Author Contributions
Conceptualization, M.M.; methodology, J.L., M.S., S.W., J.W. and S.H.; validation, J.L. and M.M.; formal analysis, J.L. and M.M.; investigation, J.L., M.S., S.W., J.W. and S.H.; resources, M.M. and H.S.; data curation, J.L.; writing—original draft preparation, J.L. and M.M.; writing—review and editing, J.L., S.H., H.S., M.S. and M.M.; visualization, J.L., J.W. and M.S.; supervision, S.H., H.S. and M.M.; project administration, M.M.; funding acquisition, M.M., H.S. and S.H. All authors have read and agreed to the published version of the manuscript.
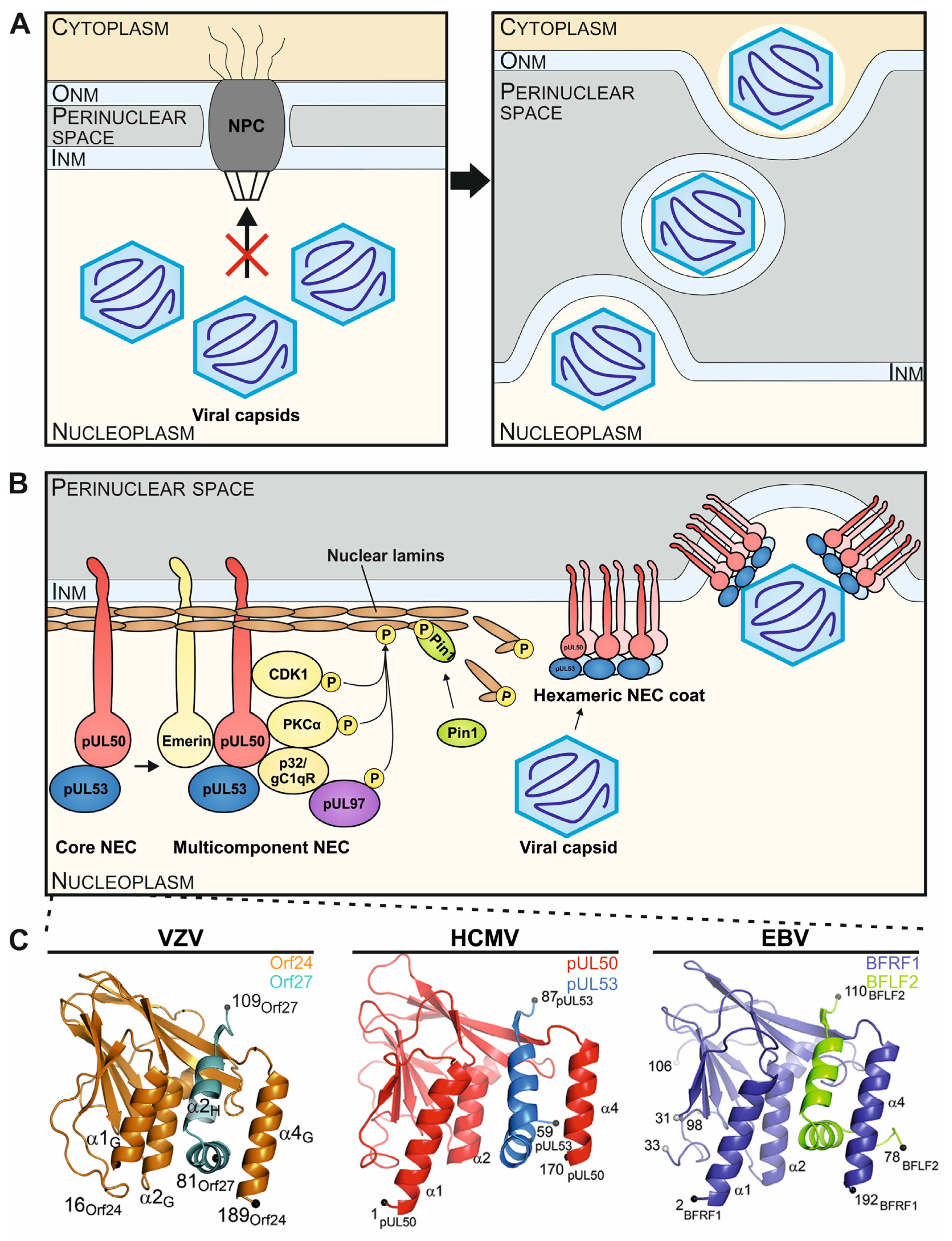
Figure 1.
Illustration of the principle and multifunctional properties of the herpesviral nuclear egress complex (NEC) through the formation of core and multicomponent NEC arrangements. (
A) Herpesviral capsids, assembled in the nucleus of infected cells, exceed the size exclusion limits of the nuclear pore complex (NPC), so that the fine-regulated process of nuclear egress evolved to enable the efficient nucleocytoplasmic transition of capsids through the nuclear envelope (INM and ONM, inner and outer nuclear membranes). (
B) A schematic representation visualizes the stepwise formation of the HCMV-specific NEC, in its heterodimeric and hexameric core versions, and the multicomponent extension, including NEC-associated proteins. The core NEC, consisting of pUL50 and pUL53, recruits several cellular and viral proteins for the temporary reorganization of the nuclear lamina. A hexameric arrangement of the core NEC facilitates the docking and egress of viral capsids through the INM into the perinuclear space (refined illustrations referring to [
20]). (
C) Crystal structures of the VZV Orf24-Orf27, EBV BFRF1-BFLF2 and HCMV pUL50-pUL53 hook-into-groove formations in ribbon representations (refined illustrations referring to [
14,
18,
19]).
Figure 1.
Illustration of the principle and multifunctional properties of the herpesviral nuclear egress complex (NEC) through the formation of core and multicomponent NEC arrangements. (
A) Herpesviral capsids, assembled in the nucleus of infected cells, exceed the size exclusion limits of the nuclear pore complex (NPC), so that the fine-regulated process of nuclear egress evolved to enable the efficient nucleocytoplasmic transition of capsids through the nuclear envelope (INM and ONM, inner and outer nuclear membranes). (
B) A schematic representation visualizes the stepwise formation of the HCMV-specific NEC, in its heterodimeric and hexameric core versions, and the multicomponent extension, including NEC-associated proteins. The core NEC, consisting of pUL50 and pUL53, recruits several cellular and viral proteins for the temporary reorganization of the nuclear lamina. A hexameric arrangement of the core NEC facilitates the docking and egress of viral capsids through the INM into the perinuclear space (refined illustrations referring to [
20]). (
C) Crystal structures of the VZV Orf24-Orf27, EBV BFRF1-BFLF2 and HCMV pUL50-pUL53 hook-into-groove formations in ribbon representations (refined illustrations referring to [
14,
18,
19]).
![Cells 11 04030 g001]()
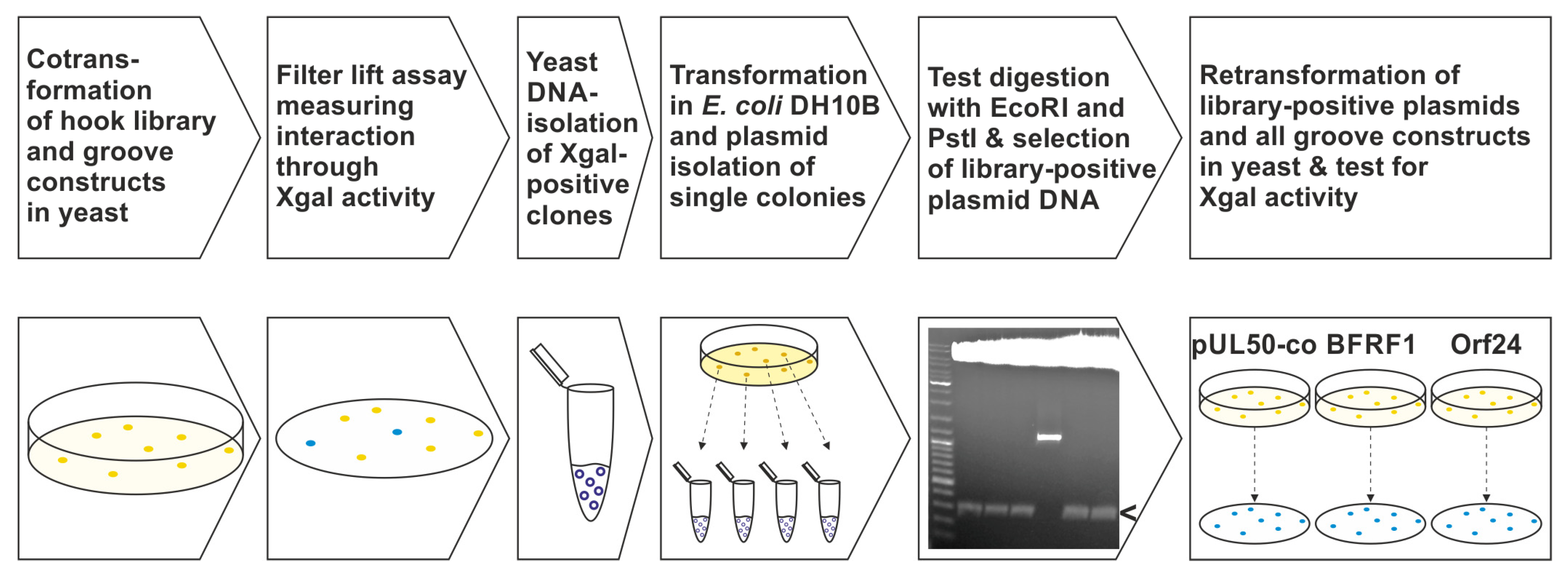
Figure 2.
The methodological approach of the Y2H screening and further validation using a randomized pUL53 hook mutagenesis library and the groove constructs of VZV, HCMV and EBV. 1st and 2nd steps: cotransformation of the library with either pUL50, pUL50-co, BFRF1 or Orf24 in the yeast strain Y153 and measurement of Xgal activity. 3rd and fourth steps: library DNA isolation of interacting clones, transformation in bacteria and plasmid isolation of single colonies. 5th step: restriction enzyme digestion of plasmid DNA for selection of library DNA with an insert size of approximately 300 bp (i.e., confirming the hook construct), indicated by the arrow. 6th step: retransformation of confirmed library plasmids along with pUL50, pUL50-co, BFRF1 or Orf24, respectively, and testing for so-called shared-hook activity.
Figure 2.
The methodological approach of the Y2H screening and further validation using a randomized pUL53 hook mutagenesis library and the groove constructs of VZV, HCMV and EBV. 1st and 2nd steps: cotransformation of the library with either pUL50, pUL50-co, BFRF1 or Orf24 in the yeast strain Y153 and measurement of Xgal activity. 3rd and fourth steps: library DNA isolation of interacting clones, transformation in bacteria and plasmid isolation of single colonies. 5th step: restriction enzyme digestion of plasmid DNA for selection of library DNA with an insert size of approximately 300 bp (i.e., confirming the hook construct), indicated by the arrow. 6th step: retransformation of confirmed library plasmids along with pUL50, pUL50-co, BFRF1 or Orf24, respectively, and testing for so-called shared-hook activity.
Figure 3.
Sequence alignment of homologous hook proteins, highlighting conserved amino acids and amino acids representing contact interfaces. Increasing levels of sequence conservation are indicated by darker shades of green in the alignment. The buried surface area at hook-into-groove contact positions in α-, β-, and γ-herpesviruses is indicated by grey squares. Darker shades of grey indicate a larger buried surface. Lower-case letters mark those residues that were not resolved in the crystal structure (refined illustration referring to [
17]). Note, that HCMV pUL53 is the only hook protein with an alanine at position 85, whereas the amino acid proline is conserved at this residue for the other hook proteins. The elements of the secondary structure are depicted schematically below the alignment. HSV-1, herpes simplex virus type 1; HSV-2, herpes simplex virus type 2; VZV, varicella zoster virus; PRV, pseudorabies virus; HCMV, human cytomegalovirus; MCMV, murine cytomegalovirus; HHV-6A, human herpesvirus 6A; HHV-6B, human herpesvirus 6B; HHV-7, human herpesvirus 7; EBV, Epstein-Barr virus; KSHV, Kaposi’s sarcoma-associated herpesvirus; MHV-68, murine herpesvirus 68.
Figure 3.
Sequence alignment of homologous hook proteins, highlighting conserved amino acids and amino acids representing contact interfaces. Increasing levels of sequence conservation are indicated by darker shades of green in the alignment. The buried surface area at hook-into-groove contact positions in α-, β-, and γ-herpesviruses is indicated by grey squares. Darker shades of grey indicate a larger buried surface. Lower-case letters mark those residues that were not resolved in the crystal structure (refined illustration referring to [
17]). Note, that HCMV pUL53 is the only hook protein with an alanine at position 85, whereas the amino acid proline is conserved at this residue for the other hook proteins. The elements of the secondary structure are depicted schematically below the alignment. HSV-1, herpes simplex virus type 1; HSV-2, herpes simplex virus type 2; VZV, varicella zoster virus; PRV, pseudorabies virus; HCMV, human cytomegalovirus; MCMV, murine cytomegalovirus; HHV-6A, human herpesvirus 6A; HHV-6B, human herpesvirus 6B; HHV-7, human herpesvirus 7; EBV, Epstein-Barr virus; KSHV, Kaposi’s sarcoma-associated herpesvirus; MHV-68, murine herpesvirus 68.
![Cells 11 04030 g003]()
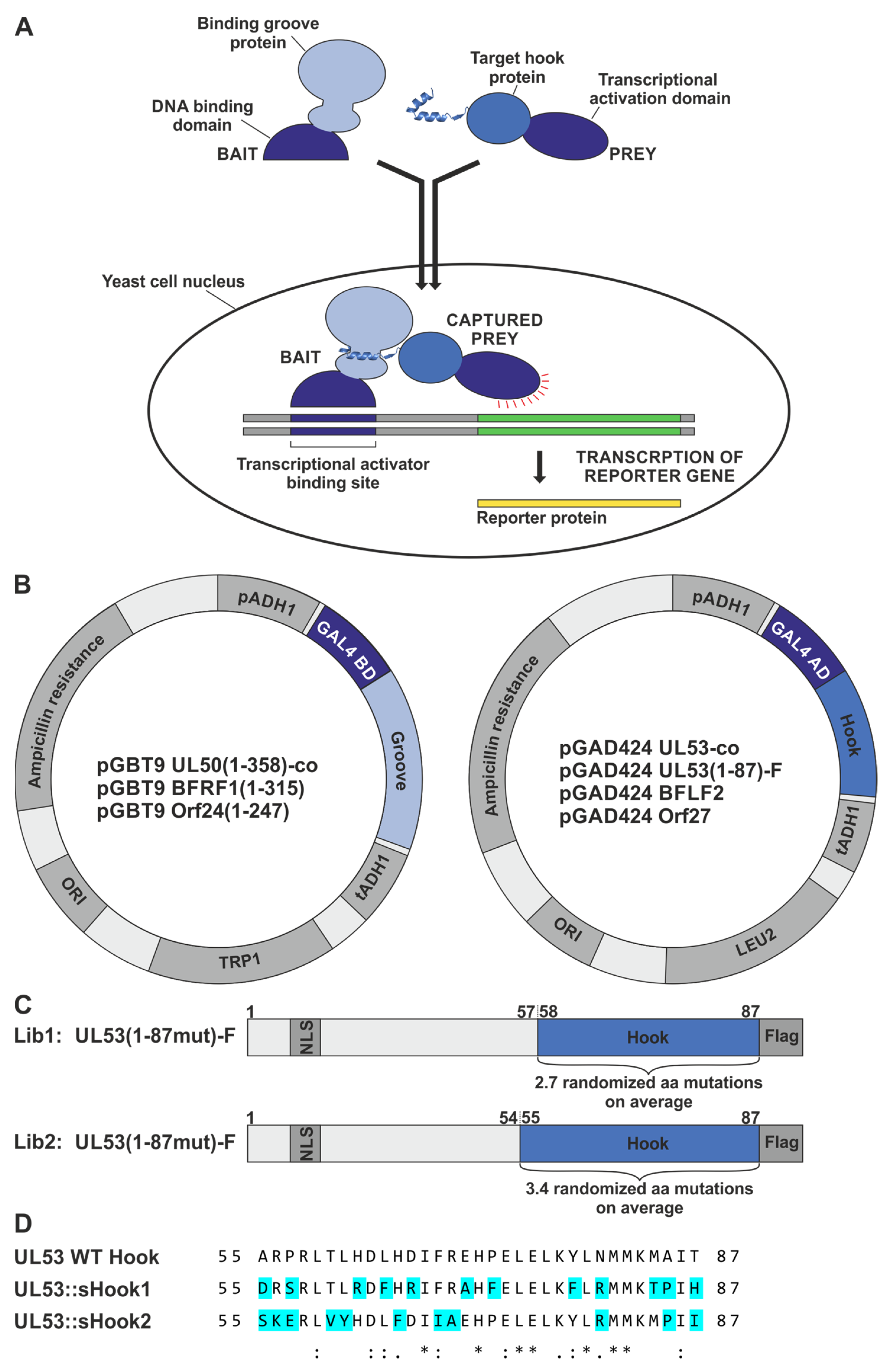
Figure 4.
Schematic overview of the Y2H principle, yeast expression constructs and design of the hook mutagenesis libraries of truncated pUL53. (
A) The Y2H system uses reporter genes to detect the interaction of proteins inside the yeast cell nucleus. The hook-into-groove interaction of a prey hook protein fused to the GAL4 activation domain (AD), with a bait groove protein fused to the GAL4 binding domain (BD), brings together the two-domain transcriptional activator, which then switches on the expression of reporter genes (illustration based on [
35]). (
B) Vector map of yeast expression constructs coding for fusion proteins of the GAL4 AD with hook proteins, here ORF UL53-co, ORF UL53(1–87)-Flag, ORF BFLF2 and Orf27, and the GAL4 BD fused to groove proteins lacking their transmembrane domain (TMD), here ORF UL50(1–358)-co, ORF BFRF1(1–315) and Orf24(1–247). The plasmids allow the selection of plasmid-positive clones for leucine (LEU2) or tryptophan (TRP1) auxotrophies in yeast, or ampicillin resistance in bacteria, respectively. Ori, origin of replication; pADH1/tADH1, transcriptional promoter/terminator region of the alcohol dehydrogenase gene 1. (
C) Design of the hook mutagenesis libraries, Lib1 (amino acid mutation rate of 2.7) and Lib2 (rate of 3.4), expressing truncated pUL53-fusion versions with nuclear localization signal (NLS) and Flag-tag indicated in dark-grey. The hook region of pUL53 amino acids 58–87 for Lib1 and 55–87 for Lib2, containing 2.7 or 3.4 randomized amino acid mutations on average, is depicted in blue. (
D) In addition to the randomized mutants of the libraries, two HCMV-EBV/VZV hybrid constructs were generated on the basis of primary sequence-predicted optimization. A comparative analysis of sequences was used to design these two hybrid constructs of a full-length-expressed pUL53 contained in the yeast vector pGAD424, in which binding-relevant amino acids of the construct sHook1 were preferentially adapted to a more EBV BFLF2-like fashion, while those of sHook2 were adapted to a more VZV Orf27-like fashion (for details, see
Section 3.1.1). *, indicates positions of fully conserved residues; :, indicates conservation between groups of strongly similar properties (i.e., scoring > 0.5 in the Gonnet PAM 250 matrix); ., indicates conservation between groups of weakly similar properties (i.e., scoring ≤ 0.5 and > 0 in the Gonnet PAM 250 matrix).
Figure 4.
Schematic overview of the Y2H principle, yeast expression constructs and design of the hook mutagenesis libraries of truncated pUL53. (
A) The Y2H system uses reporter genes to detect the interaction of proteins inside the yeast cell nucleus. The hook-into-groove interaction of a prey hook protein fused to the GAL4 activation domain (AD), with a bait groove protein fused to the GAL4 binding domain (BD), brings together the two-domain transcriptional activator, which then switches on the expression of reporter genes (illustration based on [
35]). (
B) Vector map of yeast expression constructs coding for fusion proteins of the GAL4 AD with hook proteins, here ORF UL53-co, ORF UL53(1–87)-Flag, ORF BFLF2 and Orf27, and the GAL4 BD fused to groove proteins lacking their transmembrane domain (TMD), here ORF UL50(1–358)-co, ORF BFRF1(1–315) and Orf24(1–247). The plasmids allow the selection of plasmid-positive clones for leucine (LEU2) or tryptophan (TRP1) auxotrophies in yeast, or ampicillin resistance in bacteria, respectively. Ori, origin of replication; pADH1/tADH1, transcriptional promoter/terminator region of the alcohol dehydrogenase gene 1. (
C) Design of the hook mutagenesis libraries, Lib1 (amino acid mutation rate of 2.7) and Lib2 (rate of 3.4), expressing truncated pUL53-fusion versions with nuclear localization signal (NLS) and Flag-tag indicated in dark-grey. The hook region of pUL53 amino acids 58–87 for Lib1 and 55–87 for Lib2, containing 2.7 or 3.4 randomized amino acid mutations on average, is depicted in blue. (
D) In addition to the randomized mutants of the libraries, two HCMV-EBV/VZV hybrid constructs were generated on the basis of primary sequence-predicted optimization. A comparative analysis of sequences was used to design these two hybrid constructs of a full-length-expressed pUL53 contained in the yeast vector pGAD424, in which binding-relevant amino acids of the construct sHook1 were preferentially adapted to a more EBV BFLF2-like fashion, while those of sHook2 were adapted to a more VZV Orf27-like fashion (for details, see
Section 3.1.1). *, indicates positions of fully conserved residues; :, indicates conservation between groups of strongly similar properties (i.e., scoring > 0.5 in the Gonnet PAM 250 matrix); ., indicates conservation between groups of weakly similar properties (i.e., scoring ≤ 0.5 and > 0 in the Gonnet PAM 250 matrix).
![Cells 11 04030 g004]()
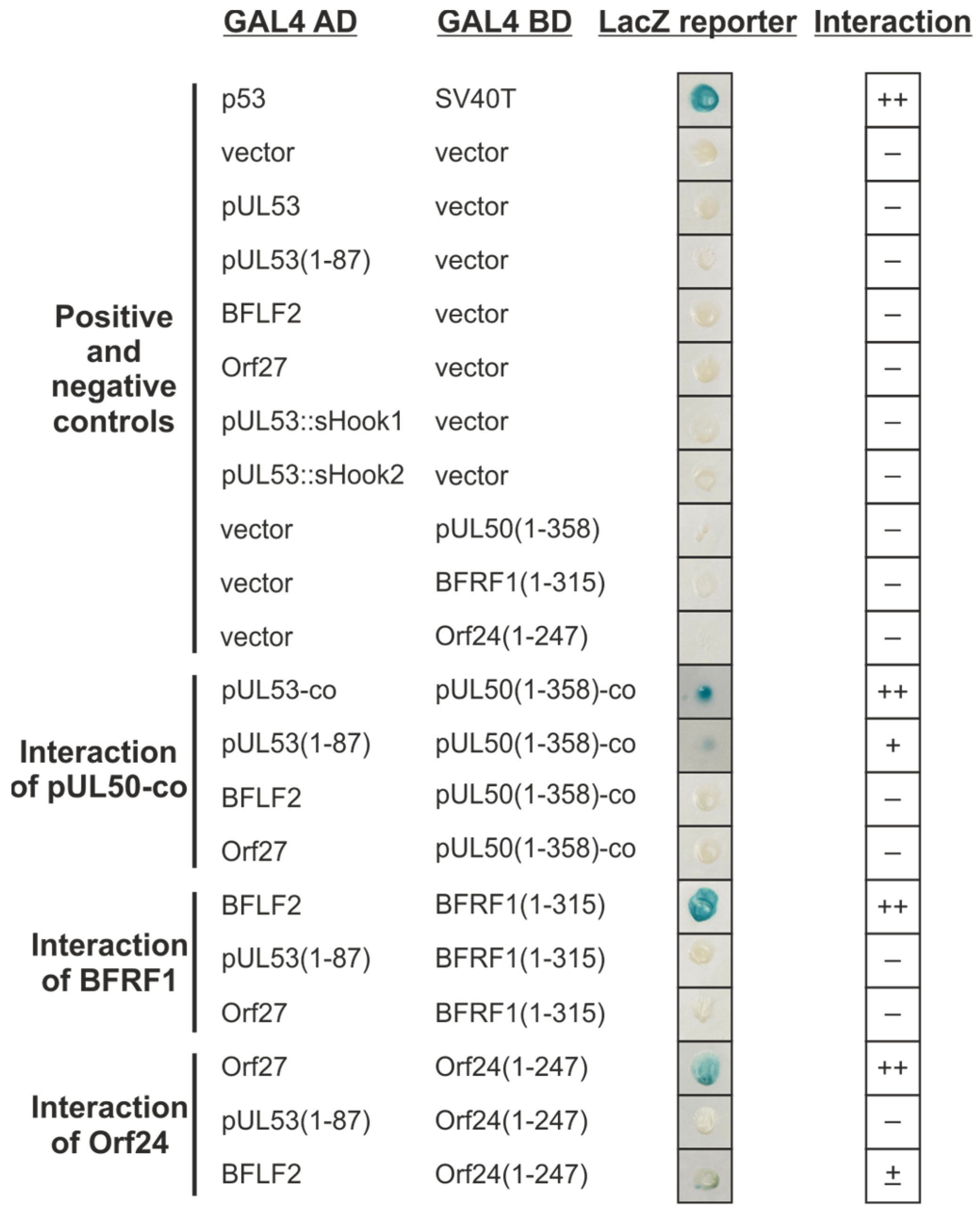
Figure 5.
Y2H analysis of interactions between the hook and groove proteins of HCMV, EBV and VZV. The indicated hook proteins fused to the GAL4 AD, were cotransformed in various combinations with groove proteins fused to the GAL4 BD, into yeast strain Y153. The ß-galactosidase expression was detected with the filter lift assay. The known interaction of cellular tumor suppressor p53 with the simian virus 40 (SV40)-derived oncoprotein SV40T served as a positive control. The cotransformation of the empty vectors pGAD424 and pGBT9 with the constructs were used as a negative control. Interaction analysis of codon-optimized pUL50-co, BFRF1 and Orf24 with codon-optimized pUL53-co, pUL53(1–87), BFLF2 and Orf27, respectively. ++, indicates ≥ 50%; +, indicates < 50% Xgal-positive yeast colonies in the filter lift assay measuring interaction; –, indicates not interacting protein pairs.
Figure 5.
Y2H analysis of interactions between the hook and groove proteins of HCMV, EBV and VZV. The indicated hook proteins fused to the GAL4 AD, were cotransformed in various combinations with groove proteins fused to the GAL4 BD, into yeast strain Y153. The ß-galactosidase expression was detected with the filter lift assay. The known interaction of cellular tumor suppressor p53 with the simian virus 40 (SV40)-derived oncoprotein SV40T served as a positive control. The cotransformation of the empty vectors pGAD424 and pGBT9 with the constructs were used as a negative control. Interaction analysis of codon-optimized pUL50-co, BFRF1 and Orf24 with codon-optimized pUL53-co, pUL53(1–87), BFLF2 and Orf27, respectively. ++, indicates ≥ 50%; +, indicates < 50% Xgal-positive yeast colonies in the filter lift assay measuring interaction; –, indicates not interacting protein pairs.
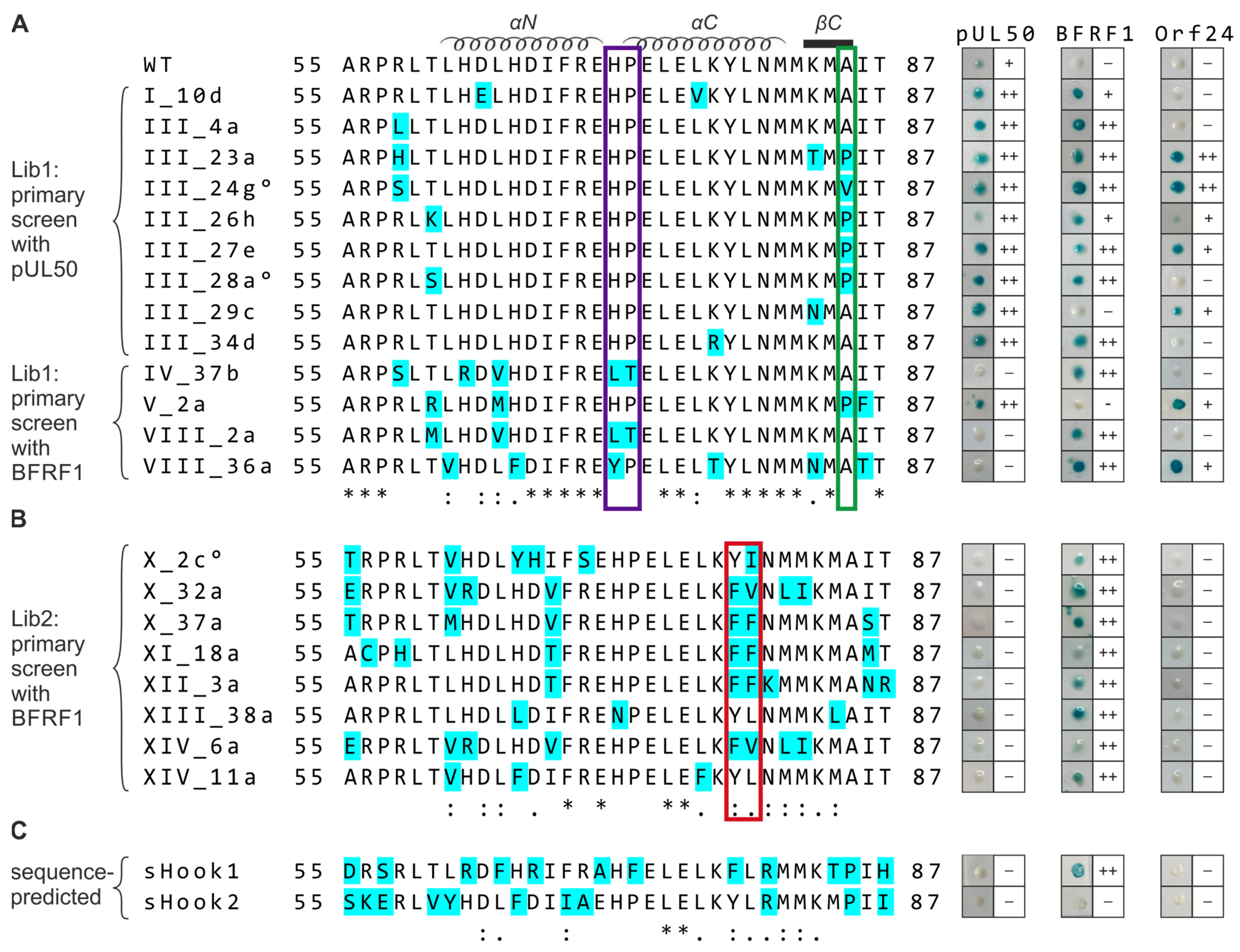
Figure 6.
Sequence alignment and Y2H data comparing the hook region of WT pUL53, library-selected and sequence-predicted clones referring to their shared-hook binding properties with three different groove proteins. Amino acid sequences of the (A,B) WT pUL53 hook region (amino acids 55–87), the library clones listed below and (C) the sequence-predicted shared-hook constructs were aligned using the online multiple sequence alignment tool ClustalW2. Mutated residues are highlighted in cyan. °, indicates further amino acid exchanges were detected upstream the hook region (i.e., A43T for III_24g, R18S for III_28a and R8C for X_2c); *, positions of fully conserved residues; :, positions conserved within groups of strongly similar properties (i.e., scoring > 0.5 in the Gonnet PAM 250 matrix); ., positions conserved within groups of weakly similar properties (i.e., scoring ≤ 0.5 and >0 in the Gonnet PAM 250 matrix). In the Y2H analysis, pUL53 library clones were identified by primary screening against HCMV pUL50 (A, upper part) or EBV BFRF1 (A, lower part, B). All primary positive clones, including sequence-predicted constructs (C, sHook1 and sHook2), were then used for a backtransformation-based confirmation step and shared-hook analysis against HCMV pUL50, EBV BFRF1, or VZV Orf24, respectively, as shown at the right. The positivity rate of blue yeast colonies detected in the Xgal filter lift assay is indicated: ++, ≥50%; +, <50%; –, not detectable.
Figure 6.
Sequence alignment and Y2H data comparing the hook region of WT pUL53, library-selected and sequence-predicted clones referring to their shared-hook binding properties with three different groove proteins. Amino acid sequences of the (A,B) WT pUL53 hook region (amino acids 55–87), the library clones listed below and (C) the sequence-predicted shared-hook constructs were aligned using the online multiple sequence alignment tool ClustalW2. Mutated residues are highlighted in cyan. °, indicates further amino acid exchanges were detected upstream the hook region (i.e., A43T for III_24g, R18S for III_28a and R8C for X_2c); *, positions of fully conserved residues; :, positions conserved within groups of strongly similar properties (i.e., scoring > 0.5 in the Gonnet PAM 250 matrix); ., positions conserved within groups of weakly similar properties (i.e., scoring ≤ 0.5 and >0 in the Gonnet PAM 250 matrix). In the Y2H analysis, pUL53 library clones were identified by primary screening against HCMV pUL50 (A, upper part) or EBV BFRF1 (A, lower part, B). All primary positive clones, including sequence-predicted constructs (C, sHook1 and sHook2), were then used for a backtransformation-based confirmation step and shared-hook analysis against HCMV pUL50, EBV BFRF1, or VZV Orf24, respectively, as shown at the right. The positivity rate of blue yeast colonies detected in the Xgal filter lift assay is indicated: ++, ≥50%; +, <50%; –, not detectable.
![Cells 11 04030 g006]()
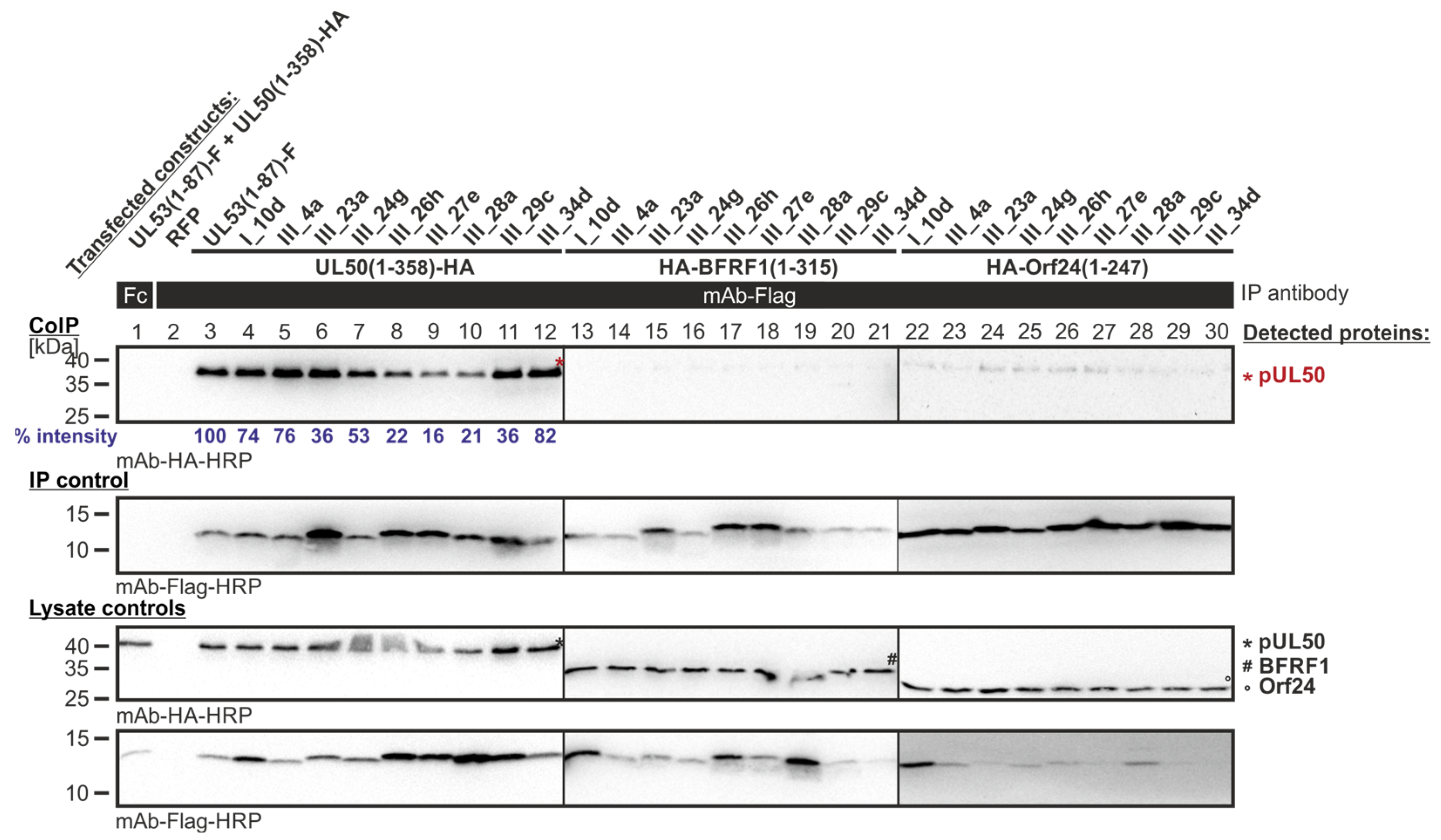
Figure 7.
CoIP-based interaction analysis of HCMV pUL50, EBV BFRF1 and VZV Orf24 with the library clones that exerted shared-hook binding activity in the primary screening of Lib1 with pUL50. 293T cells were transiently transfected with plasmids coding for the HA-tagged groove proteins, pUL50, BFRF1 or Orf24, lacking their TMD, in combination with the Flag-tagged library constructs. The wild-type interaction of truncated pUL53 with pUL50 served as a positive and RFP as a negative control. At three d p.t., cells were lysed and Flag-tagged proteins were immunoprecipitated using mAb-Flag; a chicken Fc fragment served as a specificity control. Lysate controls taken prior to the IP and CoIP samples were subjected to standard Wb analysis using tag-specific antibodies as indicated. Blue numbers: each individual CoIP band was first normalized to the corresponding IP signal and was then set in relation to the reference WT interaction (% intensity) using Aida Image Analyzer v.4.23 software (mean values of triplicate densitometric determinations are given); CoIP was performed once.
Figure 7.
CoIP-based interaction analysis of HCMV pUL50, EBV BFRF1 and VZV Orf24 with the library clones that exerted shared-hook binding activity in the primary screening of Lib1 with pUL50. 293T cells were transiently transfected with plasmids coding for the HA-tagged groove proteins, pUL50, BFRF1 or Orf24, lacking their TMD, in combination with the Flag-tagged library constructs. The wild-type interaction of truncated pUL53 with pUL50 served as a positive and RFP as a negative control. At three d p.t., cells were lysed and Flag-tagged proteins were immunoprecipitated using mAb-Flag; a chicken Fc fragment served as a specificity control. Lysate controls taken prior to the IP and CoIP samples were subjected to standard Wb analysis using tag-specific antibodies as indicated. Blue numbers: each individual CoIP band was first normalized to the corresponding IP signal and was then set in relation to the reference WT interaction (% intensity) using Aida Image Analyzer v.4.23 software (mean values of triplicate densitometric determinations are given); CoIP was performed once.
![Cells 11 04030 g007]()
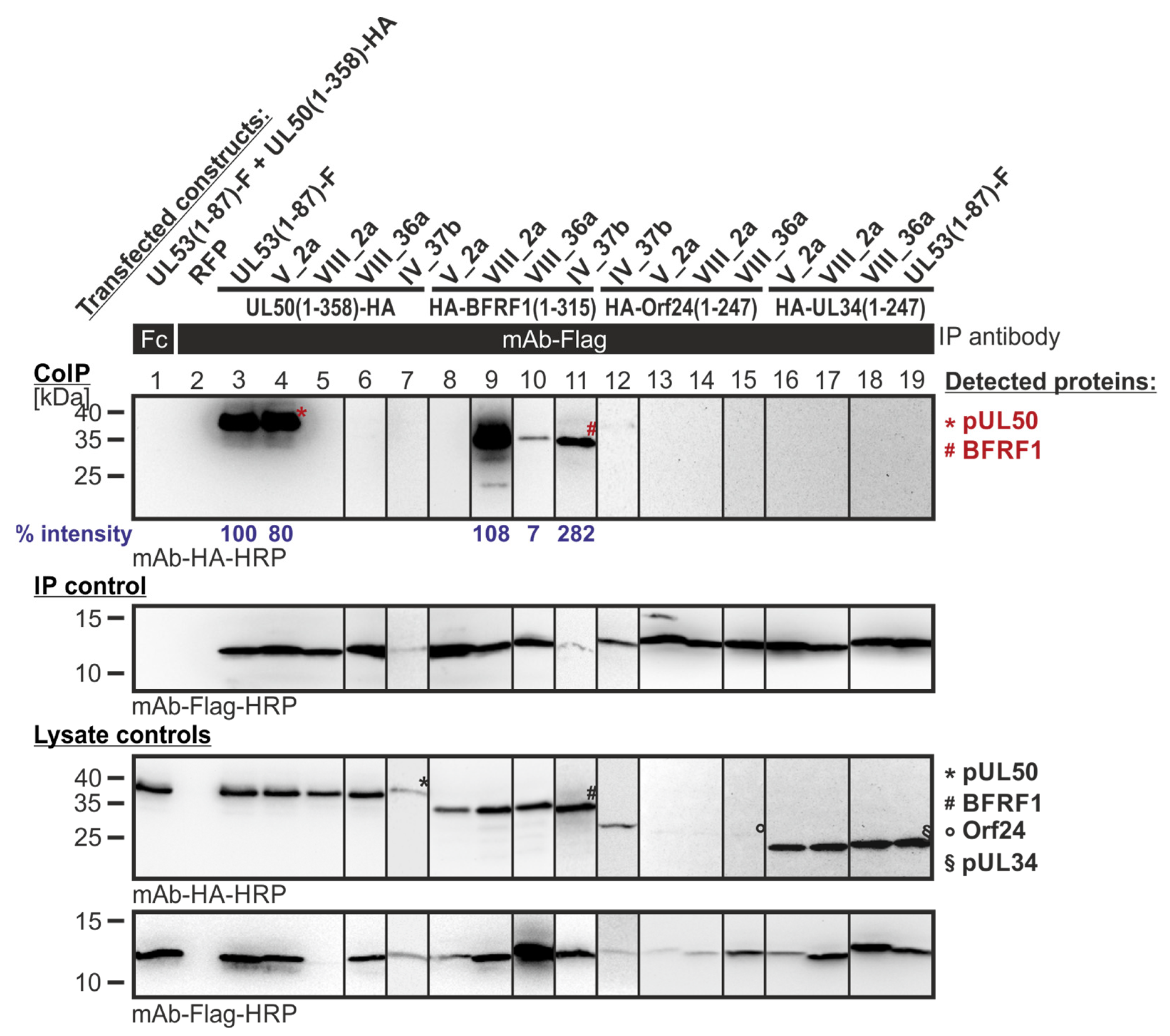
Figure 8.
CoIP-based interaction analysis of HCMV pUL50, EBV BFRF1, VZV Orf24 and HSV-1 pUL34 with the library clones that exerted shared-hook binding activity in the primary screening of Lib1 with BFRF1. 293T cells were transiently transfected with plasmids coding for the HA-tagged groove proteins, pUL50, BFRF1, Orf24, or pUL34, lacking their TMD, in combination with the Flag-tagged library constructs. The wild-type interaction of truncated pUL53 with pUL50 served as a positive and RFP as a negative control. At three d p.t., cells were lysed and Flag-tagged proteins were immunoprecipitated using mAb-Flag; a chicken Fc fragment served as a specificity control. Lysate controls taken prior to the IP and CoIP samples were subjected to standard Wb analysis using tag-specific antibodies as indicated. Blue numbers: each individual CoIP band was first normalized to the corresponding IP signal and was then set in relation to the reference WT interaction (% intensity) using Aida Image Analyzer v.4.23 software (mean values of triplicate densitometric determinations are given); CoIP was performed once.
Figure 8.
CoIP-based interaction analysis of HCMV pUL50, EBV BFRF1, VZV Orf24 and HSV-1 pUL34 with the library clones that exerted shared-hook binding activity in the primary screening of Lib1 with BFRF1. 293T cells were transiently transfected with plasmids coding for the HA-tagged groove proteins, pUL50, BFRF1, Orf24, or pUL34, lacking their TMD, in combination with the Flag-tagged library constructs. The wild-type interaction of truncated pUL53 with pUL50 served as a positive and RFP as a negative control. At three d p.t., cells were lysed and Flag-tagged proteins were immunoprecipitated using mAb-Flag; a chicken Fc fragment served as a specificity control. Lysate controls taken prior to the IP and CoIP samples were subjected to standard Wb analysis using tag-specific antibodies as indicated. Blue numbers: each individual CoIP band was first normalized to the corresponding IP signal and was then set in relation to the reference WT interaction (% intensity) using Aida Image Analyzer v.4.23 software (mean values of triplicate densitometric determinations are given); CoIP was performed once.
![Cells 11 04030 g008]()
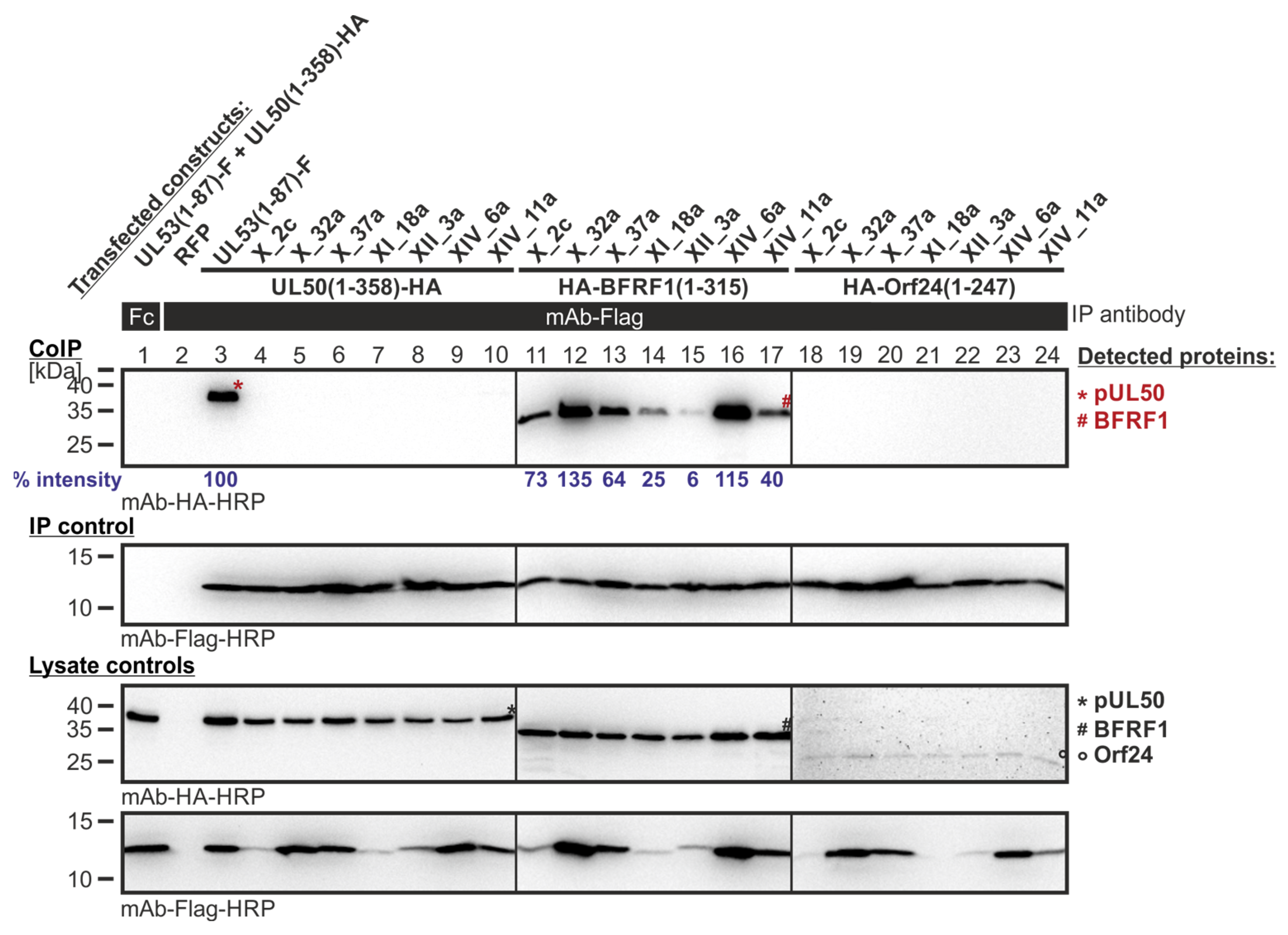
Figure 9.
CoIP-based interaction analysis of HCMV pUL50, EBV BFRF1 and VZV Orf24 with the library clones that exerted shared-hook binding activity in the primary screening of Lib2 with BFRF1. 293T cells were transiently transfected with plasmids coding for the HA-tagged groove proteins, pUL50, BFRF1, or Orf24, lacking their TMD, in combination with the Flag-tagged library constructs. The wild-type interaction of truncated pUL53 with pUL50 served as a positive and RFP as a negative control. At three d p.t., cells were lysed and Flag-tagged proteins were immunoprecipitated using mAb-Flag; a chicken Fc fragment served as a specificity control. Lysate controls taken prior to the IP and CoIP samples were subjected to standard Wb analysis using tag-specific antibodies as indicated. Blue numbers: each individual CoIP band was first normalized to the corresponding IP signal and was then set in relation to the reference WT interaction (% intensity) using Aida Image Analyzer v.4.23 software (mean values of triplicate densitometric determinations are given); CoIP was performed once.
Figure 9.
CoIP-based interaction analysis of HCMV pUL50, EBV BFRF1 and VZV Orf24 with the library clones that exerted shared-hook binding activity in the primary screening of Lib2 with BFRF1. 293T cells were transiently transfected with plasmids coding for the HA-tagged groove proteins, pUL50, BFRF1, or Orf24, lacking their TMD, in combination with the Flag-tagged library constructs. The wild-type interaction of truncated pUL53 with pUL50 served as a positive and RFP as a negative control. At three d p.t., cells were lysed and Flag-tagged proteins were immunoprecipitated using mAb-Flag; a chicken Fc fragment served as a specificity control. Lysate controls taken prior to the IP and CoIP samples were subjected to standard Wb analysis using tag-specific antibodies as indicated. Blue numbers: each individual CoIP band was first normalized to the corresponding IP signal and was then set in relation to the reference WT interaction (% intensity) using Aida Image Analyzer v.4.23 software (mean values of triplicate densitometric determinations are given); CoIP was performed once.
![Cells 11 04030 g009]()
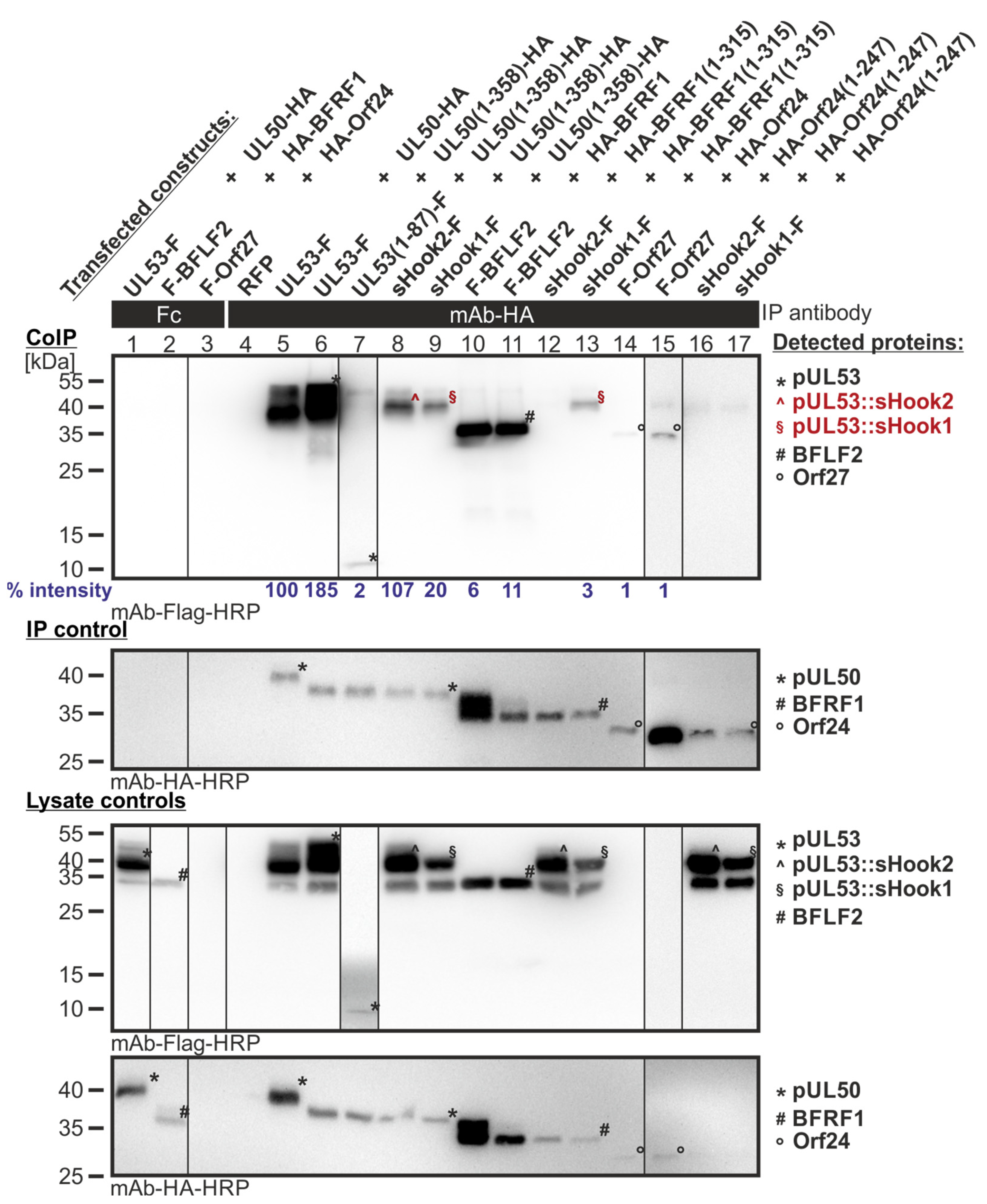
Figure 10.
CoIP-based interaction analysis of sequence-predicted shared-hook constructs with three groove proteins. 293T cells were transiently transfected with constructs coding for the HA-tagged groove proteins, pUL50, BFRF1 or Orf24, either full-length or without TMD, in combination with the Flag-tagged sequence-predicted shared-hook constructs. Positive controls were the original hook proteins pUL53, BFLF2 or Orf27 and RFP served as a negative control. At three d p.t., cells were lysed and HA-tagged proteins were immunoprecipitated using mAb-HA; a chicken Fc fragment served as a specificity control. Lysate controls taken prior to the IP and CoIP samples were subjected to standard Wb analysis using tag-specific antibodies as indicated. Interaction of the sequence-predicted shared-hook constructs is indicated with red symbols. Blue numbers: each individual CoIP band was first normalized to the corresponding IP signal and was then set in relation to the reference WT interaction (% intensity) using Aida Image Analyzer v.4.23 software (mean values of triplicate densitometric determinations are given); CoIP was performed three times.
Figure 10.
CoIP-based interaction analysis of sequence-predicted shared-hook constructs with three groove proteins. 293T cells were transiently transfected with constructs coding for the HA-tagged groove proteins, pUL50, BFRF1 or Orf24, either full-length or without TMD, in combination with the Flag-tagged sequence-predicted shared-hook constructs. Positive controls were the original hook proteins pUL53, BFLF2 or Orf27 and RFP served as a negative control. At three d p.t., cells were lysed and HA-tagged proteins were immunoprecipitated using mAb-HA; a chicken Fc fragment served as a specificity control. Lysate controls taken prior to the IP and CoIP samples were subjected to standard Wb analysis using tag-specific antibodies as indicated. Interaction of the sequence-predicted shared-hook constructs is indicated with red symbols. Blue numbers: each individual CoIP band was first normalized to the corresponding IP signal and was then set in relation to the reference WT interaction (% intensity) using Aida Image Analyzer v.4.23 software (mean values of triplicate densitometric determinations are given); CoIP was performed three times.
![Cells 11 04030 g010]()
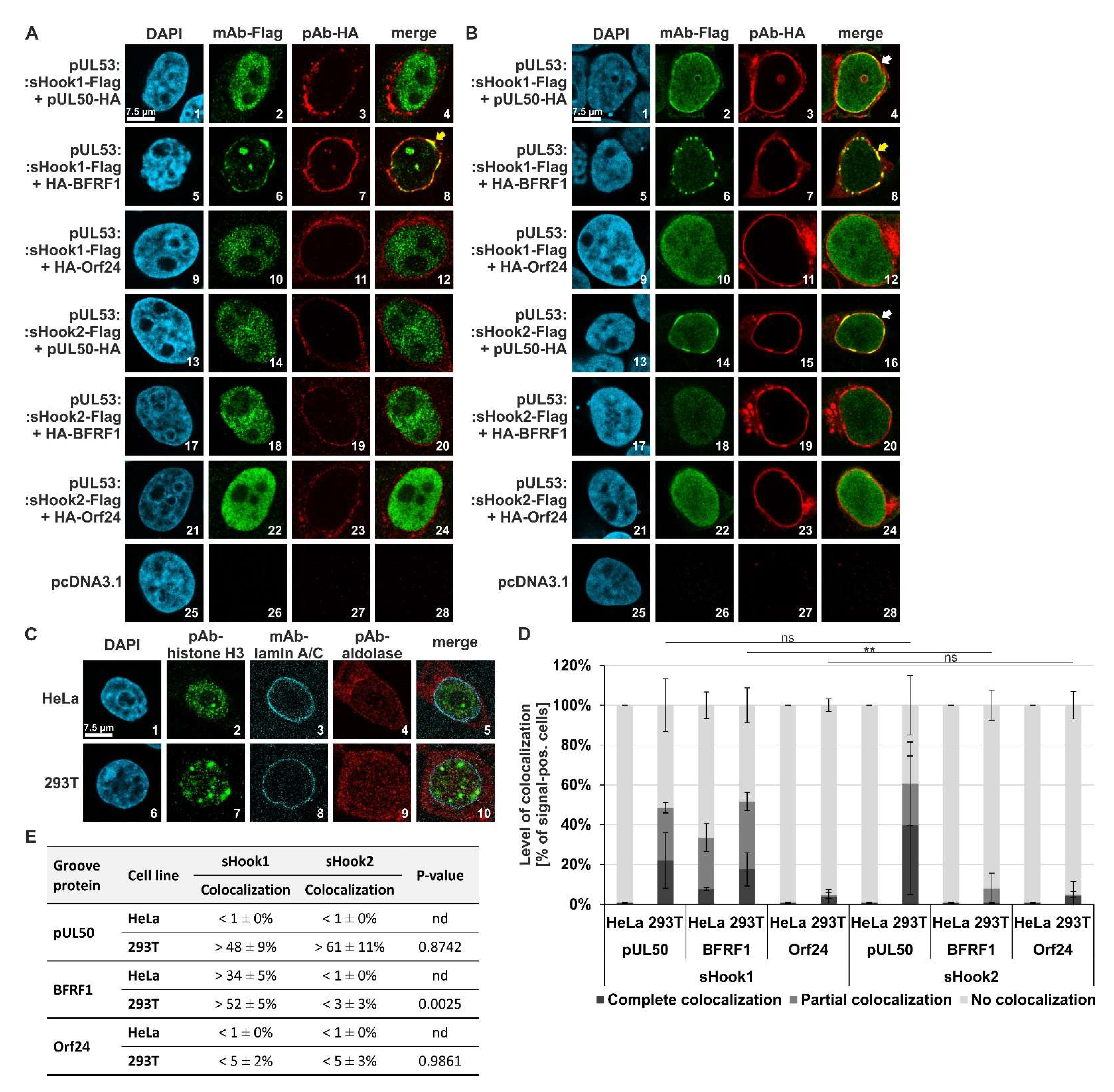
Figure 11.
Qualitative and quantitative analyses of nuclear rim colocalization patterns: coexpressed sequence-predicted hybrid constructs (HCMV/EBV pUL53 sHook1 and HCMV/VZV pUL53 sHook2) in combination with three different groove proteins (HCMV pUL50, EBV BFRF1 or VZV Orf24). (A) HeLa or (B) 293T cells were transiently cotransfected with either constructs coding for the Flag-tagged sequence-predicted hooks, sHook1 or sHook2, and the HA-tagged groove proteins pUL50, BFRF1 or Orf24. Two d p.t., cells were fixed, used for immunostaining with tag-specific antibodies and analyzed by confocal imaging. DAPI counterstaining indicated the morphology of nuclei of the respective cells. In both types of cells, the localization patterns of coexpressed construct pairs were analyzed; the scale bar in panels A–C, picture 1 marks 7.5 μm. On a qualitative level of analysis (A,B), the yellow arrows indicate complete nuclear rim colocalization, and the white arrows show partial rim colocalization. (C) Staining of subcellular localizations of marker proteins in HeLa and 293T cells, i.e., the nuclear lamina, the nucleoplasmic and cytosolic compartments. (D) Quantitation of the colocalization patterns of pUL53 sHook1 and sHook2 with groove proteins HCMV pUL50, EBV BFRF1 and VZV Orf24. Localization patterns observed in HeLa and 293T cells were quantified and classified as complete, partial or no colocalization. Mean values ± SD are given as expressed in a percentage of the entire number of counted cells. Statistical analysis was performed using an ordinary two-way ANOVA and post-hoc Tukey correction; **, p < 0.01; ns, not significant. (E) Summarized findings of colocalization obtained for the two sequence-predicted hybrid constructs, combining complete and partial colocalization as total percentages; nd, not determined.
Figure 11.
Qualitative and quantitative analyses of nuclear rim colocalization patterns: coexpressed sequence-predicted hybrid constructs (HCMV/EBV pUL53 sHook1 and HCMV/VZV pUL53 sHook2) in combination with three different groove proteins (HCMV pUL50, EBV BFRF1 or VZV Orf24). (A) HeLa or (B) 293T cells were transiently cotransfected with either constructs coding for the Flag-tagged sequence-predicted hooks, sHook1 or sHook2, and the HA-tagged groove proteins pUL50, BFRF1 or Orf24. Two d p.t., cells were fixed, used for immunostaining with tag-specific antibodies and analyzed by confocal imaging. DAPI counterstaining indicated the morphology of nuclei of the respective cells. In both types of cells, the localization patterns of coexpressed construct pairs were analyzed; the scale bar in panels A–C, picture 1 marks 7.5 μm. On a qualitative level of analysis (A,B), the yellow arrows indicate complete nuclear rim colocalization, and the white arrows show partial rim colocalization. (C) Staining of subcellular localizations of marker proteins in HeLa and 293T cells, i.e., the nuclear lamina, the nucleoplasmic and cytosolic compartments. (D) Quantitation of the colocalization patterns of pUL53 sHook1 and sHook2 with groove proteins HCMV pUL50, EBV BFRF1 and VZV Orf24. Localization patterns observed in HeLa and 293T cells were quantified and classified as complete, partial or no colocalization. Mean values ± SD are given as expressed in a percentage of the entire number of counted cells. Statistical analysis was performed using an ordinary two-way ANOVA and post-hoc Tukey correction; **, p < 0.01; ns, not significant. (E) Summarized findings of colocalization obtained for the two sequence-predicted hybrid constructs, combining complete and partial colocalization as total percentages; nd, not determined.
![Cells 11 04030 g011]()
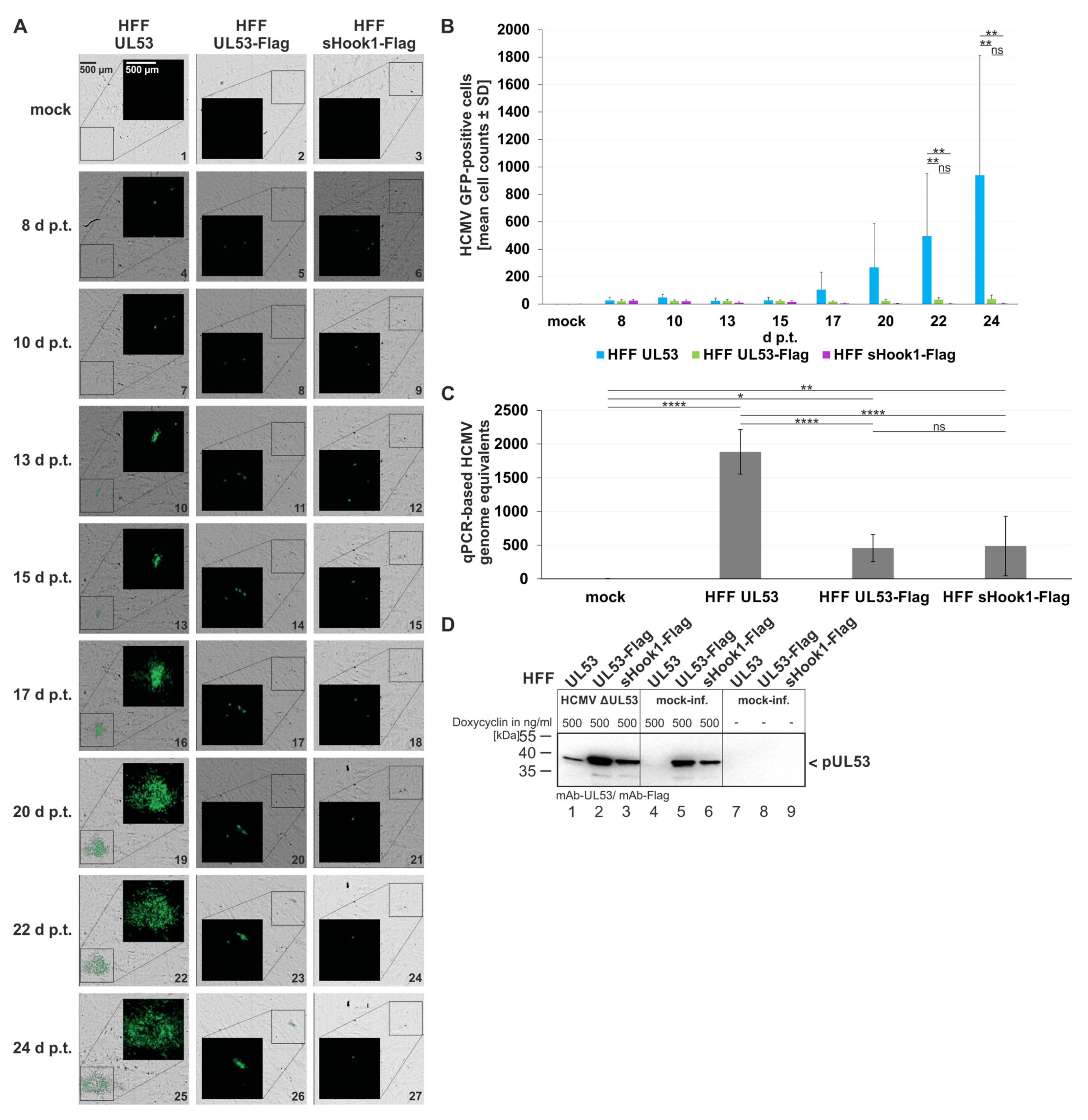
Figure 12.
Qualitative and quantitative analyses of HCMV AD169-GFP ΔUL53 virus reconstitution on the different recombinant HFF cell populations. HFFs that express pUL53, pUL53-Flag or pUL53::sHook1-Flag were transfected with the infectious bacterial artificial chromosome of HCMV ΔUL53. (A) Images of the GFP signals and the respective brightfield illuminations were taken at indicated time points; scale bar in panel A, picture 1 marks 500 μm. (B) Quantitative analysis of fluorescence-positive cells was achieved with automated counting by the CellReporterXpress® software (Molecular Devices LLC, San Jose, CA, USA). Counting parameters for positive nuclei were set to an intensity of at least 100, a minimal width of 7 and a maximal width of 30. Measurements were performed in biological sextuplicates per cell population of 5.05% area of the well and mean values ± SD are given. (C) qPCR-based assay for the determination of viral genome equivalents referring to the respective recombinant HFF populations. Viral supernatants were harvested at day 24 p.t. and subjected to IE1-specific qPCR. Calculations were performed in biological sextuplicates per cell population; mean values ± SD are shown. (B,C) Statistical analysis was performed using an ordinary two-way ANOVA and post-hoc Tukey correction; ****, p < 0.0001; **, p < 0.01; *, p < 0.1; ns, not significant; no significant differences were noted for values between 1–20 d p.t. (B). (D) Wb-based expression analysis of the conditionally expressing HFF populations. Recombinant protein expression in the three HFF populations of HFF-UL53, HFF-UL53-Flag and HFF-UL53::sHook1-Flag cells was either uninduced (-) or induced (500 ng/mL Dox). Cells were either infected with HCMV ΔUL53 or remained mock-infected. At 24 h p.t., cells were harvested and lysed. Total lysate samples were subjected to standard Wb analysis using tag-specific or protein-specific monoclonal antibodies as indicated.
Figure 12.
Qualitative and quantitative analyses of HCMV AD169-GFP ΔUL53 virus reconstitution on the different recombinant HFF cell populations. HFFs that express pUL53, pUL53-Flag or pUL53::sHook1-Flag were transfected with the infectious bacterial artificial chromosome of HCMV ΔUL53. (A) Images of the GFP signals and the respective brightfield illuminations were taken at indicated time points; scale bar in panel A, picture 1 marks 500 μm. (B) Quantitative analysis of fluorescence-positive cells was achieved with automated counting by the CellReporterXpress® software (Molecular Devices LLC, San Jose, CA, USA). Counting parameters for positive nuclei were set to an intensity of at least 100, a minimal width of 7 and a maximal width of 30. Measurements were performed in biological sextuplicates per cell population of 5.05% area of the well and mean values ± SD are given. (C) qPCR-based assay for the determination of viral genome equivalents referring to the respective recombinant HFF populations. Viral supernatants were harvested at day 24 p.t. and subjected to IE1-specific qPCR. Calculations were performed in biological sextuplicates per cell population; mean values ± SD are shown. (B,C) Statistical analysis was performed using an ordinary two-way ANOVA and post-hoc Tukey correction; ****, p < 0.0001; **, p < 0.01; *, p < 0.1; ns, not significant; no significant differences were noted for values between 1–20 d p.t. (B). (D) Wb-based expression analysis of the conditionally expressing HFF populations. Recombinant protein expression in the three HFF populations of HFF-UL53, HFF-UL53-Flag and HFF-UL53::sHook1-Flag cells was either uninduced (-) or induced (500 ng/mL Dox). Cells were either infected with HCMV ΔUL53 or remained mock-infected. At 24 h p.t., cells were harvested and lysed. Total lysate samples were subjected to standard Wb analysis using tag-specific or protein-specific monoclonal antibodies as indicated.
![Cells 11 04030 g012]()
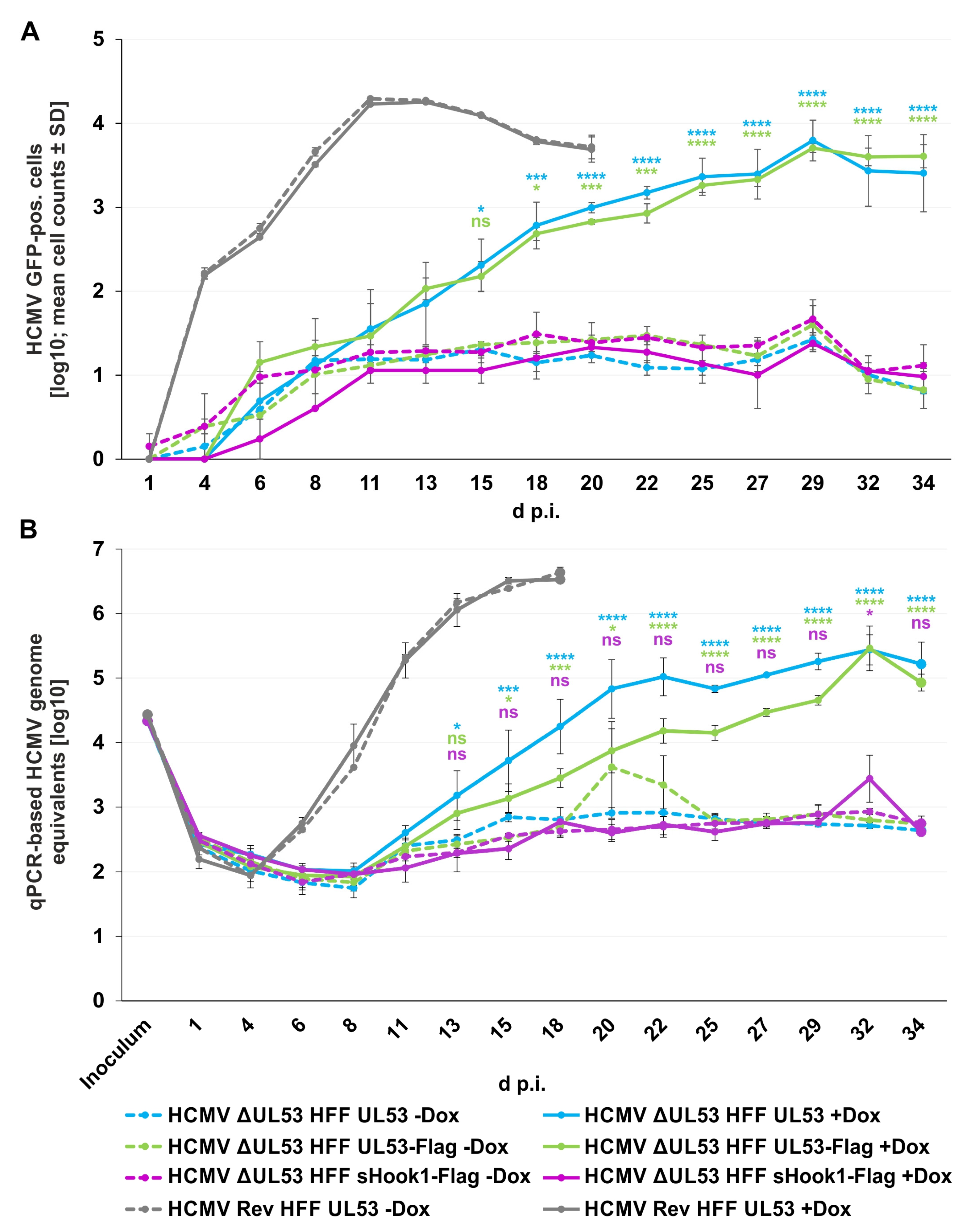
Figure 13.
Viral replication kinetics of HCMV ΔUL53 and its revertant (HCMV Rev) determined by quantitation of GFP-positive cells and HCMV-specific qPCR on the different recombinant HFF populations. 80,000 inducibly expressing HFFs in 24-well plates were infected with HCMV ΔUL53 or HCMV Rev at a viral dose of 5 × 106 genome copies. pUL53, pUL53-Flag or pUL53::sHook1-Flag protein expression was either Dox-induced (+Dox) or remained non-induced (−Dox). (A) The number of HCMV-infected cells was measured by detection of GFP signal-positive cells at indicated time points with the CellReporterXpress® software using the ImageXpress® Pico device. Values represent 25.04 % of the area of a well and are given as a mean value ± SD of two independently infected wells. (B) Viral supernatants were harvested at indicated time points and viral genome equivalents were determined by qPCR. Each value represents the mean ± SD of two independent biological replicates, each measured twice. (A,B) Statistical analysis was performed using an ordinary two-way ANOVA and post-hoc Sidak correction; ****, p < 0.0001; ***, p < 0.001; *, p < 0.1; ns, not significant; no significant differences were noted for values between 1–13 d p.i. (A) or 1–11 d p.i. (B), respectively.
Figure 13.
Viral replication kinetics of HCMV ΔUL53 and its revertant (HCMV Rev) determined by quantitation of GFP-positive cells and HCMV-specific qPCR on the different recombinant HFF populations. 80,000 inducibly expressing HFFs in 24-well plates were infected with HCMV ΔUL53 or HCMV Rev at a viral dose of 5 × 106 genome copies. pUL53, pUL53-Flag or pUL53::sHook1-Flag protein expression was either Dox-induced (+Dox) or remained non-induced (−Dox). (A) The number of HCMV-infected cells was measured by detection of GFP signal-positive cells at indicated time points with the CellReporterXpress® software using the ImageXpress® Pico device. Values represent 25.04 % of the area of a well and are given as a mean value ± SD of two independently infected wells. (B) Viral supernatants were harvested at indicated time points and viral genome equivalents were determined by qPCR. Each value represents the mean ± SD of two independent biological replicates, each measured twice. (A,B) Statistical analysis was performed using an ordinary two-way ANOVA and post-hoc Sidak correction; ****, p < 0.0001; ***, p < 0.001; *, p < 0.1; ns, not significant; no significant differences were noted for values between 1–13 d p.i. (A) or 1–11 d p.i. (B), respectively.
![Cells 11 04030 g013]()
Table 1.
Screening procedure to identify pUL53 clones from Lib1 that exert shared-hook binding activity in the Y2H system.
Table 1.
Screening procedure to identify pUL53 clones from Lib1 that exert shared-hook binding activity in the Y2H system.
| | HCMV pUL53 Library (Lib1) Screened for Interaction with: |
|---|
| | HCMV pUL50 #
wt │ co | EBV BFRF1 | VZV Orf24 |
|---|
| Y153 cell number/0.5 µg library DNA | ~2.87 × 107 | ~3.64 × 107 | ~3.49 × 107 |
| Interacting pUL53 library clones in primary screening * | 20 │ 28 | 16 | 3 |
| Recovered in E. coli DH10B § | 18 │ 27 | 16 | 2 |
| Positive yeast retransformants with pUL50 * | 17 │ 26 | 5 | 0 |
| Positive yeast retransformants with BFRF1 * | 8 | 4 | 0 |
| Positive yeast retransformants with Orf24 * | 5 | 2 | 0 |
Table 2.
Screening procedure to identify pUL53 clones from Lib2 that exert shared-hook binding activity in the Y2H system.
Table 2.
Screening procedure to identify pUL53 clones from Lib2 that exert shared-hook binding activity in the Y2H system.
| | HCMV pUL53 Library (Lib2) Screened for Interaction with EBV BFRF1 |
|---|
| Y153 cell number/0.5 µg library DNA | ~5.09 × 107 |
| Interacting pUL53 library clones in primary screening * | 35 |
| Recovered in E. coli DH10B § | 31 |
| Positive yeast retransformants with pUL50 * | 2 |
| Positive yeast retransformants with BFRF1 * | 9 |
| Positive yeast retransformants with Orf24 * | 0 |


 and
and 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}