
HDAC6 Inhibition Alleviates Ischemia- and Cisplatin-Induced Acute Kidney Injury by Promoting Autophagy
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Antibodies and Reagents
2.2. In Vitro Model of Renal IRI and Cisplatin in BUMPT Cell and Examination of Apoptosis
2.3. Animals and Treatment
2.4. Renal Function Assay
2.5. Plasmids and Lentiviral Transduction
2.6. Cell Viability Assay
2.7. Analysis of Autophagy Dynamics in CAG-RFP-EGFP-LC3 Autophagy Reporter
2.8. Histology and TUNEL Staining
2.9. Immunohistochemistry (IHC) and Immunofluorescence
2.10. Immunoblot Analysis
2.11. Statistical Analysis
3. Results
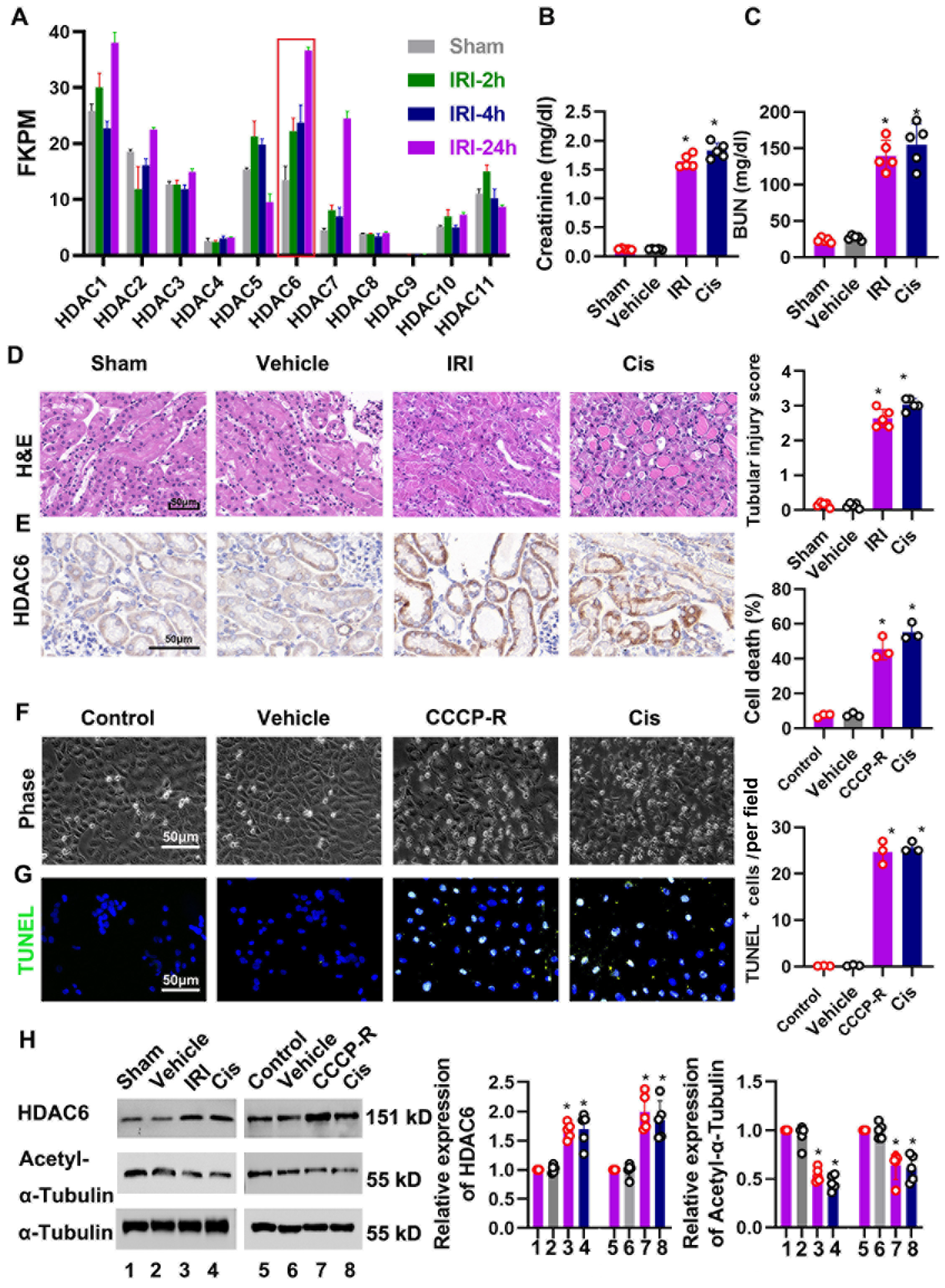
3.1. HDAC6 Expression Is Increased after Ischemia- and Cisplatin-Induced Acute Kidney Injury
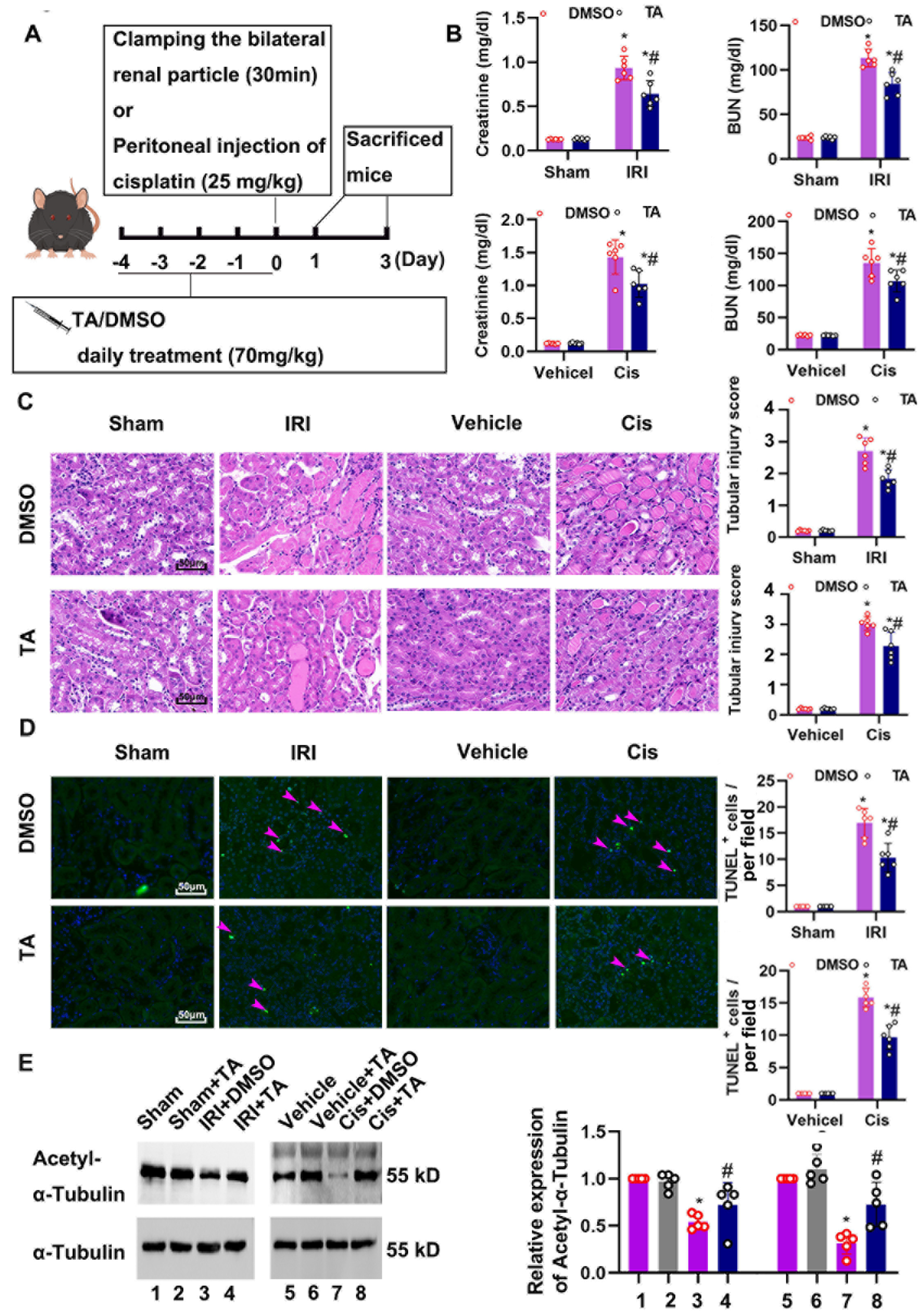
3.2. HDAC6 Inhibition Attenuated Ischemia- and Cisplatin-Induced Acute Kidney Injury
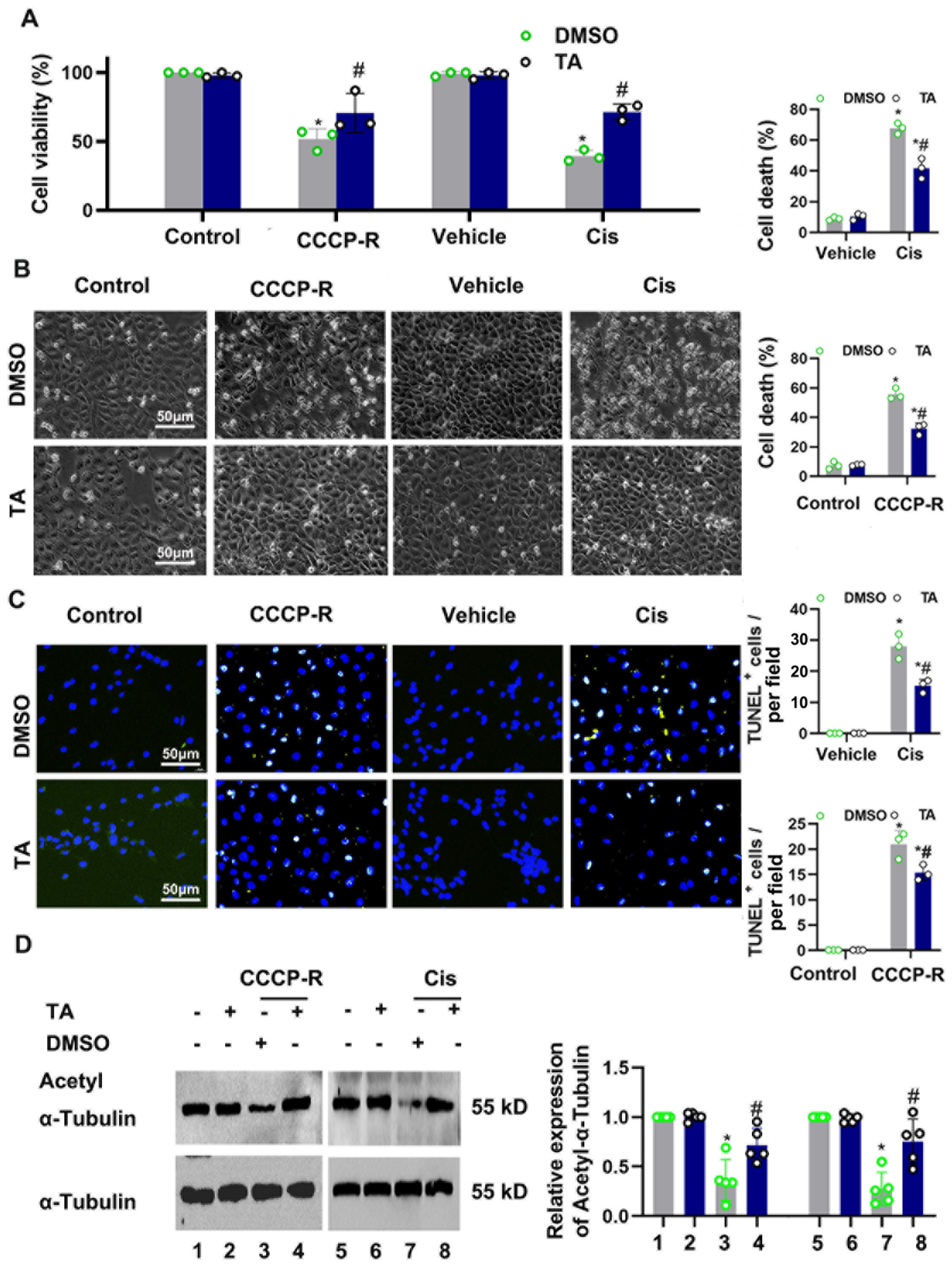
3.3. HDAC6 Inhibition Ameliorated CCCP-and Cisplatin-Induced Tubular Cell Death
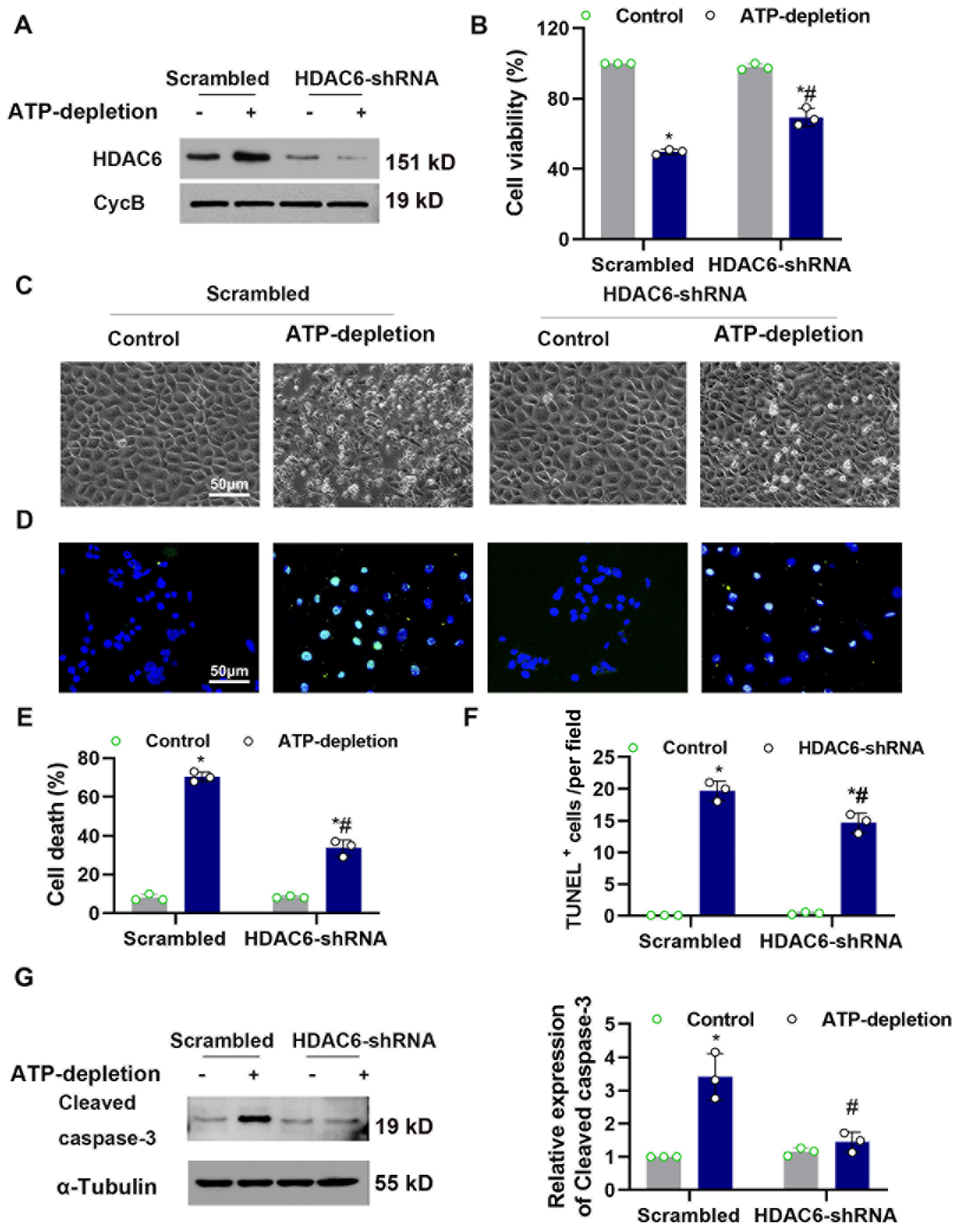
3.4. Knocked-Down HDAC6 Ameliorated CCCP-Induced Tubular Cell Death
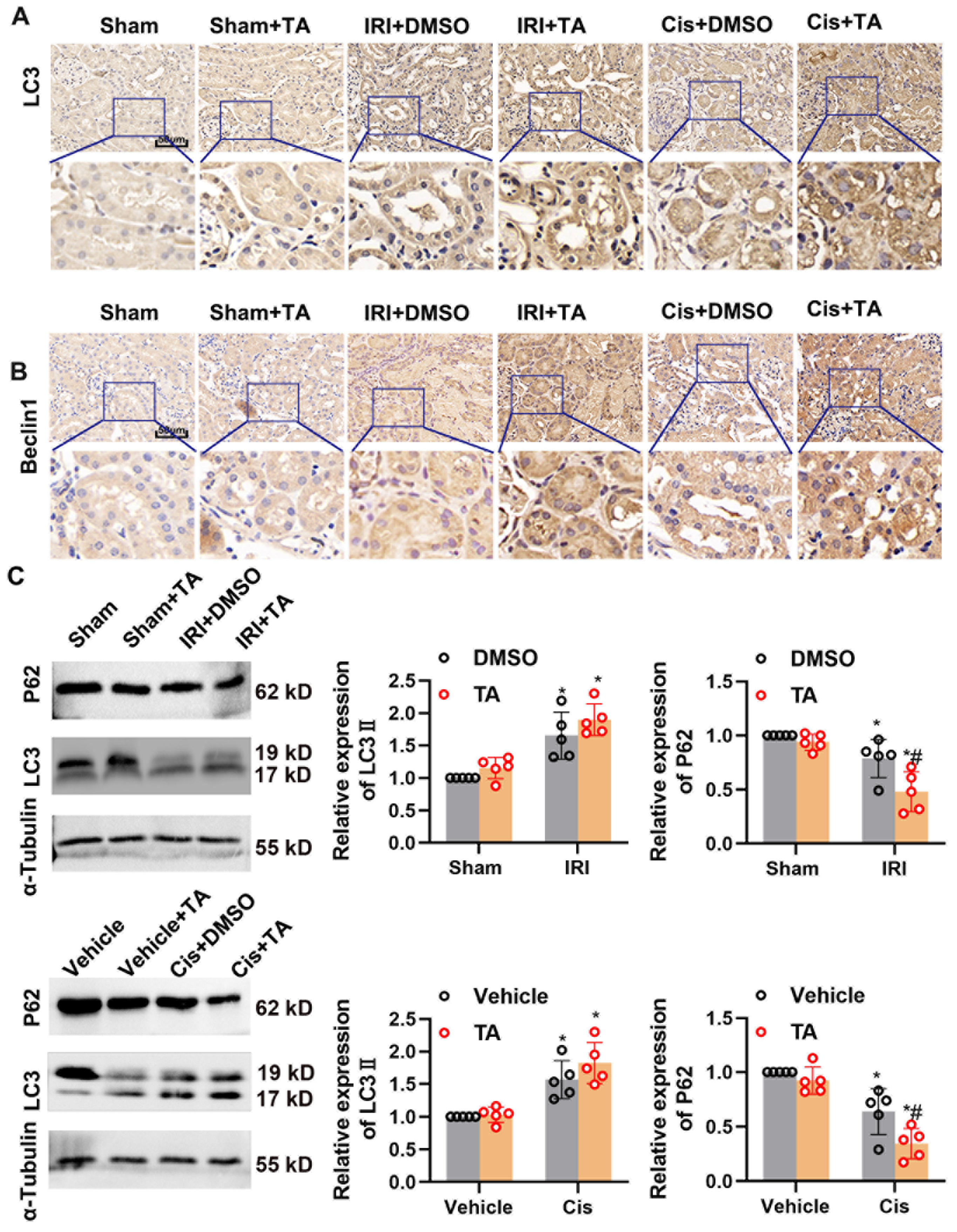
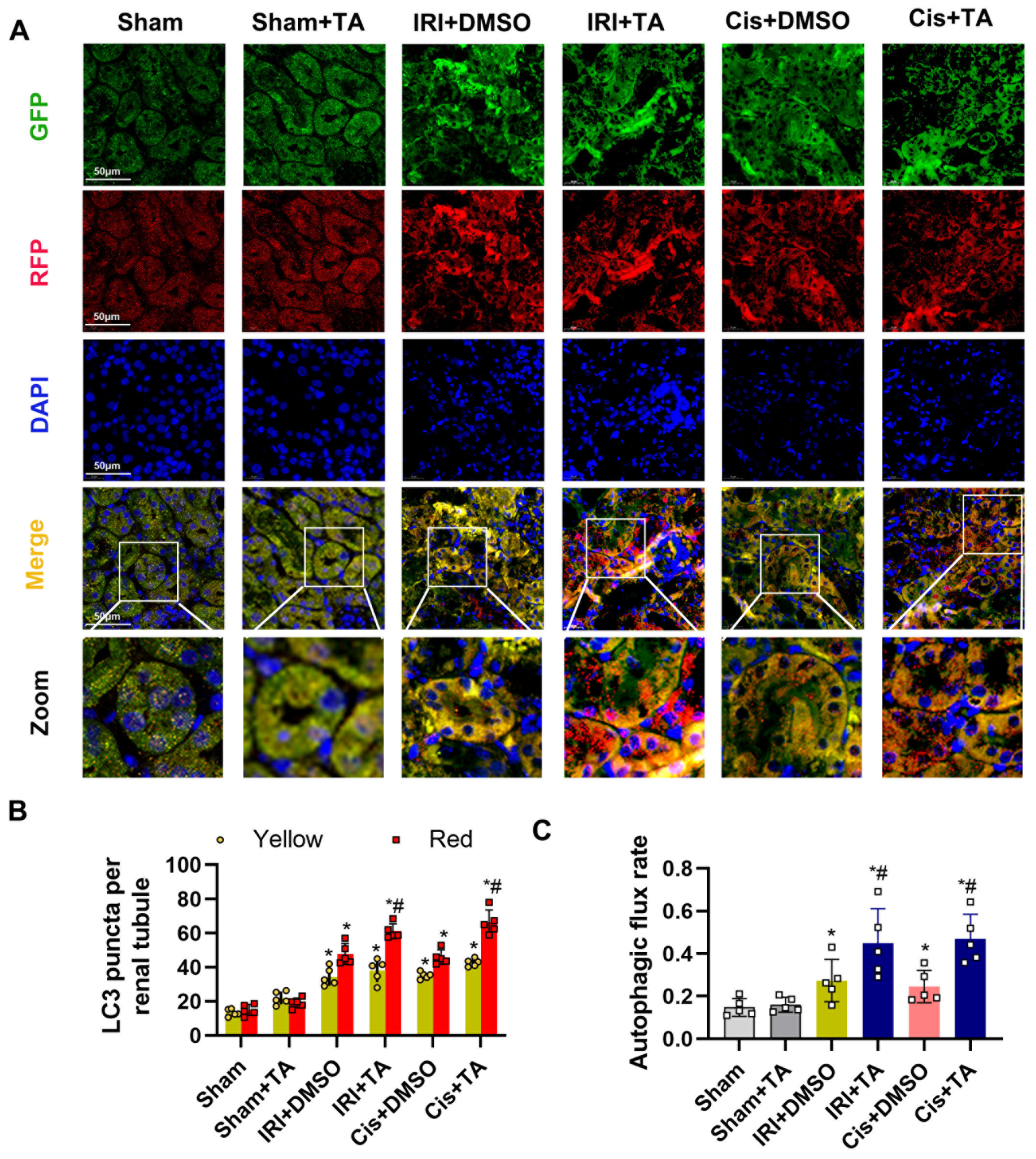
3.5. HDAC6 Inhibition Promotes Autophagy in Ischemia- and Cisplatin-Induced AKI
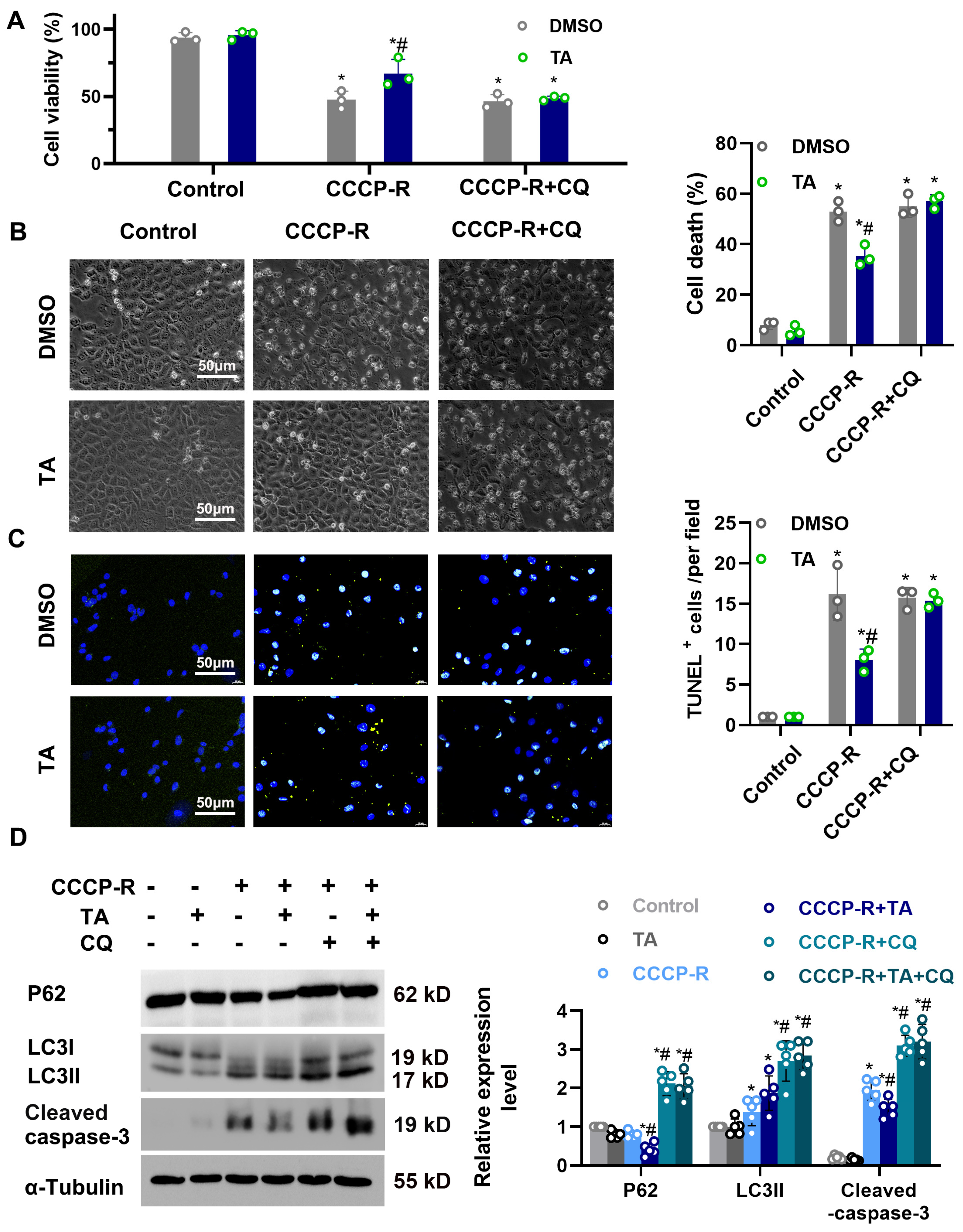
3.6. Chloroquine Suppresses the Protective Effects of HDAC6 Inhibition on Cellular Viability during CCCP Treatment
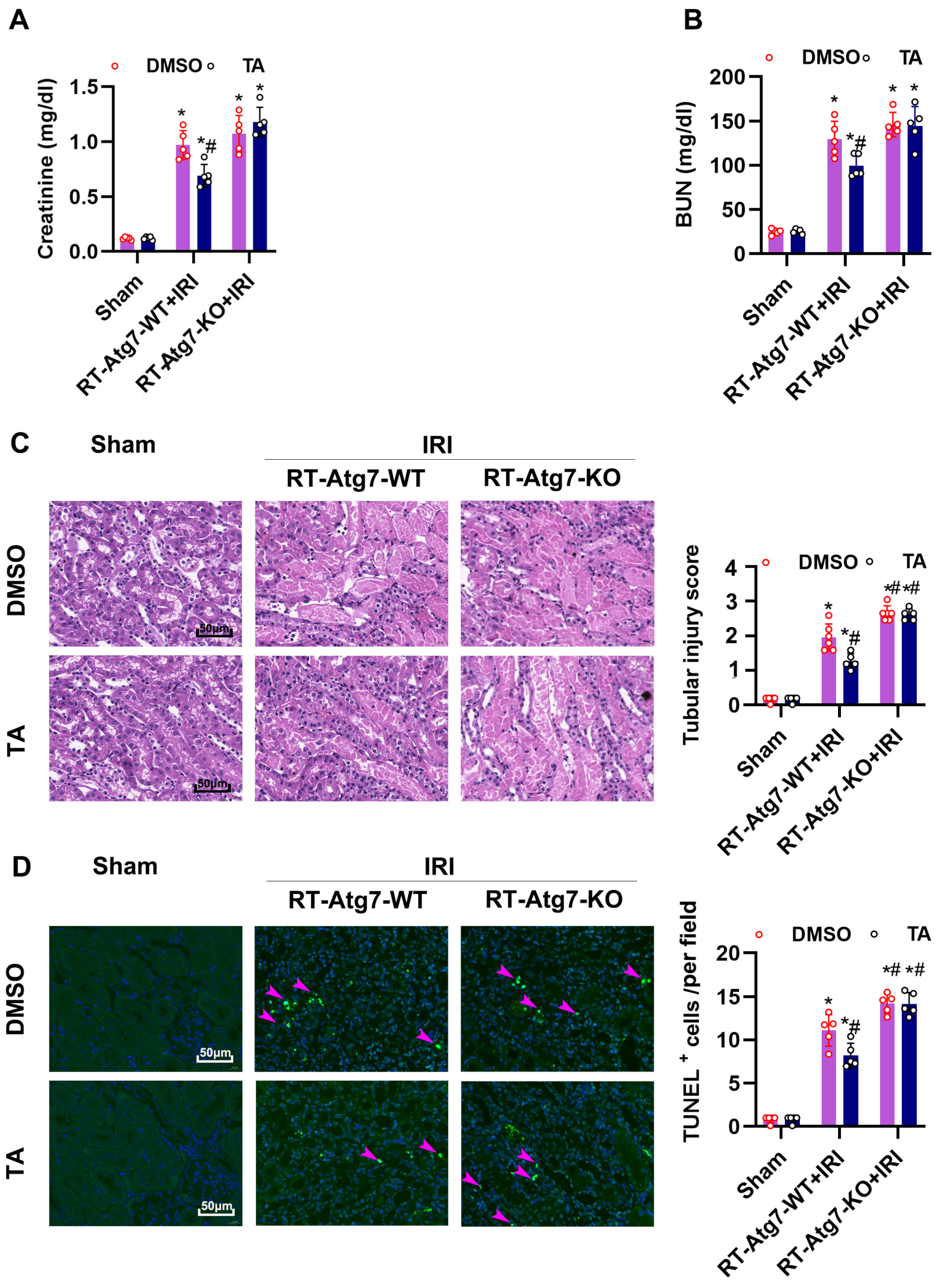
3.7. Ischemia-Induced Autophagy Is Inhibited in Renal Tubules in RT-Atg7-KO Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kellum, J.A.; Romagnani, P.; Ashuntantang, G.; Ronco, C.; Zarbock, A.; Anders, H.J. Acute kidney injury. Nat. Rev. Dis. Primers 2021, 7, 52. [Google Scholar] [CrossRef]
- Pickkers, P.; Darmon, M.; Hoste, E.; Joannidis, M.; Legrand, M.; Ostermann, M.; Prowle, J.R.; Schneider, A.; Schetz, M. Acute kidney injury in the critically ill: An updated review on pathophysiology and management. Intensive Care Med. 2021, 47, 835–850. [Google Scholar] [CrossRef]
- Ronco, C.; Bellomo, R.; Kellum, J.A. Acute kidney injury. Lancet 2019, 394, 1949–1964. [Google Scholar] [CrossRef]
- Zuk, A.; Bonventre, J.V. Acute Kidney Injury. Annu. Rev. Med. 2016, 67, 293–307. [Google Scholar] [CrossRef] [Green Version]
- Volarevic, V.; Djokovic, B.; Jankovic, M.G.; Harrell, C.R.; Fellabaum, C.; Djonov, V.; Arsenijevic, N. Molecular mechanisms of cisplatin-induced nephrotoxicity: A balance on the knife edge between renoprotection and tumor toxicity. J. Biomed. Sci. 2019, 26, 25. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.B.; Lee, D.S.; Padanilam, B.J.; Kim, J. Repeated Administration of Cisplatin Transforms Kidney Fibroblasts through G2/M Arrest and Cellular Senescence. Cells 2022, 11, 3472. [Google Scholar] [CrossRef]
- Kim, M.J.; Moon, D.; Jung, S.; Lee, J.; Kim, J. Cisplatin nephrotoxicity is induced via poly(ADP-ribose) polymerase activation in adult zebrafish and mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2020, 318, R843–R854. [Google Scholar] [CrossRef]
- Kitada, M.; Koya, D. Autophagy in metabolic disease and ageing. Nat. Rev. Endocrinol. 2021, 17, 647–661. [Google Scholar] [CrossRef]
- Tang, C.; Livingston, M.J.; Liu, Z.; Dong, Z. Autophagy in kidney homeostasis and disease. Nat. Rev. Nephrol. 2020, 16, 489–508. [Google Scholar] [CrossRef]
- Festa, B.P.; Chen, Z.; Berquez, M.; Debaix, H.; Tokonami, N.; Prange, J.A.; Hoek, G.V.D.; Alessio, C.; Raimondi, A.; Nevo, N.; et al. Impaired autophagy bridges lysosomal storage disease and epithelial dysfunction in the kidney. Nat. Commun. 2018, 9, 161. [Google Scholar] [CrossRef]
- Yang, Z.; Klionsky, D.J. An overview of the molecular mechanism of autophagy. Curr. Top. Microbiol. Immunol. 2009, 335, 1–32. [Google Scholar]
- Nakatogawa, H.; Ichimura, Y.; Ohsumi, Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 2007, 130, 165–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, M.; Wei, Q.; Dong, G.; Komatsu, M.; Su, Y.; Dong, Z. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int. 2012, 82, 1271–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parmigiani, R.B.; Xu, W.S.; Venta-Perez, G.; Erdjument-Bromage, H.; Yaneva, M.; Tempst, P.; Marks, P.A. HDAC6 is a specific deacetylase of peroxiredoxins and is involved in redox regulation. Proc. Natl. Acad. Sci. USA 2008, 105, 9633–9638. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Zhang, C.; Hassan, S.; Liu, X.; Song, F.; Chen, K.; Zhang, W.; Yang, J. Histone deacetylase 6 in cancer. J. Hematol. Oncol. 2018, 11, 111. [Google Scholar] [CrossRef] [Green Version]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.F.; Yao, T.P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Miyake, Y.; Keusch, J.J.; Wang, L.; Saito, M.; Hess, D.; Wang, X.; Melancon, B.J.; Helquist, P.; Gut, H.; Matthias, P. Structural insights into HDAC6 tubulin deacetylation and its selective inhibition. Nat. Chem. Biol. 2016, 12, 748–754. [Google Scholar] [CrossRef]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination Therapy With Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi. Front. Oncol. 2018, 8, 92. [Google Scholar] [CrossRef] [Green Version]
- Trzeciakiewicz, H.; Ajit, D.; Tseng, J.H.; Chen, Y.; Ajit, A.; Tabassum, Z.; Lobrovich, R.; Peterson, C.; Riddick, N.V.; Itano, M.S.; et al. An HDAC6-dependent surveillance mechanism suppresses tau-mediated neurodegeneration and cognitive decline. Nat. Commun. 2020, 11, 5522. [Google Scholar] [CrossRef]
- Ke, B.; Chen, Y.; Tu, W.; Ye, T.; Fang, X.; Yang, L. Inhibition of HDAC6 activity in kidney diseases: A new perspective. Mol. Med. 2018, 24, 33. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Ueno, T.; Iwata, J.; Murata, S.; Tanida, I.; Ezaki, J.; Mizushima, N.; Ohsumi, Y.; Uchiyama, Y.; et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 2005, 169, 425–434. [Google Scholar] [CrossRef]
- Zhu, J.; Zhang, Y.; Shi, L.; Xia, Y.; Zha, H.; Li, H.; Song, Z. RP105 protects against ischemic and septic acute kidney injury via suppressing TLR4/NF-kappaB signaling pathways. Int. Immunopharmacol. 2022, 109, 108904. [Google Scholar] [CrossRef]
- Liu, J.; Livingston, M.J.; Dong, G.; Tang, C.; Su, Y.; Wu, G.; Yin, X.M.; Dong, Z. Histone deacetylase inhibitors protect against cisplatin-induced acute kidney injury by activating autophagy in proximal tubular cells. Cell Death Dis. 2018, 9, 322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyndman, K.A. Histone Deacetylases in Kidney Physiology and Acute Kidney Injury. Semin. Nephrol. 2020, 40, 138–147. [Google Scholar] [CrossRef]
- Wei, Q.; Dong, Z. Mouse model of ischemic acute kidney injury: Technical notes and tricks. Am. J. Physiol. Renal Physiol. 2012, 303, F1487–F1494. [Google Scholar] [CrossRef] [Green Version]
- Livingston, M.J.; Wang, J.; Zhou, J.; Wu, G.; Ganley, I.G.; Hill, J.A.; Yin, X.M.; Dong, Z. Clearance of damaged mitochondria via mitophagy is important to the protective effect of ischemic preconditioning in kidneys. Autophagy 2019, 15, 2142–2162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, T.; Qi, C.; Lai, Y.; Xie, J.; Wang, H.; Zhang, L.; Lin, T.; Jv, M.; Li, J.; Wang, Y.; et al. HDAC6-mediated alpha-tubulin deacetylation suppresses autophagy and enhances motility of podocytes in diabetic nephropathy. J. Cell. Mol. Med. 2020, 24, 11558–11572. [Google Scholar] [CrossRef]
- Tang, J.; Shi, Y.; Liu, N.; Xu, L.; Zang, X.; Li, P.; Zhang, J.; Zheng, X.; Qiu, A.; Zhuang, S. Blockade of histone deacetylase 6 protects against cisplatin-induced acute kidney injury. Clin. Sci. 2018, 132, 339–359. [Google Scholar] [CrossRef]
- Livingston, M.J.; Shu, S.; Fan, Y.; Li, Z.; Jiao, Q.; Yin, X.M.; Venkatachalam, M.A.; Dong, Z. Tubular cells produce FGF2 via autophagy after acute kidney injury leading to fibroblast activation and renal fibrosis. Autophagy, 2022; 1–22, Online ahead of print. [Google Scholar] [CrossRef]
- Levine, M.H.; Wang, Z.; Bhatti, T.R.; Wang, Y.; Aufhauser, D.D.; McNeal, S.; Liu, Y.; Cheraghlou, S.; Han, R.; Wang, L.; et al. Class-specific histone/protein deacetylase inhibition protects against renal ischemia reperfusion injury and fibrosis formation. Am. J. Transplant. 2015, 15, 965–973. [Google Scholar] [CrossRef]
- Xiang, X.; Dong, G.; Zhu, J.; Zhang, G.; Dong, Z. Inhibition of HDAC3 protects against kidney cold storage/transplantation injury and allograft dysfunction. Clin. Sci. 2022, 136, 45–60. [Google Scholar] [CrossRef]
- Hai, Y.; Christianson, D.W. Histone deacetylase 6 structure and molecular basis of catalysis and inhibition. Nat. Chem. Biol. 2016, 12, 741–747. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Huang, R.; Guo, F.; Liang, Y.; Xiang, J.; Lei, S.; Shi, M.; Li, L.; Liu, J.; Feng, Y.; et al. Selective Histone Deacetylase 6 Inhibitor 23BB Alleviated Rhabdomyolysis-Induced Acute Kidney Injury by Regulating Endoplasmic Reticulum Stress and Apoptosis. Front. Pharmacol. 2018, 9, 274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.X.; Ma, W.J.; He, L.Y.; Zhang, C.H.; Zhang, C.; Wang, Y.; Chen, C.N.; Shen, D.Y.; Gao, H.M.; Guo, R.R.; et al. Macrophage migration inhibitory factor (MIF) acetylation protects neurons from ischemic injury. Cell Death Dis. 2022, 13, 466. [Google Scholar] [CrossRef] [PubMed]
- Periyasamy-Thandavan, S.; Jiang, M.; Wei, Q.; Smith, R.; Yin, X.M.; Dong, Z. Autophagy is cytoprotective during cisplatin injury of renal proximal tubular cells. Kidney Int. 2008, 74, 631–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, S.; Livingston, M.; Hao, J.; Li, L.; Mei, C.; Dong, Z. Autophagy is activated to protect against endotoxic acute kidney injury. Sci. Rep. 2016, 6, 22171. [Google Scholar] [CrossRef] [Green Version]
- Magupalli, V.G.; Negro, R.; Tian, Y.; Hauenstein, A.V.; Di Caprio, G.; Skillern, W.; Deng, Q.; Orning, P.; Alam, H.B.; Maliga, Z.; et al. HDAC6 mediates an aggresome-like mechanism for NLRP3 and pyrin inflammasome activation. Science 2020, 369, eaas8995. [Google Scholar] [CrossRef]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Wan, W. Acetylation in the regulation of autophagy. Autophagy, 2022; 1–9, Online ahead of print. [Google Scholar] [CrossRef]
- Sun, J.; Tai, S.; Tang, L.; Yang, H.; Chen, M.; Xiao, Y.; Li, X.; Zhu, Z.; Zhou, S. Acetylation Modification during Autophagy and Vascular Aging. Front. Physiol. 2021, 12, 598267. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhao, L.; Cui, L.; Huang, Y.; Ye, J.; Zhang, Q.; Jiang, X.; Zhang, D.; Huang, Y. Decreased alpha-tubulin acetylation induced by an acidic environment impairs autophagosome formation and leads to rat cardiomyocyte injury. J. Mol. Cell. Cardiol. 2019, 127, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Geeraert, C.; Ratier, A.; Pfisterer, S.G.; Perdiz, D.; Cantaloube, I.; Rouault, A.; Pattingre, S.; Proikas-Cezanne, T.; Codogno, P.; Poüs, C. Starvation-induced hyperacetylation of tubulin is required for the stimulation of autophagy by nutrient deprivation. J. Biol. Chem. 2010, 285, 24184–24194. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, L.; Song, Z.; Li, C.; Deng, F.; Xia, Y.; Huang, J.; Wu, X.; Zhu, J. HDAC6 Inhibition Alleviates Ischemia- and Cisplatin-Induced Acute Kidney Injury by Promoting Autophagy. Cells 2022, 11, 3951. https://doi.org/10.3390/cells11243951
Shi L, Song Z, Li C, Deng F, Xia Y, Huang J, Wu X, Zhu J. HDAC6 Inhibition Alleviates Ischemia- and Cisplatin-Induced Acute Kidney Injury by Promoting Autophagy. Cells. 2022; 11(24):3951. https://doi.org/10.3390/cells11243951
Chicago/Turabian StyleShi, Lang, Zhixia Song, Chenglong Li, Fangjing Deng, Yao Xia, Jing Huang, Xiongfei Wu, and Jiefu Zhu. 2022. "HDAC6 Inhibition Alleviates Ischemia- and Cisplatin-Induced Acute Kidney Injury by Promoting Autophagy" Cells 11, no. 24: 3951. https://doi.org/10.3390/cells11243951