.J. Microbiota Long-Term Dynamics and Prediction of Acute Graft-versus-Host-Disease in Pediatric Allogeneic Stem Cell Transplantation. Microbiome 2021, 9, 148. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Jin, H.; Zhou, L.; Zhang, X.; Fan, Z.; Dai, M.; Lin, Q.; Huang, F.; Xuan, L.; Zhang, H.; et al. Intestinal Microbiota at Engraftment Influence Acute Graft-versus-Host Disease via the Treg/Th17 Balance in Allo-HSCT Recipients. Front. Immunol. 2018, 9, 669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Xu, S.; Ren, Z.; Jiang, J.; Zheng, S. Gut Microbiota and Allogeneic Transplantation. J. Transl. Med. 2015, 13, 275. [Google Scholar] [CrossRef] [PubMed]
- Ilett, E.E.; Jørgensen, M.; Noguera-Julian, M.; Nørgaard, J.C.; Daugaard, G.; Helleberg, M.; Paredes, R.; Murray, D.D.; Lundgren, J.; MacPherson, C.; et al. Associations of the Gut Microbiome and Clinical Factors with Acute GVHD in Allogeneic HSCT Recipients. Blood Adv. 2020, 4, 5797–5809. [Google Scholar] [CrossRef]
- Liang, Q.; Bible, P.W.; Liu, Y.; Zou, B.; Wei, L. DeepMicrobes: Taxonomic Classification for Metagenomics with Deep Learning. NAR Genom. Bioinform. 2020, 2, lqaa009. [Google Scholar] [CrossRef] [Green Version]
- Wood, D.E.; Lu, J.; Langmead, B. Improved Metagenomic Analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef]
- Menzel, P.; Ng, K.L.; Krogh, A. Fast and Sensitive Taxonomic Classification for Metagenomics with Kaiju. Nat. Commun. 2016, 7, 11257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2019. [Google Scholar]
- Bacigalupo, A.; Ballen, K.; Rizzo, D.; Giralt, S.; Lazarus, H.; Ho, V.; Apperley, J.; Slavin, S.; Pasquini, M.; Sandmaier, B.M.; et al. Defining the Intensity of Conditioning Regimens: Working Definitions. Biol. Blood Marrow Transplant. 2009, 15, 1628–1633. [Google Scholar] [CrossRef] [Green Version]
- Smirnova, E.; Huzurbazar, S.; Jafari, F. PERFect: PERmutation Filtering Test for Microbiome Data. Biostatistics 2019, 20, 615–631. [Google Scholar] [CrossRef]
- Melsted, P.; Halldórsson, B.V. KmerStream: Streaming Algorithms for k-Mer Abundance Estimation. Bioinformatics 2014, 30, 3541–3547. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Anderson, M.J.; Walsh, D.C.I. PERMANOVA, ANOSIM, and the Mantel Test in the Face of Heterogeneous Dispersions: What Null Hypothesis Are You Testing? Ecol. Monogr. 2013, 83, 557–574. [Google Scholar] [CrossRef]
- Barua, S.; Islam, M.M.; Yao, X.; Murase, K. MWMOTE--Majority Weighted Minority Oversampling Technique for Imbalanced Data Set Learning. IEEE Trans. Knowl. Data Eng. 2014, 26, 405–425. [Google Scholar] [CrossRef]
- De Cáceres, M.; Legendre, P. Associations between Species and Groups of Sites: Indices and Statistical Inference. Ecology 2009, 90, 3566–3574. [Google Scholar] [CrossRef]
- Dufrêne, M.; Legendre, P. Species Assemblages and Indicator Species: The Need for a Flexible Asymmetrical Approach. Ecol. Monogr. 1997, 67, 345–366. [Google Scholar] [CrossRef]
- De Cáceres, M.; Legendre, P.; Moretti, M. Improving Indicator Species Analysis by Combining Groups of Sites. Oikos 2010, 119, 1674–1684. [Google Scholar] [CrossRef]
- Kira, K.; Rendell, L.A. A Practical Approach to Feature Selection. In Machine Learning Proceedings 1992; Elsevier: Amsterdam, The Netherlands, 1992; pp. 249–256. [Google Scholar]
- Robnik-Šikonja, M. Experiments with Cost-Sensitive Feature Evaluation. In European Conference on Machine Learning; Springer: Berlin/Heidelberg, Germany, 2003; pp. 325–336. [Google Scholar]
- Robnik-Šikonja, M.; Kononenko, I. Theoretical and Empirical Analysis of ReliefF and RReliefF. Mach. Learn. 2003, 53, 23–69. [Google Scholar] [CrossRef] [Green Version]
- Przepiorka, D.; Weisdorf, D.; Martin, P.; Klingemann, H.G.; Beatty, P.; Hows, J.; Thomas, E.D. 1994 Consensus Conference on Acute GVHD Grading. Bone Marrow Transplant. 1995, 15, 825–828. [Google Scholar]
- Glucksberg, H.R.; Storb, R.; Fefer, A.; Buckner, C.D.; Neiman, P.E.; Clift, R.A.; Lerner, K.G.; Thomas, E.D. Clinical Manifestations of Graft-versus-Host Disease in Human Recipients of Marrow from HL-A-Matched Sibling Donor, S. Transplantation 1974, 18, 295–304. [Google Scholar] [CrossRef]
- Han, L.; Zhao, K.; Li, Y.; Han, H.; Zhou, L.; Ma, P.; Fan, Z.; Sun, H.; Jin, H.; Jiang, Z.; et al. A Gut Microbiota Score Predicting Acute Graft-versus-Host Disease Following Myeloablative Allogeneic Hematopoietic Stem Cell Transplantation. Am. J. Transplant. 2020, 20, 1014–1027. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.; Chappell, G.T.; Mazzoli, A.; Tewari, M.; Choi, S.W.; Wiens, J. Predicting Acute Graft-Versus-Host Disease Using Machine Learning and Longitudinal Vital Sign Data From Electronic Health Records. JCO Clin. Cancer Inform. 2020, 4, 128–135. [Google Scholar] [CrossRef]
- Nakasone, H.; Fukuda, T.; Kanda, J.; Mori, T.; Yano, S.; Kobayashi, T.; Miyamura, K.; Eto, T.; Kanamori, H.; Iwato, K.; et al. Impact of Conditioning Intensity and TBI on Acute GVHD after Hematopoietic Cell Transplantation. Bone Marrow Transplant. 2015, 50, 559–565. [Google Scholar] [CrossRef] [Green Version]
- Nassereddine, S.; Rafei, H.; Elbahesh, E.; Tabbara, I. Acute Graft versus Host Disease: A Comprehensive Review. Anticancer Res. 2017, 37, 1547–1555. [Google Scholar] [CrossRef] [Green Version]
- Jørgensen, M.; Nørgaard, J.C.; Ilett, E.E.; Marandi, R.Z.; Noguera-Julian, M.; Paredes, R.; Murray, D.D.; Lundgren, J.; MacPherson, C.R.; Sengeløv, H. Metabolic Potential of the Gut Microbiome Is Significantly Impacted by Conditioning Regimen in Allogeneic Hematopoietic Stem Cell Transplantation Recipients. Int. J. Mol. Sci. 2022, 23, 11115. [Google Scholar] [CrossRef]
- Grandgirard, J.; Poinsot, D.; Krespi, L.; Nénon, J.P.; Cortesero, A.M. Logratio Analysis and Compositional Distance. Entomol. Exp. Appl. 2002, 103, 239–248. [Google Scholar] [CrossRef]
- Payen, M.; Nicolis, I.; Robin, M.; Michonneau, D.; Delannoye, J.; Mayeur, C.; Kapel, N.; Berçot, B.; Butel, M.J.; Le Goff, J.; et al. Functional and Phylogenetic Alterations in Gut Microbiome Are Linked to Graft-versus-Host Disease Severity. Blood Adv. 2020, 4, 1824–1832. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lin, Y.; Li, X.; Xu, X.; Zhao, Y.; Xu, L.; Gao, Y.; Li, Y.; Tan, Y.; Qian, P.; et al. Tyrosine Supplement Ameliorates Murine AGVHD by Modulation of Gut Microbiome and Metabolome. EBioMedicine 2020, 61, 103048. [Google Scholar] [CrossRef] [PubMed]
- Biagi, E.; Zama, D.; Nastasi, C.; Consolandi, C.; Fiori, J.; Rampelli, S.; Turroni, S.; Centanni, M.; Severgnini, M.; Peano, C.; et al. Gut Microbiota Trajectory in Pediatric Patients Undergoing Hematopoietic SCT. Bone Marrow Transplant. 2015, 50, 992–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morjaria, S.; Schluter, J.; Taylor, B.P.; Littmann, E.R.; Carter, R.A.; Fontana, E.; Peled, J.U.; van den Brink, M.R.M.; Xavier, J.B.; Taur, Y. Antibiotic-Induced Shifts in Fecal Microbiota Density and Composition during Hematopoietic Stem Cell Transplantation. Infect. Immun. 2019, 87, e00206-19. [Google Scholar] [CrossRef] [Green Version]
- Beye, M.; Bakour, S.; Traore, S.I.; Raoult, D.; Fournier, P.E. “Lascolabacillus Massiliensis”: A New Species Isolated from the Human Gut. New Microbes New Infect. 2016, 11, 91–92. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Base Level | Alternate Level | β Coefficient 95% CI | β (SE) | z | p | VIF |
|---|---|---|---|---|---|---|---|
| (Intercept) | −3.517 (−6.829 −0.777) | 1.533 | −2.295 | 0.022 | |||
| Conditioning | non-myeloablative | myeloablative | 1.759 (0.069 3.694) | 0.913 | 1.926 | 0.054 | 5.921 |
| Cyclophosphamide | none | used | −0.476 (−2.662 1.638) | 1.07 | −0.445 | 0.656 | 3.243 |
| Disease group | acute leukemia | other hematological malignancies | −0.235 (−1.037 0.587) | 0.411 | −0.571 | 0.568 | 1.154 |
| DR relationship | related | unrelated | 0.318 (−0.566 1.293) | 0.469 | 0.677 | 0.498 | 1.065 |
| Donor sex | male | female | 0.606 (−0.358 1.59) | 0.494 | 1.228 | 0.219 | 1.702 |
| Karnofsky D0 | <90 | 90≥ | 0.166 (−0.631 0.943) | 0.399 | 0.417 | 0.677 | 1.075 |
| Radiation group | none | irradiated | 1.204 (−0.479 3.126) | 0.907 | 1.327 | 0.184 | 4.983 |
| Recipient age | <60 | 60≥ | 0.186 (−0.662 1.072) | 0.439 | 0.423 | 0.672 | 1.388 |
| Recipient sex | male | female | −0.29 (−1.278 0.655) | 0.49 | −0.592 | 0.554 | 1.709 |
| Transplant source | bone marrow | peripheral blood | 0.441 (−1.237 2.294) | 0.88 | 0.501 | 0.617 | 1.896 |
| Variable | Df | Sum Sq | R2 | F | p |
|---|---|---|---|---|---|
| aGvHD group (aGvHD vs. NaGvHD) | 1 | 5932 | 0.011 | 2.001 | 0.045 * |
| Disease group (acute leukemia vs. other hematological malignancies) | 1 | 28,525 | 0.052 | 9.621 | <0.001 *** |
| Recipient age (<60 vs. 60≥) | 1 | 4183 | 0.008 | 1.411 | 0.147 |
| Recipient sex (male vs. female) | 1 | 11,783 | 0.021 | 3.974 | 0.014 * |
| Residual | 167 | 495,129 | 0.899 | ||
| Total | 171 | 550,634 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zargari Marandi, R.; Jørgensen, M.; Ilett, E.E.; Nørgaard, J.C.; Noguera-Julian, M.; Paredes, R.; Lundgren, J.D.; Sengeløv, H.; MacPherson, C.R. Pre-Transplant Prediction of Acute Graft-versus-Host Disease Using the Gut Microbiome. Cells 2022, 11, 4089. https://doi.org/10.3390/cells11244089
Zargari Marandi R, Jørgensen M, Ilett EE, Nørgaard JC, Noguera-Julian M, Paredes R, Lundgren JD, Sengeløv H, MacPherson CR. Pre-Transplant Prediction of Acute Graft-versus-Host Disease Using the Gut Microbiome. Cells. 2022; 11(24):4089. https://doi.org/10.3390/cells11244089
Chicago/Turabian StyleZargari Marandi, Ramtin, Mette Jørgensen, Emma Elizabeth Ilett, Jens Christian Nørgaard, Marc Noguera-Julian, Roger Paredes, Jens D. Lundgren, Henrik Sengeløv, and Cameron Ross MacPherson. 2022. "Pre-Transplant Prediction of Acute Graft-versus-Host Disease Using the Gut Microbiome" Cells 11, no. 24: 4089. https://doi.org/10.3390/cells11244089