
Combined Noncoding RNA-mRNA Regulomics Signature in Reprogramming and Pluripotency in iPSCs
 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
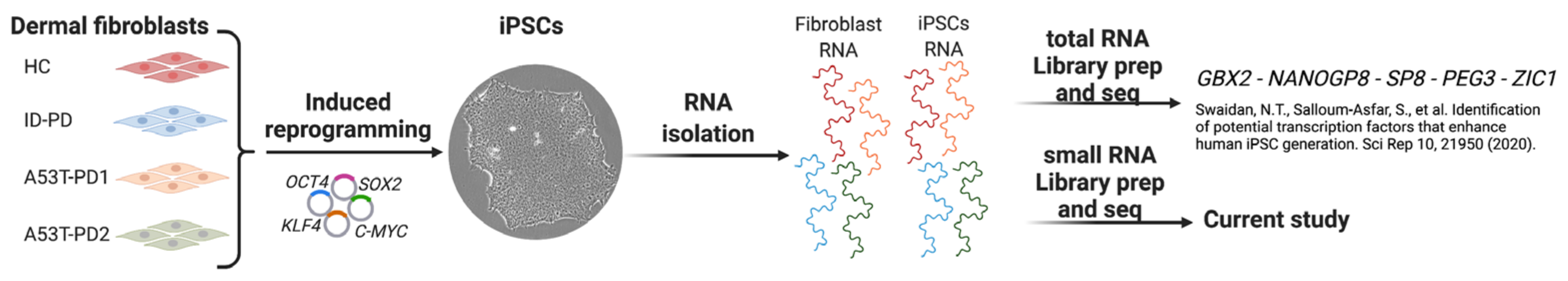
2.1. RNA Samples
2.2. Small RNA Library Preparation and Sequencing
2.3. Sequencing Read Mapping and Small RNA Annotation
2.4. sncRNAs Analysis Using WiND Workflow
2.5. Differential Expression Analysis: CLC Genomics Workbench Version 20.0.4
2.6. miRNA Profiling Comparison and Functional Enrichment Tests
2.7. Evaluation of Other sncRNAs Expressions in Fibroblasts and Their iPSCs
3. Results
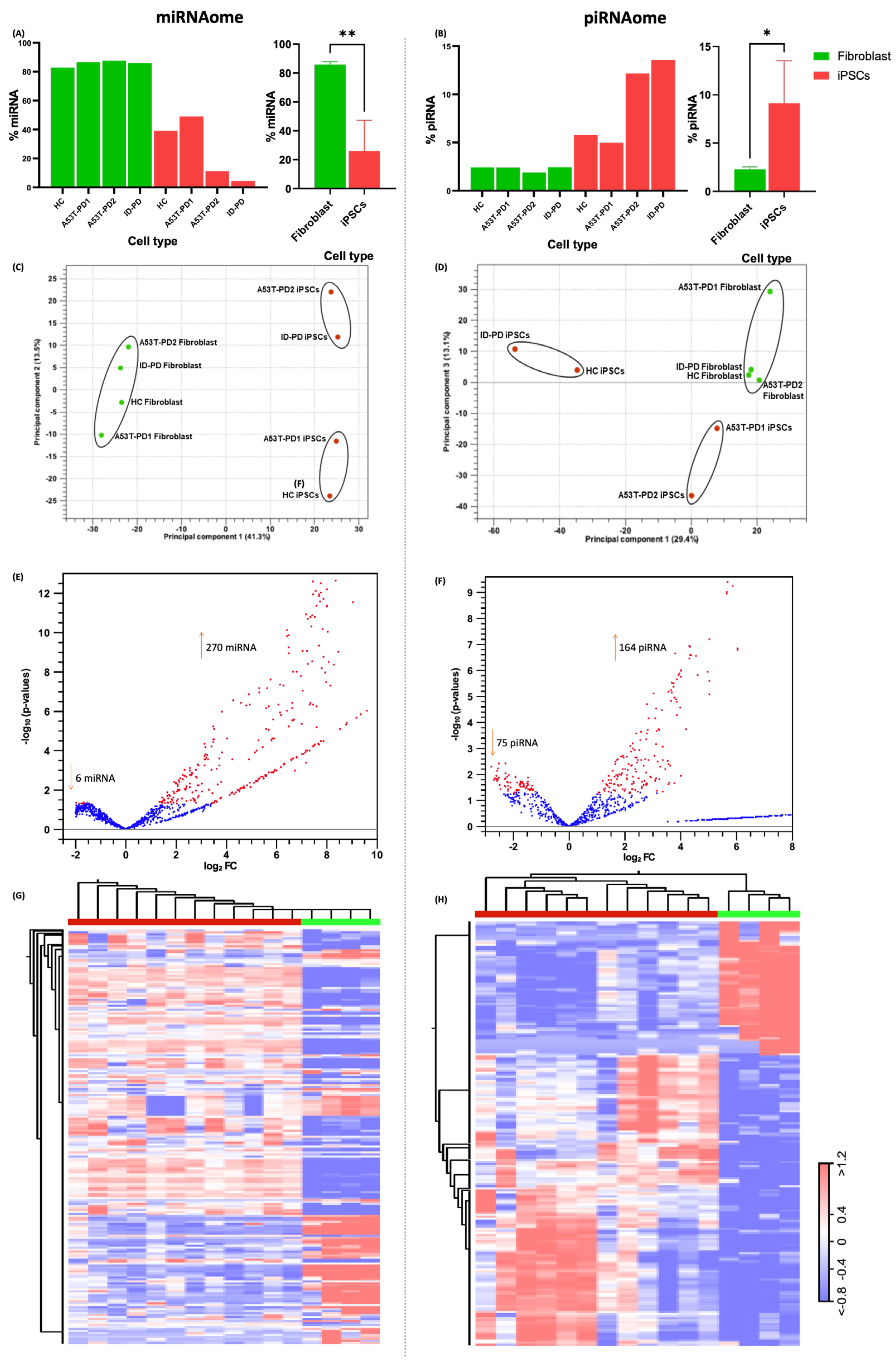
3.1. Small RNA Sequencing Data Preprocessing and Comparison of Fibroblasts and iPSCs
3.2. miRNA and piRNA Profiles Are Differentially Expressed in Fibroblasts vs. Their Reprogrammed iPSCs
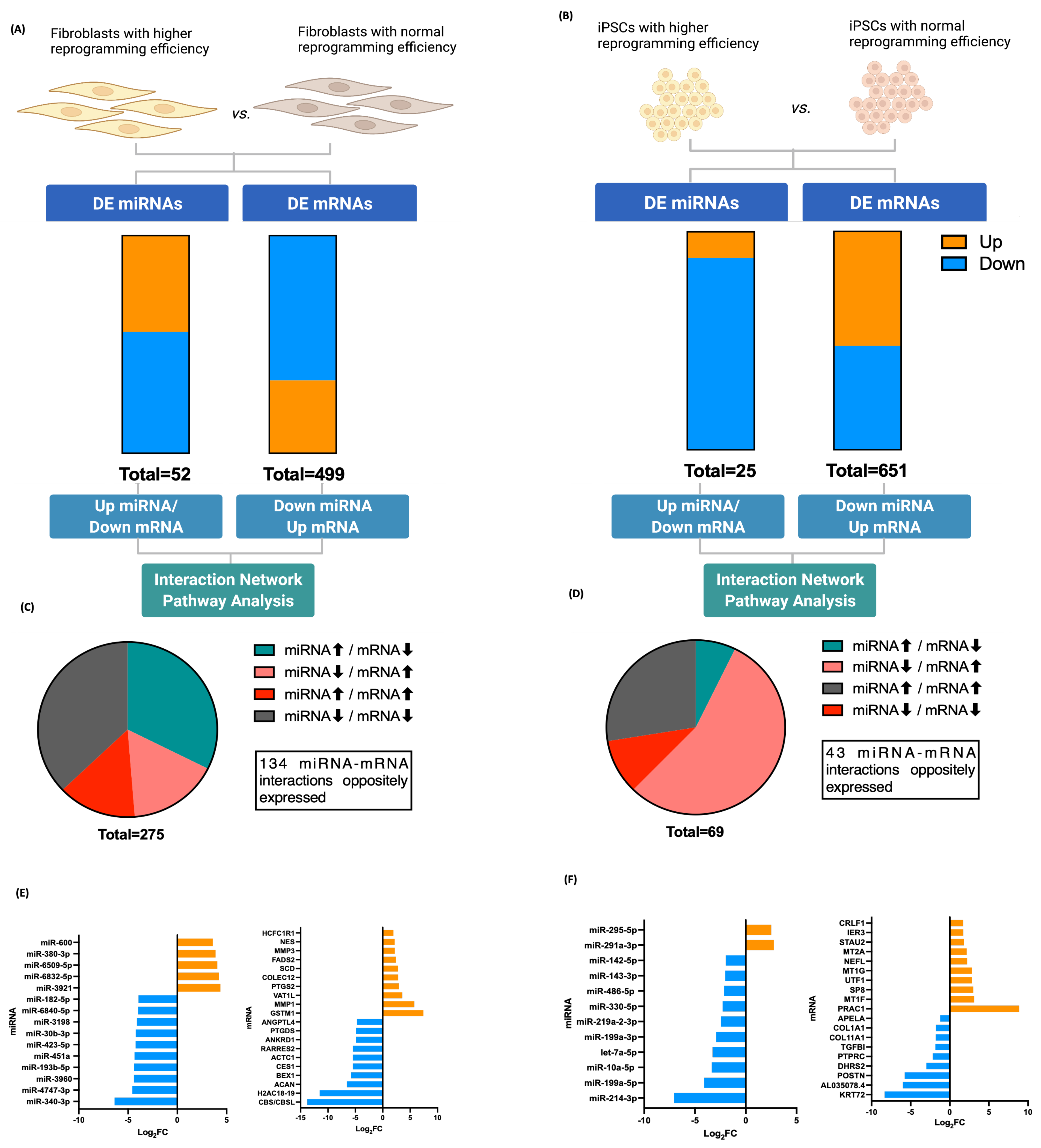
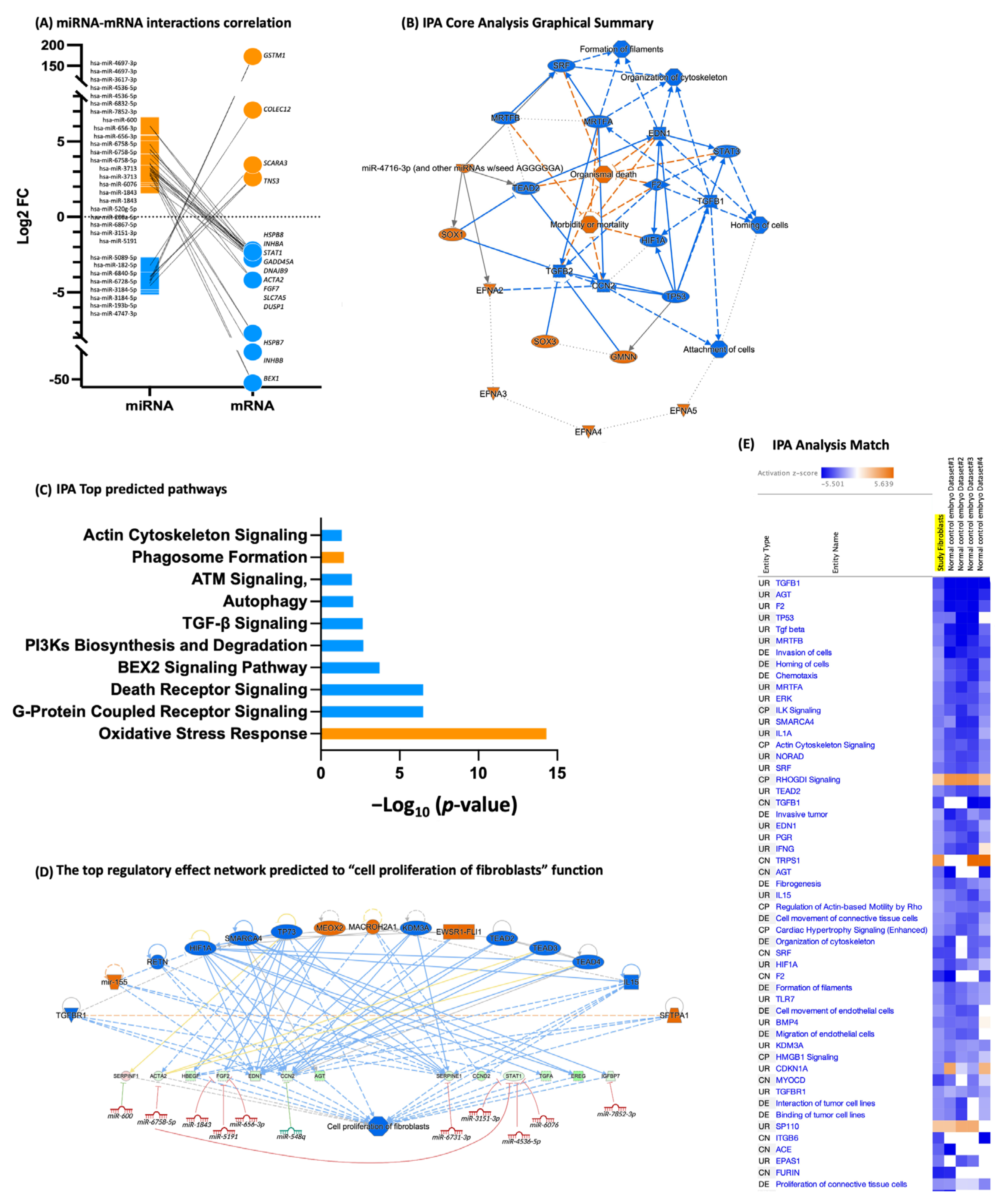
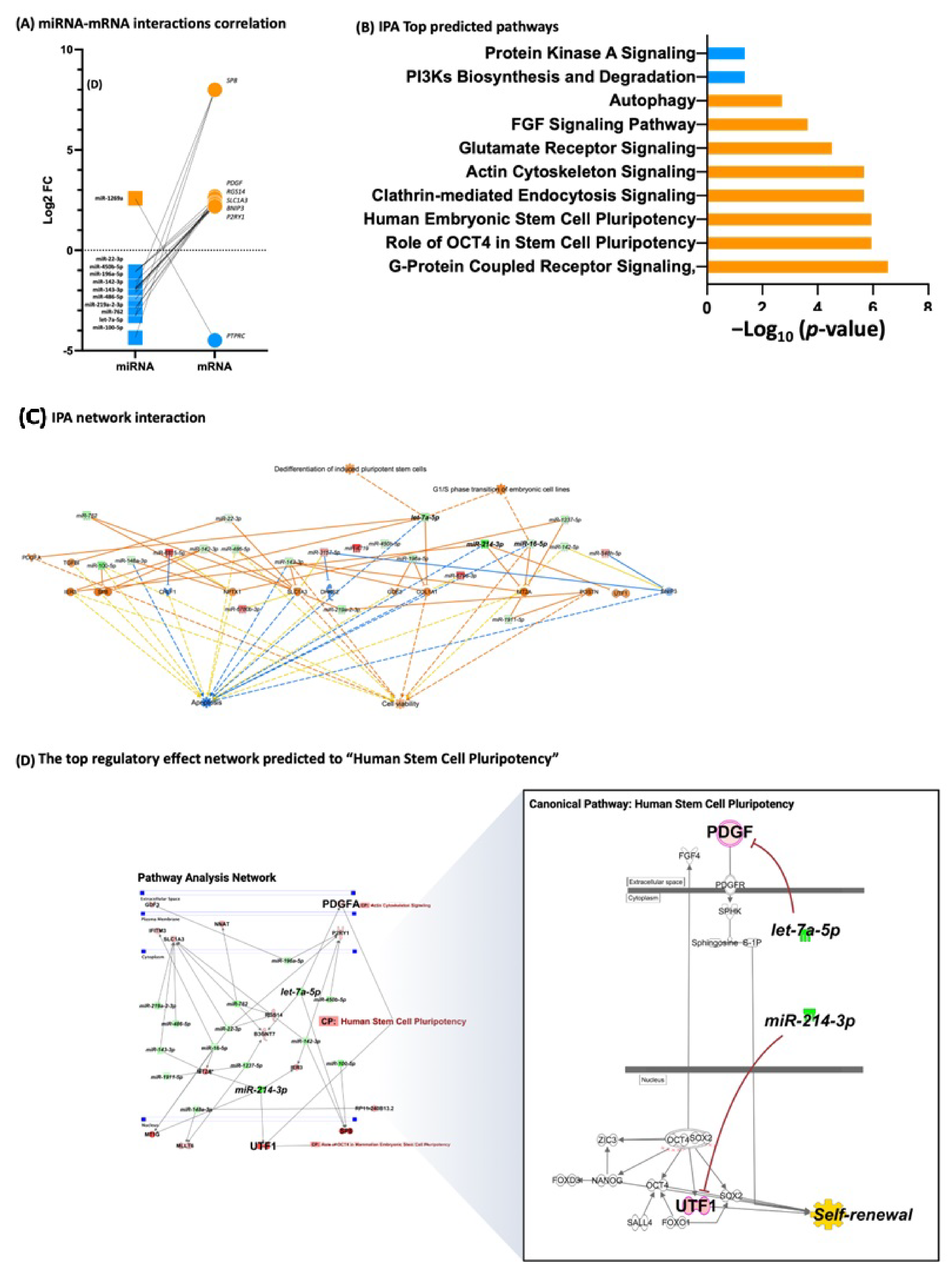
3.3. Fibroblasts Differentially Expressed miRNA, miRNA Target Prediction, and Interaction with Transcriptome
3.4. iPSCs Differentially Expressed miRNA, miRNA Target Prediction, and Interaction with Transcriptome
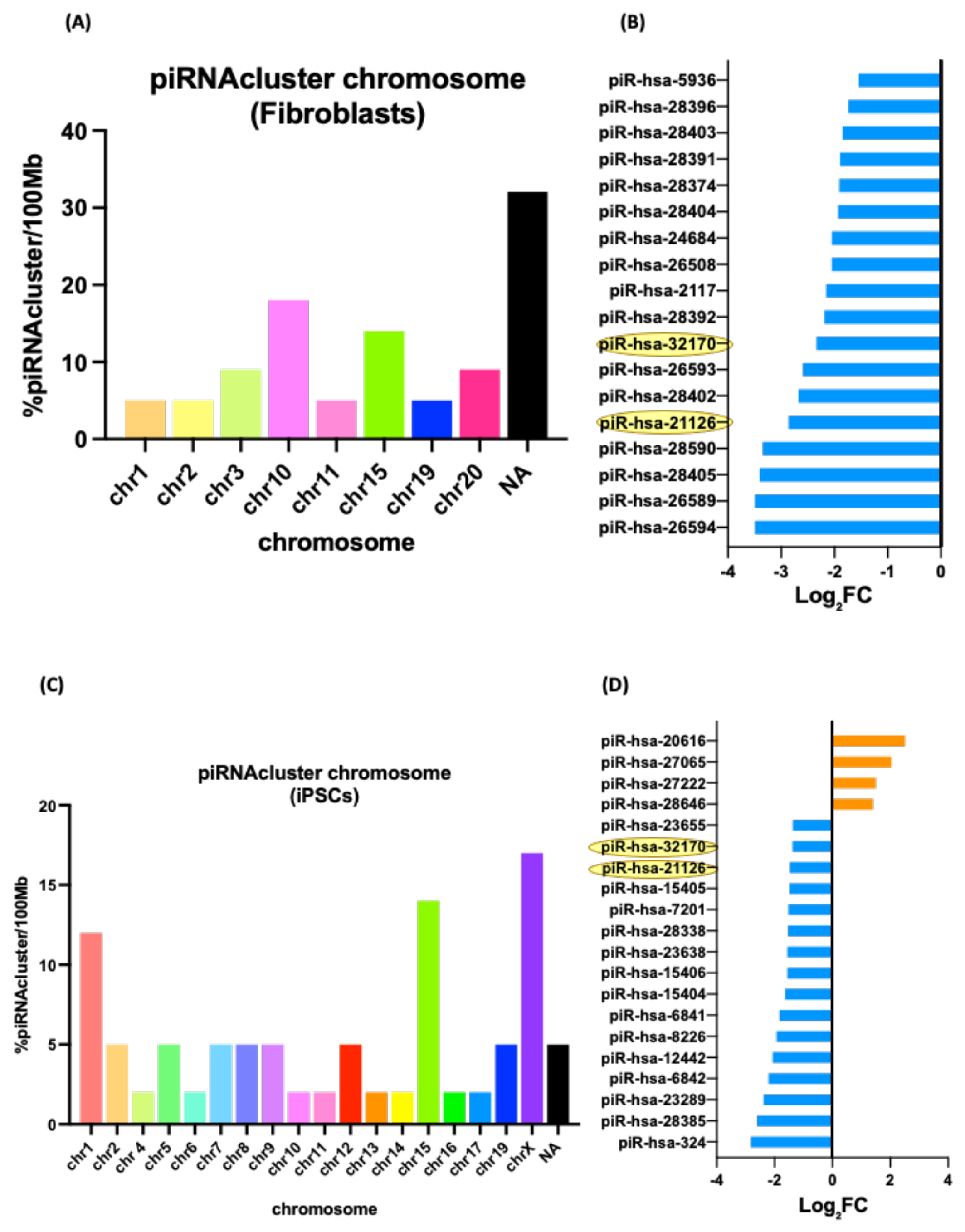
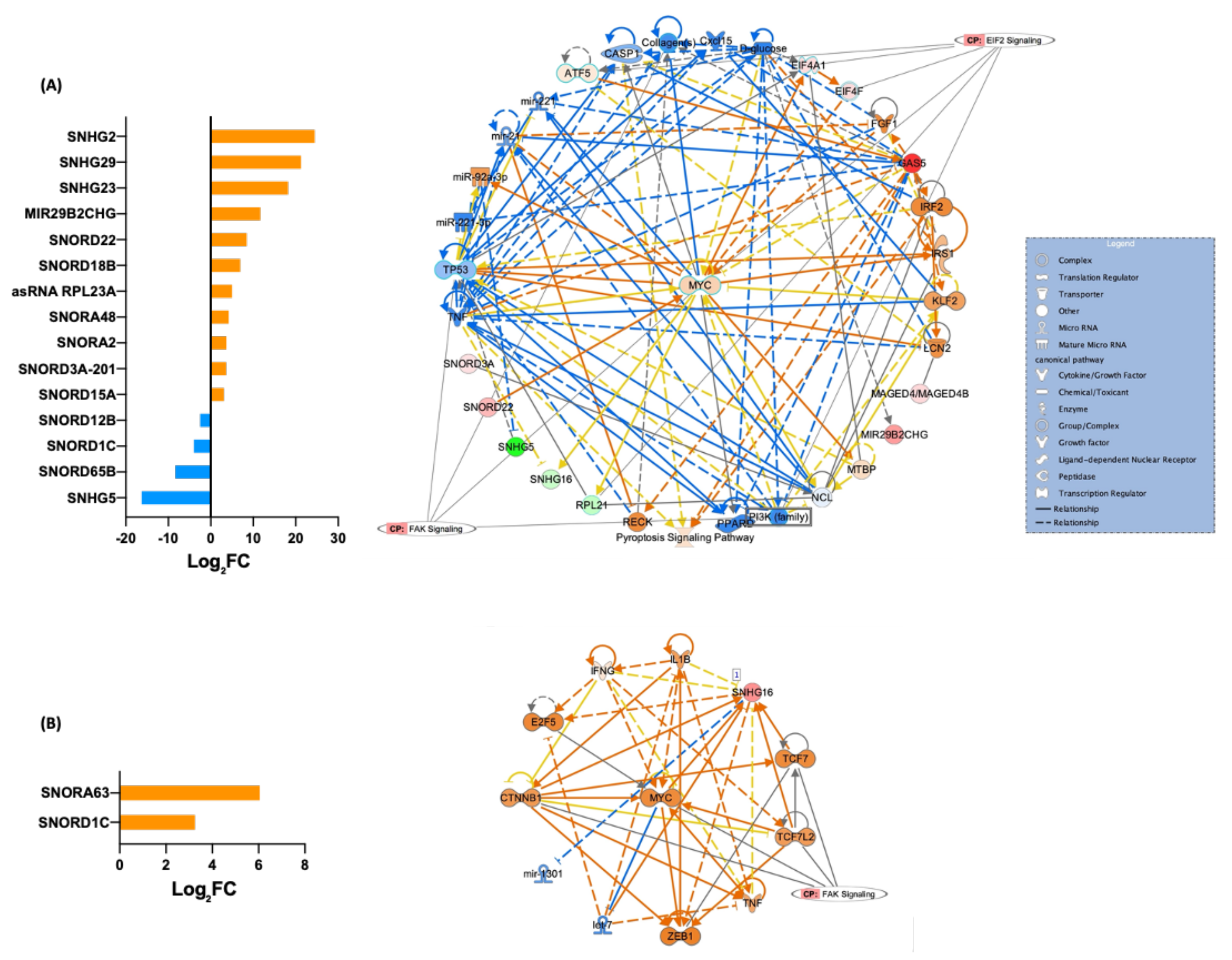
3.5. Differentially Expressed piRNAs and Other ncRNAs in Fibroblasts and Their iPSCs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Gorecka, J.; Kostiuk, V.; Fereydooni, A.; Gonzalez, L.; Luo, J.; Dash, B.; Isaji, T.; Ono, S.; Liu, S.; Lee, S.R.; et al. The potential and limitations of induced pluripotent stem cells to achieve wound healing. Stem Cell Res. Ther. 2019, 10, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karagiannis, P.; Takahashi, K.; Saito, M.; Yoshida, Y.; Okita, K.; Watanabe, A.; Inoue, H.; Yamashita, J.K.; Todani, M.; Nakagawa, M.; et al. Induced pluripotent stem cells and their use in human models of disease and development. Physiol. Rev. 2019, 99, 79–114. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, B. Reprogramming barriers and enhancers: Strategies to enhance the efficiency and kinetics of induced pluripotency. Cell Regen. 2015, 4, 4–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaikkonen, M.U.; Lam, M.T.; Glass, C.K. Non-coding RNAs as regulators of gene expression and epigenetics. Cardiovasc. Res. 2011, 90, 430–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Wu, W.; Chen, Q.; Chen, M. Non-coding RNAs and their integrated networks. J. Integr. Bioinform. 2019, 16, 20190027. [Google Scholar] [CrossRef]
- Kanwal, S.; Guo, X.; Ward, C.; Volpe, G.; Qin, B.; Esteban, M.A.; Bao, X. Role of long Non-coding RNAs in reprogramming to induced pluripotency. Genom. Proteom. Bioinform. 2020, 18, 16–25. [Google Scholar] [CrossRef]
- Artmann, S.; Jung, K.; Bleckmann, A.; Beißbarth, T. Detection of Simultaneous Group Effects in MicroRNA Expression and Related Target Gene Sets. PLoS ONE 2012, 7, e38365. [Google Scholar] [CrossRef] [Green Version]
- Ozata, D.M.; Gainetdinov, I.; Zoch, A.; O’Carroll, D.; Zamore, P.D. PIWI-interacting RNAs: Small RNAs with big functions. Nat. Rev. Genet. 2019, 20, 89–108. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Fan, C.; Zhao, B. Differential expression of piRNAs in reprogrammed pluripotent stem cells from mouse embryonic fibroblasts. IUBMB Life 2019, 71, 1906–1915. [Google Scholar] [CrossRef]
- Kondetimmanahalli, R.; Gharpure, K.M.; Wu, S.Y.; Lopez-Berestein, G.; Sood, A.K. Noncoding RNAs: Novel Targets in Anticancer Drug Development. In Cancer and Noncoding RNAs; Elsevier: Amsterdam, The Netherlands, 2018; pp. 447–459. [Google Scholar]
- Liang, J.; Wen, J.; Huang, Z.; Chen, X.-P.; Zhang, B.-X.; Chu, L. Small nucleolar RNAs: Insight into their function in cancer. Front. Oncol. 2019, 9, 587. [Google Scholar] [CrossRef]
- Yang, C.-S.; Rana, T.M. Learning the molecular mechanisms of the reprogramming factors: Let’s start from microRNAs. Mol. BioSystems 2013, 9, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Heinrich, E.-M.; Dimmeler, S. MicroRNAs and stem cells: Control of pluripotency, reprogramming, and lineage commitment. Circ. Res. 2012, 110, 1014–1022. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Guo, X.; Hong, W.; Liu, Q.; Wei, T.; Lu, C.; Gao, L.; Ye, D.; Zhou, Y.; Chen, J.; et al. Critical regulation of miR-200/ZEB2 pathway in Oct4/Sox2-induced mesenchymal-to-epithelial transition and induced pluripotent stem cell generation. Proc. Natl. Acad. Sci. USA 2013, 110, 2858–2863. [Google Scholar] [CrossRef] [Green Version]
- Li, M.A.; He, L. microRNAs as novel regulators of stem cell pluripotency and somatic cell reprogramming. Bioessays 2012, 34, 670–680. [Google Scholar] [CrossRef] [Green Version]
- Mallanna, S.K.; Rizzino, A. Emerging roles of microRNAs in the control of embryonic stem cells and the generation of induced pluripotent stem cells. Dev. Biol. 2010, 344, 16–25. [Google Scholar] [CrossRef] [Green Version]
- González, F.; Boué, S.; Belmonte, J.C.I. Methods for making induced pluripotent stem cells: Reprogramming a la carte. Nat. Rev. Genet. 2011, 12, 231–242. [Google Scholar] [CrossRef]
- Sherstyuk, V.V.; Medvedev, S.P.; Zakian, S.M. Noncoding RNAs in the regulation of pluripotency and reprogramming. Stem Cell Rev. Rep. 2018, 14, 58–70. [Google Scholar] [CrossRef]
- Fong, Y.W.; Ho, J.J.; Inouye, C.; Tjian, R. The dyskerin ribonucleoprotein complex as an OCT4/SOX2 coactivator in embryonic stem cells. Elife 2014, 3, e03573. [Google Scholar] [CrossRef]
- Swaidan, N.T.; Salloum-Asfar, S.; Palangi, F.; Errafii, K.; Soliman, N.H.; Aboughalia, A.T.; Wali, A.H.S.; Abdulla, S.A.; Emara, M.M. Identification of potential transcription factors that enhance human iPSC generation. Sci. Rep. 2020, 10, 21950. [Google Scholar] [CrossRef]
- Geles, K.; Palumbo, D.; Sellitto, A.; Giurato, G.; Cianflone, E.; Marino, F.; Torella, D.; Cappa, V.M.; Nassa, G.; Tarallo, R.; et al. WIND (Workflow for pIRNAs aNd beyonD): A strategy for in-depth analysis of small RNA-seq data. F1000Research 2021, 10, 1. [Google Scholar] [CrossRef]
- Love, M.; Anders, S.; Huber, W. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Belinda, P.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lun, A.T.; Smyth, G.K. From reads to genes to pathways: Differential expression analysis of RNA-Seq experiments using Rsubread and the edgeR quasi-likelihood pipeline. F1000Research 2016, 5, 1438. [Google Scholar]
- Salloum-Asfar, S.; Elsayed, A.; Elhag, S.; Abdulla, S. Circulating Non-Coding RNAs as a Signature of Autism Spectrum Disorder Symptomatology. Int. J. Mol. Sci. 2021, 22, 6549. [Google Scholar] [CrossRef]
- Anokye-Danso, F.; Trivedi, C.M.; Juhr, D.; Gupta, M.; Cui, Z.; Tian, Y.; Zhang, Y.; Yang, W.; Gruber, P.J.; Epstein, J.A.; et al. Highly efficient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency. Cell Stem Cell 2011, 8, 376–388. [Google Scholar] [CrossRef] [Green Version]
- Clancy, J.L.; Patel, H.R.; Hussein, S.M.I.; Tonge, P.D.; Cloonan, N.; Corso, A.J.; Li, M.; Lee, D.-S.; Shin, J.-Y.; Wong, J.J.L.; et al. Small RNA changes en route to distinct cellular states of induced pluripotency. Nat. Commun. 2014, 5, 5522. [Google Scholar] [CrossRef] [Green Version]
- Henzler, C.M.; Li, Z.; Dang, J.; Arcila, M.L.; Zhou, H.; Liu, J.; Chang, K.-Y.; Bassett, D.S.; Rana, T.M.; Kosik, K.S. Staged miRNA re-regulation patterns during reprogramming. Genome Biol. 2013, 14, R149. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Wei, X.; Wang, M.; Zhang, R.; Fu, Y.; Xing, M.; Hua, Q.; Xie, X. Proliferation rate of somatic cells affects reprogramming efficiency. J. Biol. Chem. 2013, 288, 9767–9778. [Google Scholar] [CrossRef] [Green Version]
- Pakshir, P.; Alizadehgiashi, M.; Wong, B.; Coelho, N.; Chen, X.; Gong, Z.; Shenoy, V.B.; McCulloch, C.A.; Hinz, B. Dynamic fibroblast contractions attract remote macrophages in fibrillar collagen matrix. Nat. Commun. 2019, 10, 1850. [Google Scholar] [CrossRef] [Green Version]
- Razak, S.R.A.; Baba, Y.; Nakauchi, H.; Otsu, M.; Watanabe, S. DNA methylation is involved in the expression of miR-142-3p in fibroblasts and induced pluripotent stem cells. Stem Cells Int. 2014, 2014, 101349. [Google Scholar]
- Gu, K.-L.; Zhang, Q.; Yan, Y.; Li, T.T.; Duan, F.F.; Hao, J.; Wang, X.W.; Shi, M.; Wu, D.R.; Guo, W.T.; et al. Pluripotency-associated miR-290/302 family of microRNAs promote the dismantling of naive pluripotency. Cell Res. 2016, 26, 350–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, H.; Ni, N.; Zhang, D.; Wang, Y.; Tang, Z.; Sun, N.; Ju, Y.; Dai, X.; Zhang, Y.; Liu, Y.; et al. miR-762 regulates the proliferation and differentiation of retinal progenitor cells by targeting NPDC1. Cell Cycle 2020, 19, 1754–1767. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Yang, F.; Yang, M.; An, W.; Maguire, E.M.; Chen, Q.; Xiao, R.; Wu, W.; Zhang, L.; Wang, W.; et al. miR-214-3p-Sufu-GLI1 is a novel regulatory axis controlling inflammatory smooth muscle cell differentiation from stem cells and neointimal hyperplasia. Stem Cell Res. Ther. 2020, 11, 465. [Google Scholar] [CrossRef] [PubMed]
- Girard, A.; Sachidanandam, R.; Hannon, G.J.; Carmell, M.A. A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature 2006, 442, 199–202. [Google Scholar] [CrossRef]
- Lee, J.; Kim, M.S.; Park, S.H.; Jang, Y.K. Tousled-like kinase 1 is a negative regulator of core transcription factors in murine embryonic stem cells. Sci. Rep. 2018, 8, 334. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.; Mao, W.; Ren, W.; Xu, F.; Zhu, Q.; Lu, C.; Lin, Z.; Zhang, Z.; Chu, Y.; Liu, R.; et al. Ubiquitin-protein ligase E3C maintains non-small-cell lung cancer stemness by targeting AHNAK-p53 complex. Cancer Lett. 2018, 443, 125–134. [Google Scholar] [CrossRef]
- Penzo, M.; Clima, R.; Trerè, D.; Montanaro, L. Separated siamese twins: Intronic small nucleolar rnas and matched host genes may be altered in conjunction or separately in multiple cancer types. Cells 2020, 9, 387. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| piRNA | Predicted Target | Predicted Target Name | Predicted Pathway |
|---|---|---|---|
| piR-hsa-26594 | GOLGA2P11 | GOLGA2 Pseudogene 11 | Protein serine/threonine kinase activity/Ubiquitin-protein transferase activity/Regulation of chromatin assembly or disassembly |
| piR-hsa-28405 | TP53TG1 | TP53 Target 1 lncRNA | |
| piR-hsa-28402 | LINC00661 | Long Intergenic Non-Protein Coding RNA 661 | |
| piR-hsa-32170 | TLK1 | Serine/Threonine-Protein Kinase Tousled-Like 1 | |
| UBE3C | Ubiquitin Protein Ligase E3C | ||
| piR-hsa-2117 | ARMCX5 | Armadillo Repeat Containing X-Linked 5 | |
| DOK6 | Downstream of Tyrosine Kinase 6 |
| piRNA | Predicted Target | Predicted Target Name | Predicted Pathway |
|---|---|---|---|
| piR-hsa-324 | ZNF318 | Zinc Finger Protein 318 | Regulation of transcription/multicellular organism development/DNA-binding transcription factor activity |
| piR-hsa-23289 | POMGNT2 | Protein O-Linked Mannose N-Acetylglucosaminyltransferase 2 | |
| TJP1 | Tight Junction Protein 1 | ||
| piR-hsa-12442 | FTH1P18 | Ferritin heavy chain like 18 | |
| NBPF4, 6 & 7 | NBPF Member 4, 6 and 7 | ||
| PRRG1 | Proline-Rich and Gla Domain 1 | ||
| piR-hsa-8226 | LOC642852 | Long Intergenic Non-Protein Coding RNA 205 | |
| piR-hsa-6841 | MYH3 & 4 | Myosin Heavy Chain 3 and 4 | |
| piR-hsa-32170 | TLK1 | Serine/Threonine-Protein Kinase Tousled-Like 1 | |
| UBE3C | Ubiquitin Protein Ligase E3C | ||
| piR-hsa-23655 | CHURC1 | Churchill Domain Containing 1 | |
| piR-hsa-28646 | LINC01015 | Long Intergenic Non-Protein Coding RNA 1015 | |
| ZNF668 | Zinc Finger Protein 668 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salloum-Asfar, S.; Abdulla, S.A.; Taha, R.Z.; Thompson, I.R.; Emara, M.M. Combined Noncoding RNA-mRNA Regulomics Signature in Reprogramming and Pluripotency in iPSCs. Cells 2022, 11, 3833. https://doi.org/10.3390/cells11233833
Salloum-Asfar S, Abdulla SA, Taha RZ, Thompson IR, Emara MM. Combined Noncoding RNA-mRNA Regulomics Signature in Reprogramming and Pluripotency in iPSCs. Cells. 2022; 11(23):3833. https://doi.org/10.3390/cells11233833
Chicago/Turabian StyleSalloum-Asfar, Salam, Sara A. Abdulla, Rowaida Z. Taha, I. Richard Thompson, and Mohamed M. Emara. 2022. "Combined Noncoding RNA-mRNA Regulomics Signature in Reprogramming and Pluripotency in iPSCs" Cells 11, no. 23: 3833. https://doi.org/10.3390/cells11233833