

Molecular Cluster Mining of Adrenocortical Carcinoma via Multi-Omics Data Analysis Aids Precise Clinical Therapy
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. Molecular Subtypes were Identified Using Multi-Omics Analysis
2.3. Characteristics of Genetic Variations among Subtypes
2.4. Comparison of Signaling Pathway Activation and Immune Infiltration
2.5. Prediction of Immunotherapy and Chemotherapy Treatment
2.6. Statistical Analysis
3. Results
3.1. Establishment of Molecular Subtypes
3.2. Signaling Pathway Activation in ACC Subtypes
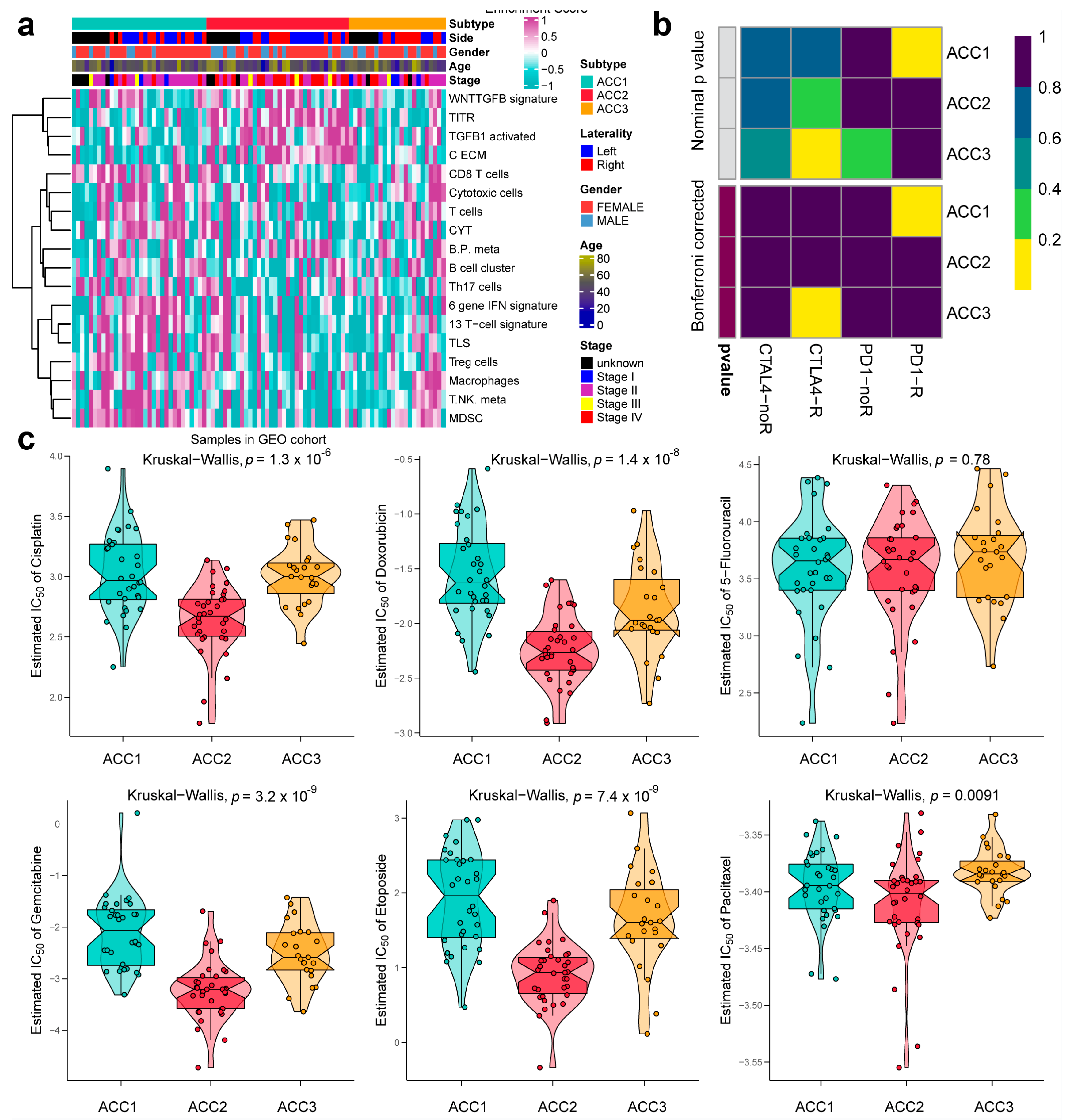
3.3. ACC1 Patients May Benefit More from Anti-PD-1 Therapy, and Chemotherapy Is More Suitable for ACC2 Patients
3.4. Extra Validation for Molecular Subtypes in GEO Cohorts
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lehmann, T.; Wrzesinski, T. The molecular basis of adrenocortical cancer. Cancer Genet. 2012, 205, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Else, T.; Kim, A.C.; Sabolch, A.; Raymond, V.M.; Kandathil, A.; Caoili, E.M.; Jolly, S.; Miller, B.S.; Giordano, T.J.; Hammer, G.D. Adrenocortical carcinoma. Endocr. Rev. 2014, 35, 282–326. [Google Scholar] [CrossRef] [Green Version]
- Michalkiewicz, E.; Sandrini, R.; Figueiredo, B.; Miranda, E.C.; Caran, E.; Oliveira-Filho, A.G.; Marques, R.; Pianovski, M.A.; Lacerda, L.; Cristofani, L.M.; et al. Clinical and outcome characteristics of children with adrenocortical tumors: A report from the International Pediatric Adrenocortical Tumor Registry. J. Clin. Oncol. 2004, 22, 838–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fassnacht, M.; Johanssen, S.; Quinkler, M.; Bucsky, P.; Willenberg, H.S.; Beuschlein, F.; Terzolo, M.; Mueller, H.-H.; Hahner, S.; Allolio, B.; et al. Limited prognostic value of the 2004 international union against cancer staging classification for adrenocortical carcinoma: Proposal for a revised Tnm classification. Cancer 2009, 115, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Fassnacht, M.; Kroiss, M.; Allolio, B. Update in adrenocortical carcinoma. J. Clin. Endocrinol. Metab. 2013, 98, 4551–4564. [Google Scholar] [CrossRef] [PubMed]
- Datta, J.; Roses, R.E. Surgical management of adrenocortical carcinoma: An evidence-based approach. Surg. Oncol. Clin. N. Am. 2016, 25, 153–170. [Google Scholar] [CrossRef] [PubMed]
- Nekic, A.B.; Knezevic, N.; Tomsic, K.Z.; Kraljevic, I.; Balasko, A.; Polovina, T.S.; Solak, M.; Dusek, T.; Kastelan, D.; Croatian ACC Study Group. The effect of surgeon expertise on the outcome of patients with adrenocortical carcinoma. J. Pers. Med. 2022, 12, 100. [Google Scholar] [CrossRef] [PubMed]
- Berruti, A.; Grisanti, S.; Pulzer, A.; Claps, M.; Daffara, F.; Loli, P.; Mannelli, M.; Boscaro, M.; Arvat, E.; Tiberio, G.; et al. Long-term outcomes of adjuvant mitotane therapy in patients with radically resected adrenocortical carcinoma. J. Clin. Endocrinol. Metab. 2017, 102, 1358–1365. [Google Scholar] [CrossRef] [PubMed]
- Jouinot, A.; Armignacco, R.; Assie, G. Genomics of benign adrenocortical tumors. J. Steroid Biochem. Mol. Biol. 2019, 193, 105414. [Google Scholar] [CrossRef]
- Giordano, T.J.; Thomas, D.G.; Kuick, R.; Lizyness, M.; Misek, D.E.; Smith, A.L.; Sanders, D.; Aljundi, R.T.; Gauger, P.G.; Thompson, N.W.; et al. Distinct transcriptional profiles of adrenocortical tumors uncovered by DNA microarray analysis. Am. J. Pathol. 2003, 162, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Qi, Y.; Zhu, Y.; Ning, G.; Huang, Y. Molecular markers and targeted therapies for adrenocortical carcinoma. Clin. Endocrinol. 2014, 80, 159–168. [Google Scholar] [CrossRef]
- Kim, A.C.; Reuter, A.L.; Zubair, M.; Else, T.; Serecky, K.; Bingham, N.C.; Lavery, G.G.; Parker, K.L.; Hammer, G.D. Targeted disruption of beta-catenin in Sf1-expressing cells impairs development and maintenance of the adrenal cortex. Development 2008, 135, 2593–2602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragazzon, B.; Libé, R.; Gaujoux, S.; Assié, G.; Fratticci, A.; Launay, P.; Clauser, E.; Bertagna, X.; Tissier, F.; de Reyniès, A.; et al. Transcriptome analysis reveals that P53 and {Beta}-catenin alterations occur in a group of aggressive adrenocortical cancers. Cancer Res. 2010, 70, 8276–8281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libé, R.; Groussin, L.; Tissier, F.; Elie, C.; René-Corail, F.; Fratticci, A.; Jullian, E.; Beck-Peccoz, P.; Bertagna, X.; Gicquel, C.; et al. Somatic Tp53 mutations are relatively rare among adrenocortical cancers with the frequent 17p13 loss of heterozygosity. Clin. Cancer Res. 2007, 13, 844–850. [Google Scholar] [CrossRef] [Green Version]
- Zheng, S.; Cherniack, A.D.; Dewal, N.; Moffitt, R.A.; Danilova, L.; Murray, B.A.; Lerario, A.M.; Else, T.; Knijnenburg, T.A.; Ciriello, G.; et al. Comprehensive pan-genomic characterization of adrenocortical carcinoma. Cancer Cell 2016, 29, 723–736. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Liu, B.; Wang, W.; Sun, C.; Che, J.; Yuan, X.; Zhai, C. Lung cancer stage prediction using multi-omics data. Comput. Math. Methods Med. 2022, 2022, 2279044. [Google Scholar] [CrossRef]
- Wishart, D. Metabolomics and the multi-omics view of cancer. Metabolites 2022, 12, 154. [Google Scholar] [CrossRef]
- Ding, M.Q.; Chen, L.; Cooper, G.F.; Young, J.D.; Lu, X. Precision oncology beyond targeted therapy: Combining omics data with machine learning matches the majority of cancer cells to effective therapeutics. Mol. Cancer Res. 2018, 16, 269–278. [Google Scholar] [CrossRef] [Green Version]
- Francescatto, M.; Chierici, M.; Rezvan Dezfooli, S.; Zandona, A.; Jurman, G.; Furlanello, C. Multi-omics integration for neuroblastoma clinical endpoint prediction. Biol. Direct. 2018, 13, 5. [Google Scholar] [CrossRef]
- Meng, J.; Guan, Y.; Wang, B.; Chen, L.; Chen, J.; Zhang, M.; Liang, C. Risk subtyping and prognostic assessment of prostate cancer based on consensus genes. Commun. Biol. 2022, 5, 233. [Google Scholar] [CrossRef]
- Demeure, M.J.; Coan, K.E.; Grant, C.S.; Komorowski, R.A.; Stephan, E.; Sinari, S.; Mount, D.; Bussey, K.J. Pttg1 overexpression in adrenocortical cancer is associated with poor survival and represents a potential therapeutic target. Surgery 2013, 154, 1405–1416, discussion 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legendre, C.R.; Demeure, M.J.; Whitsett, T.; Gooden, G.C.; Bussey, K.J.; Jung, S.; Waibhav, T.; Kim, S.; Salhia, B. Pathway implications of aberrant global methylation in adrenocortical cancer. PLoS ONE 2016, 11, e0150629. [Google Scholar] [CrossRef] [PubMed]
- Heaton, J.H.; Wood, M.A.; Kim, A.C.; Lima, L.O.; Barlaskar, F.M.; Almeida, M.Q.; Fragoso, M.C.; Kuick, R.; Lerario, A.M.; Simon, D.P.; et al. Progression to adrenocortical tumorigenesis in mice and humans through insulin-like growth factor 2 and beta-catenin. Am. J. Pathol. 2012, 181, 1017–1033. [Google Scholar] [CrossRef] [PubMed]
- Assié, G.; Letouzé, E.; Fassnacht, M.; Jouinot, A.; Luscap, W.; Barreau, O.; Omeiri, H.; Rodriguez, S.; Perlemoine, K.; René-Corail, F.; et al. Integrated genomic characterization of adrenocortical carcinoma. Nat. Genet. 2014, 46, 607–612. [Google Scholar] [CrossRef]
- Lu, X.; Meng, J.; Zhou, Y.; Jiang, L.; Yan, F. Movics: An R Package for multi-omics integration and visualization in cancer subtyping. Bioinformatics 2020, 36, 5539–5541. [Google Scholar] [CrossRef]
- Chalise, P.; Fridley, B.L. Integrative clustering of multi-level omic data based on non-negative matrix factorization algorithm. PLoS ONE 2017, 12, e0176278. [Google Scholar] [CrossRef] [Green Version]
- Hastie, T.; Tibshirani, R.; Walther, G. Estimating the number of data clusters via the gap statistic. J. R. Stat. Soc. B 2001, 63, 411–423. [Google Scholar]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef] [Green Version]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The molecular signatures database (Msigdb) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Meng, J.; Lu, X.; Zhou, Y.; Zhang, M.; Ge, Q.; Zhou, J.; Hao, Z.; Gao, S.; Yan, F.; Liang, C. Tumor immune microenvironment-based classifications of bladder cancer for enhancing the response rate of immunotherapy. Mol. Ther. Oncolytics 2021, 20, 410–421. [Google Scholar] [CrossRef]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Trevino, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Possemato, R.; Marks, K.M.; Shaul, Y.D.; Pacold, M.E.; Kim, D.; Birsoy, K.; Sethumadhavan, S.; Woo, H.-K.; Jang, H.G.; Jha, A.K.; et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 2011, 476, 346–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.-L.; Roh, W.; Reuben, A.; Cooper, Z.A.; Spencer, C.N.; Prieto, P.A.; Miller, J.P.; Bassett, R.L.; Gopalakrishnan, V.; Wani, K.; et al. Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discov. 2016, 6, 827–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshida, Y.; Brunet, J.P.; Tamayo, P.; Golub, T.R.; Mesirov, J.P. Subclass mapping: Identifying common subtypes in independent disease data sets. PLoS ONE 2007, 2, e1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshida, Y. Nearest template prediction: A single-sample-based flexible class prediction with confidence assessment. PLoS ONE 2010, 5, e15543. [Google Scholar] [CrossRef]
- Schneider, B.; Fukunaga, A.; Murry, D.; Yoder, C.; Fife, K.; Foster, A.; Rosenberg, L.; Kelich, S.; Li, L.; Sweeney, C. A Phase I, pharmacokinetic and pharmacodynamic dose escalation trial of weekly paclitaxel with interferon-alpha2b in patients with solid tumors. Cancer Chemother. Pharmacol. 2007, 59, 261–268. [Google Scholar] [CrossRef]
- Yu, X.; Zhu, D.; Luo, B.; Kou, W.; Cheng, Y.; Zhu, Y. Ifngamma enhances ferroptosis by increasing Jakstat pathway activation to suppress Slca711 expression in adrenocortical carcinoma. Oncol. Rep. 2022, 47, 97. [Google Scholar] [CrossRef]
- Hermsen, I.G.C.; Haak, H.R.; De Krijger, R.R.; Kerkhofs, T.M.A.; Feelders, R.A.; De Herder, W.W.; Wilmink, H.; Smit, J.W.A.; Gelderblom, H.; de Miranda, N.; et al. Mutational analyses of epidermal growth factor receptor and downstream pathways in adrenocortical carcinoma. Eur. J. Endocrinol. 2013, 169, 51–58. [Google Scholar] [CrossRef] [Green Version]
- Tömböl, Z.; Szabó, P.M.; Molnár, V.; Wiener, Z.; Tölgyesi, G.; Horányi, J.; Riesz, P.; Reismann, P.; Patócs, A.; Likó, I.; et al. Integrative molecular bioinformatics study of human adrenocortical tumors: Microrna, tissue-specific target prediction, and pathway analysis. Endocr. Relat. Cancer 2009, 16, 895–906. [Google Scholar] [CrossRef] [Green Version]
- Terzolo, M.; Zaggia, B.; Allasino, B.; De Francia, S. Practical treatment using mitotane for adrenocortical carcinoma. Curr. Opin. Endocrinol. Diabetes Obes. 2014, 21, 159–165. [Google Scholar] [CrossRef]
- Suzuki, S.; Minamidate, T.; Shiga, A.; Ruike, Y.; Ishiwata, K.; Naito, K.; Ishida, A.; Deguchi, H.; Fujimoto, M.; Koide, H.; et al. Steroid metabolites for diagnosing and predicting clinicopathological features in cortisol-producing adrenocortical carcinoma. BMC Endocr. Disord. 2020, 20, 173. [Google Scholar] [CrossRef]
- Liang, F.; Kapoun, A.M.; Lam, A.; Damm, D.L.; Quan, D.; O’Connell, M.; Protter, A.A. B-type natriuretic peptide inhibited angiotensin Ii-stimulated cholesterol biosynthesis, cholesterol transfer, and steroidogenesis in primary human adrenocortical cells. Endocrinology 2007, 148, 3722–3729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavoie, J.M.; Csizmok, V.; Williamson, L.M.; Culibrk, L.; Wang, G.; Marra, M.A.; Laskin, J.; Jones, S.J.M.; Renouf, D.J.; Kollmannsberger, C.K. Whole-genome and transcriptome analysis of advanced adrenocortical cancer highlights multiple alterations affecting epigenome and DNA repair pathways. Cold Spring Harb. Mol. Case Stud. 2022, 8, a006148. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Zhou, Y.; Lu, X.; Bian, Z.; Chen, Y.; Zhou, J.; Zhang, L.; Hao, Z.; Zhang, M.; Liang, C. Immune response drives outcomes in prostate cancer: Implications for immunotherapy. Mol. Oncol. 2021, 15, 1358–1375. [Google Scholar] [CrossRef]
- Zhang, X.; Lan, Y.; Xu, J.; Quan, F.; Zhao, E.; Deng, C.; Luo, T.; Xu, L.; Liao, G.; Yan, M.; et al. Cellmarker: A manually curated resource of cell markers in human and mouse. Nucleic Acids Res. 2019, 47, D721–D728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of Pd-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by Pd-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.T.; Sun, Z.J. Turning cold tumors into hot tumors by improving T-cell infiltration. Theranostics 2021, 11, 5365–5386. [Google Scholar] [CrossRef]
- Fassnacht, M.; Dekkers, O.M.; Else, T.; Baudin, E.; Berruti, A.; de Krijger, R.R.; Haak, H.R.; Mihai, R.; Assie, G.; Terzolo, M. European society of endocrinology clinical practice guidelines on the management of adrenocortical carcinoma in adults, in collaboration with the European Network for the study of adrenal tumors. Eur. J. Endocrinol. 2018, 179, G1–G46. [Google Scholar] [CrossRef] [PubMed]
- Raymond, V.M.; Everett, J.N.; Furtado, L.V.; Gustafson, S.L.; Jungbluth, C.R.; Gruber, S.B.; Hammer, G.D.; Stoffel, E.M.; Greenson, J.K.; Giordano, T.J.; et al. Adrenocortical carcinoma is a lynch syndrome-associated cancer. J. Clin. Oncol. 2013, 31, 3012–3018. [Google Scholar] [CrossRef] [PubMed]
- Wasserman, J.D.; Novokmet, A.; Eichler-Jonsson, C.; Ribeiro, R.C.; Rodriguez-Galindo, C.; Zambetti, G.P.; Malkin, D. Prevalence and functional consequence of Tp53 mutations in pediatric adrenocortical carcinoma: A children’s oncology group study. J. Clin. Oncol. 2015, 33, 602–609. [Google Scholar] [CrossRef] [Green Version]
- Raymond, V.M.; Else, T.; Everett, J.N.; Long, J.M.; Gruber, S.B.; Hammer, G.D. Prevalence of germline Tp53 mutations in a prospective series of unselected patients with adrenocortical carcinoma. J. Clin. Endocrinol. Metab. 2013, 98, E119–E125. [Google Scholar] [CrossRef] [PubMed]
- Ripley, R.T.; Kemp, C.D.; Davis, J.L.; Langan, R.C.; Royal, R.E.; Libutti, S.K.; Steinberg, S.M.; Wood, B.; Kammula, U.S.; Fojo, T.; et al. Liver resection and ablation for metastatic adrenocortical carcinoma. Ann. Surg. Oncol. 2011, 18, 1972–1979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fassnacht, M.; Libe, R.; Kroiss, M.; Allolio, B. Adrenocortical carcinoma: A clinician’s update. Nat. Rev. Endocrinol. 2011, 7, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Stigliano, A.; Chiodini, I.; Giordano, R.; Faggiano, A.; Canu, L.; Della Casa, S.; Loli, P.; Luconi, M.; Mantero, F.; Terzolo, M. Management of adrenocortical carcinoma: A consensus statement of the Italian society of endocrinology (Sie). J. Endocrinol. Investig. 2016, 39, 103–121. [Google Scholar] [CrossRef]
- Geraghty, M.T.; Kearns, W.G.; Pearson, P.L.; Valle, D. Isolation and characterization of an ornithine aminotransferase-related sequence (Oatl3) mapping to 10q26. Genomics 1993, 17, 510–513. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, E.; Olson, S.H.; Ahn, J.; Bundy, B.; Nishikawa, H.; Qian, F.; Jungbluth, A.A.; Frosina, D.; Gnjatic, S.; Ambrosone, C.; et al. Intraepithelial Cd8+ tumor-infiltrating lymphocytes and a high Cd8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 18538–18543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, W.T.; Adams, S.F.; Tahirovic, E.; Hagemann, I.S.; Coukos, G. Prognostic significance of tumor-infiltrating T cells in ovarian cancer: A meta-analysis. Gynecol. Oncol. 2012, 124, 192–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, R.Y.; Francois, A.; McGray, A.R.; Miliotto, A.; Odunsi, K. Compensatory upregulation of Pd-1, Lag-3, and Ctla-4 limits the efficacy of single-agent checkpoint blockade in metastatic ovarian cancer. Oncoimmunology 2017, 6, e1249561. [Google Scholar] [CrossRef] [PubMed]
- Jouinot, A.; Assié, G.; Libe, R.; Fassnacht, M.; Papathomas, T.; Barreau, O.; De La Villeon, B.; Faillot, S.; Hamzaoui, N.; Neou, M.; et al. DNA methylation is an independent prognostic marker of survival in adrenocortical cancer. J. Clin. Endocrinol. Metab. 2017, 102, 923–932. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [Green Version]
- Ho, A.S.; Kannan, K.; Roy, D.M.; Morris, L.G.T.; Ganly, I.; Katabi, N.; Ramaswami, D.; Walsh, L.; Eng, S.; Huse, J.T.; et al. The mutational landscape of adenoid cystic carcinoma. Nat. Genet. 2013, 45, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Özata, D.M.; Caramuta, S.; Velázquez-Fernández, D.; Akçakaya, P.; Xie, H.; Höög, A.; Zedenius, J.; Bäckdahl, M.; Larsson, C.; Lui, W.-O. The role of microrna deregulation in the pathogenesis of adrenocortical carcinoma. Endocr. Relat. Cancer 2011, 18, 643–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juhlin, C.C.; Goh, G.; Healy, J.; Fonseca, A.L.; Scholl, U.I.; Stenman, A.; Kunstman, J.; Brown, T.C.; Overton, J.D.; Mane, S.M.; et al. Whole-exome sequencing characterizes the landscape of somatic mutations and copy number alterations in adrenocortical carcinoma. J. Clin. Endocrinol. Metab. 2015, 100, E493–E502. [Google Scholar] [CrossRef]
- Zhang, C.; Qu, Y.; Xiao, H.; Xiao, W.; Liu, J.; Gao, Y.; Li, M.; Liu, J. Lncrna Snhg3 promotes clear cell renal cell carcinoma proliferation and migration by upregulating Top2a. Exp. Cell Res. 2019, 384, 111595. [Google Scholar] [CrossRef]
- Li, T.; Xing, Y.; Yang, F.; Sun, Y.; Zhang, S.; Wang, Q.; Zhang, W. Lncrna Snhg3 sponges Mir-577 to up-regulate smurf1 expression in prostate cancer. Cancer Med. 2020, 9, 3852–3862. [Google Scholar] [CrossRef] [PubMed]
- Achermann, J.C.; Vilain, E.J. Nr0b1-Related Adrenal Hypoplasia Congenita. In Genereviews®; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Larsen, S.J.; Rottger, R.; Schmidt, H.; Baumbach, J.E. Coli gene regulatory networks are inconsistent with gene expression data. Nucleic Acids Res. 2019, 47, 85–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rana, P.; Thai, P.; Dinh, T.; Ghosh, P. Relevant and non-redundant feature selection for cancer classification and subtype detection. Cancers 2021, 13, 4297. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HR | 95% CI | p Value | |
|---|---|---|---|
| TCGA-ACC Cohort | |||
| Age | 1.013 | (0.986–1.041) | 0.351 |
| Gender, male vs. female | 1.455 | (0.614–3.451) | 0.394 |
| Laterality, right vs. left | 1.533 | (0.664–3.54) | 0.317 |
| Stage | |||
| Stage II vs. stage I | 2.883 | (0.301–27.628) | 0.358 |
| Stage III vs. stage I | 6.28 | (0.641–61.55) | 0.115 |
| Stage IV vs. stage I | 16.164 | (1.49–175.301) | 0.022 |
| Stage unknow vs. stage I | 2.451 | (0.117–51.349) | 0.564 |
| ACC subtype | |||
| ACC2 vs. ACC1 | 45.146 | (7.393–275.694) | 0 |
| ACC3 vs. ACC1 | 4.661 | (0.877–24.779) | 0.071 |
| GEO cohort | |||
| Age | 1.01 | (0.99–1.031) | 0.31 |
| Gender, male vs. female | 1.236 | (0.649–2.354) | 0.518 |
| Laterality | |||
| Right vs. left | 1.15 | (0.54–2.45) | 0.717 |
| Unknow vs. left | 1.2 | (0.51–2.822) | 0.677 |
| Stage | |||
| Stage II vs. stage I | 2.765 | (0.351–21.81) | 0.334 |
| Stage III vs. stage I | 8.223 | (0.909–74.351) | 0.061 |
| Stage IV vs. stage I | 11.723 | (1.504–91.399) | 0.019 |
| Stage unknow vs. stage I | 4.067 | (0.453–36.548) | 0.21 |
| ACC subtype | |||
| ACC2 vs. ACC1 | 4.959 | (2.241–10.97) | 0 |
| ACC3 vs. ACC1 | 2.578 | (1.048–6.341) | 0.039 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guan, Y.; Yue, S.; Chen, Y.; Pan, Y.; An, L.; Du, H.; Liang, C. Molecular Cluster Mining of Adrenocortical Carcinoma via Multi-Omics Data Analysis Aids Precise Clinical Therapy. Cells 2022, 11, 3784. https://doi.org/10.3390/cells11233784
Guan Y, Yue S, Chen Y, Pan Y, An L, Du H, Liang C. Molecular Cluster Mining of Adrenocortical Carcinoma via Multi-Omics Data Analysis Aids Precise Clinical Therapy. Cells. 2022; 11(23):3784. https://doi.org/10.3390/cells11233784
Chicago/Turabian StyleGuan, Yu, Shaoyu Yue, Yiding Chen, Yuetian Pan, Lingxuan An, Hexi Du, and Chaozhao Liang. 2022. "Molecular Cluster Mining of Adrenocortical Carcinoma via Multi-Omics Data Analysis Aids Precise Clinical Therapy" Cells 11, no. 23: 3784. https://doi.org/10.3390/cells11233784