


Terminal Uridylyltransferases TUT4/7 Regulate microRNA and mRNA Homeostasis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. RNA Extraction
2.3. RT-qPCR
2.4. Cell Proliferation Assay
2.5. Cell Adhesion Assay
2.6. Western Blotting
2.7. RNA Sequencing and Data Analysis
3. Results
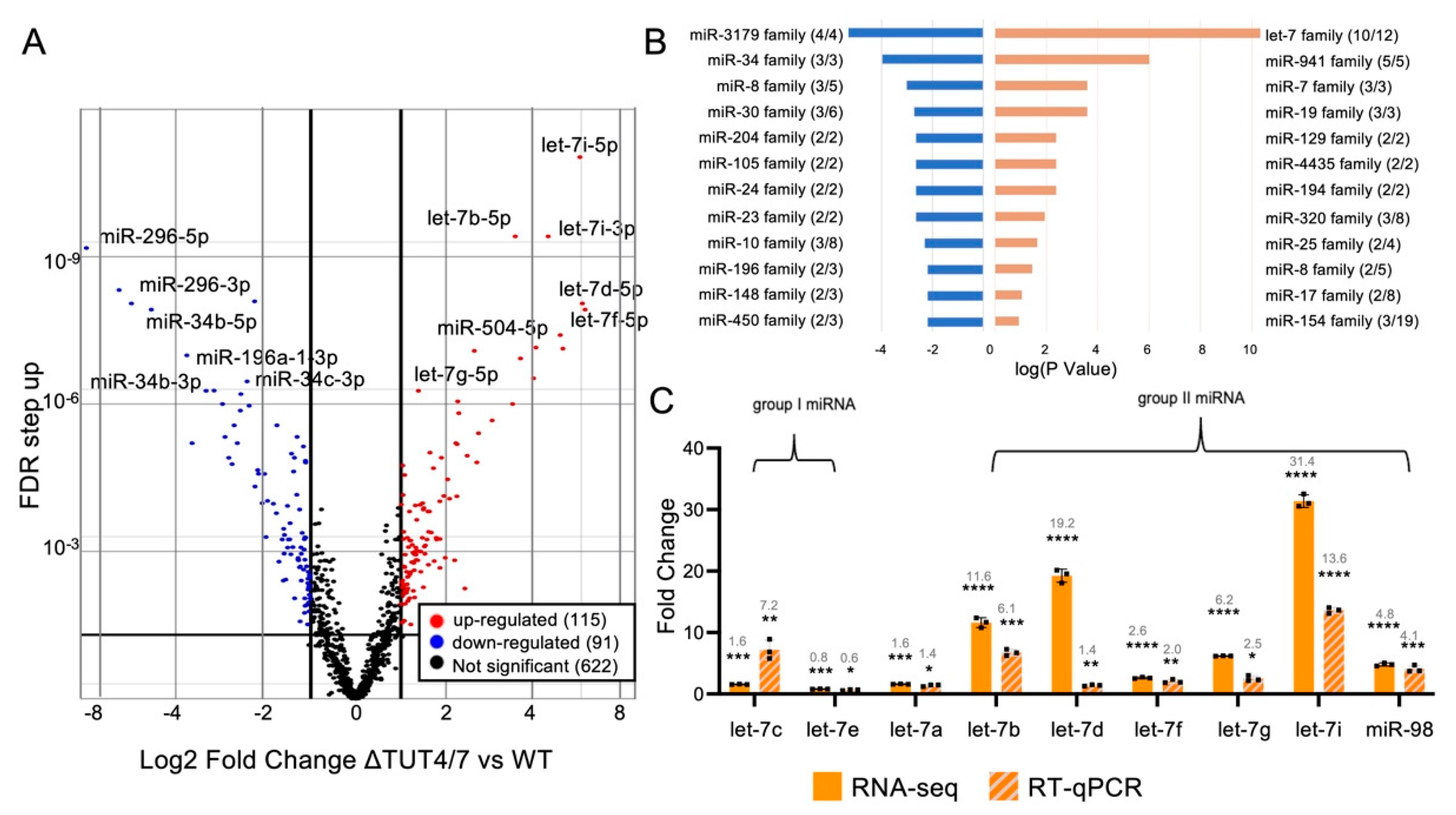
3.1. TUT4/7 Regulate miRNA Abundance
3.1.1. Several miRNA Families Are Increased in Abundance in ΔTUT4/7 Cells
3.1.2. The miRNA Families Decreased in Abundance in ΔTUT4/7 Cells
3.1.3. TUT4/7 Deletion Increases Expression of Group II miRNAs
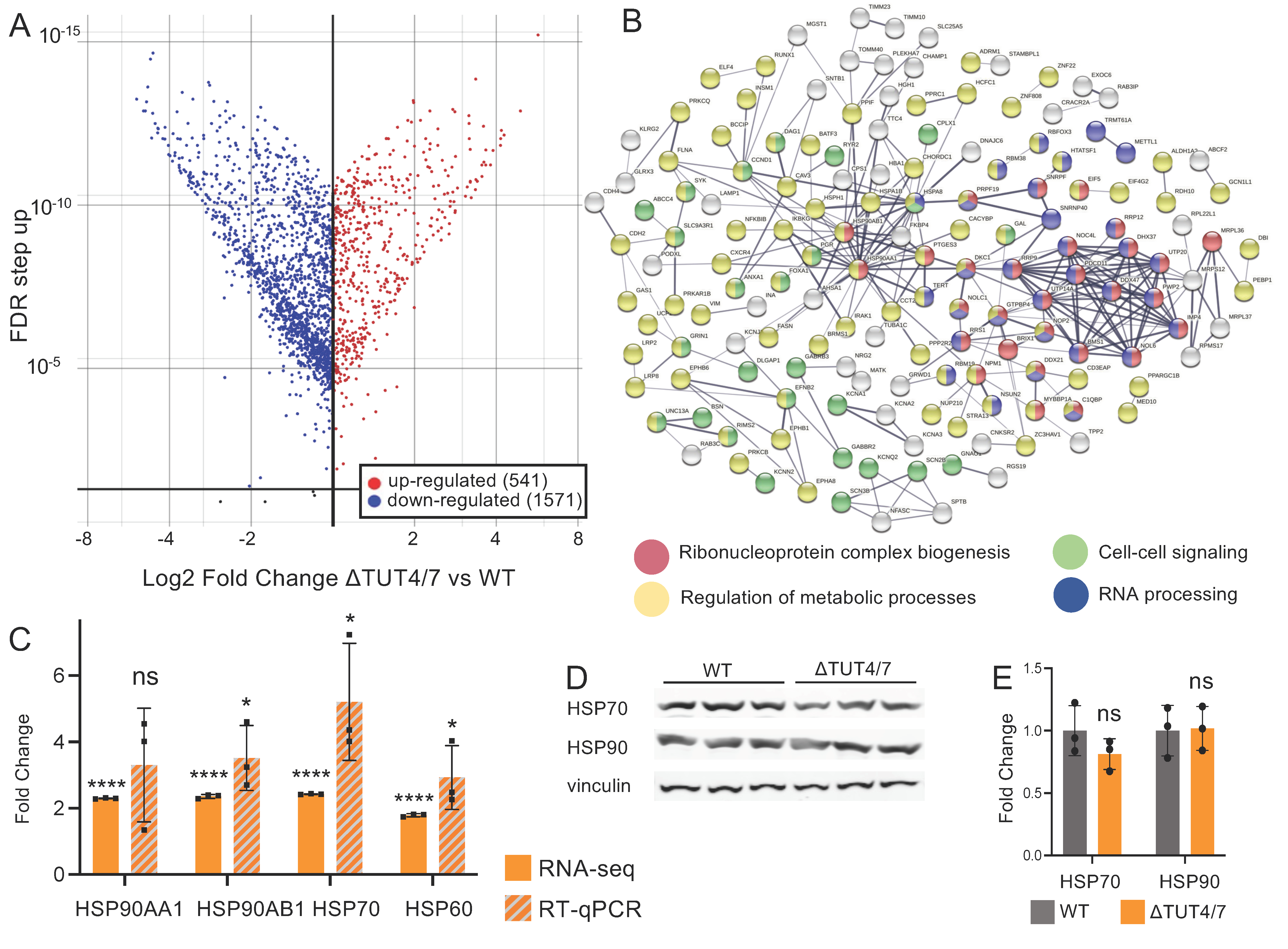
3.2. TUT4/7 Are Global Regulators of Gene Expression Levels
3.2.1. Messenger RNAs Can Be Upregulated in ΔTUT4/7 Cells
3.2.2. Messenger RNAs Can Be Downregulated in ΔTUT4/7 Cells
3.2.3. TUT4/7 Deletion Alters Cellular Phenotypes
3.3. The miRNA–mRNA Network in ΔTUT4/7 Cells
3.4. TUT4/7 Deletion Directly and Indirectly Regulates Protein Phosphorylation and Signaling
3.4.1. TUT4/7 Deletion Reduces AKT1 Levels
3.4.2. Epidermal Growth Factor Stimulation and TUT4/7 Promote AKT Phosphorylation
4. Discussion
4.1. TUT4/7 Regulate Global RNA Turnover
4.2. TUT4/7 Regulate the miRNA–mRNA Network
4.3. TUT4/7 Deletion Disrupts Cell Proliferation and Adhesion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Frederick, M.I.; Heinemann, I.U. Regulation of RNA stability at the 3′ end. Biol Chem. 2020, 402, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Rissland, O.S.; Mikulasova, A.; Norbury, C.J. Efficient RNA Polyuridylation by Noncanonical Poly(A) Polymerases. Mol. Cell. Biol. 2007, 27, 3612–3624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rissland, O.S.; Norbury, C.J. Decapping is preceded by 3′ uridylation in a novel pathway of bulk mRNA turnover. Nat. Struct. Mol. Biol. 2009, 16, 616–623. [Google Scholar] [CrossRef] [Green Version]
- Chung, C.Z.; Jaramillo, E.J.; Ellis, M.J.; Bour, D.Y.N.; Seidl, E.L.; Jo, D.H.S.; Turk, A.M.; Mann, M.R.; Bi, Y.; Haniford, D.B.; et al. RNA surveillance by uridylation-dependent RNA decay in Schizosaccharomyces pombe. Nucleic Acids Res. 2019, 47, 3045–3057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, M.-J.; West, S.; Norbury, C.J. The human cytoplasmic RNA terminal U-transferase ZCCHC11 targets histone mRNAs for degradation. RNA 2010, 17, 39–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ustianenko, D.; Pasulka, J.; Feketova, Z.; Bednarik, L.; Zigackova, D.; Fortova, A.; Zavolan, M.; Vanacova, S. TUT-DIS3L2 is a mammalian surveillance pathway for aberrant structured non-coding RNAs. EMBO J. 2016, 35, 2179–2191. [Google Scholar] [CrossRef]
- Le Pen, J.; Jiang, H.; Di Domenico, T.; Kneuss, E.; Kosałka, J.; Leung, C.; Morgan, M.; Much, C.; Rudolph, K.L.M.; Enright, A.J.; et al. Terminal uridylyltransferases target RNA viruses as part of the innate immune system. Nat. Struct. Mol. Biol. 2018, 25, 778–786. [Google Scholar] [CrossRef]
- Warkocki, Z.; Krawczyk, P.S.; Adamska, D.; Bijata, K.; Garcia-Perez, J.L.; Dziembowski, A. Uridylation by TUT4/7 Restricts Retrotransposition of Human LINE-1s. Cell. 2018, 174, 1537–1548.e29. [Google Scholar] [CrossRef] [Green Version]
- Hagan, J.; Piskounova, E.; Gregory, R.I. Lin28 recruits the TUTase Zcchc11 to inhibit let-7 maturation in mouse embryonic stem cells. Nat. Struct. Mol. Biol. 2009, 16, 1021–1025. [Google Scholar] [CrossRef] [Green Version]
- Heo, I.; Joo, C.; Kim, Y.-K.; Ha, M.; Yoon, M.-J.; Cho, J.; Yeom, K.-H.; Han, J.; Kim, V.N. TUT4 in Concert with Lin28 Suppresses MicroRNA Biogenesis through Pre-MicroRNA Uridylation. Cell 2009, 138, 696–708. [Google Scholar] [CrossRef]
- Piskounova, E.; Polytarchou, C.; Thornton, J.E.; Lapierre, R.J.; Pothoulakis, C.; Hagan, J.P.; Iliopoulos, D.; Gregory, R.I. Lin28A and lin28B Inhibit let-7 microRNA biogenesis by distinct mechanisms. Cell 2011, 147, 1066–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, M.P.; Liu, X.; Whangbo, J.; McCrossan, G.; Sanborn, K.B.; Basar, E.; Walch, M.; Lieberman, J. Apoptosis Triggers Specific, Rapid, and Global mRNA Decay with 3′ Uridylated Intermediates Degraded by DIS3L2. Cell Rep. 2015, 11, 1079–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.; Ha, M.; Chang, H.; Kwon, S.C.; Simanshu, D.K.; Patel, D.J.; Kim, V.N. Uridylation by TUT4 and TUT7 Marks mRNA for Degradation. Cell 2014, 159, 1365–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.; Yeo, J.; Kim, J.-G.; Kim, H.; Lim, J.; Lee, M.; Kim, H.H.; Ohk, J.; Jeon, H.-Y.; Lee, H.; et al. Terminal Uridylyltransferases Execute Programmed Clearance of Maternal Transcriptome in Vertebrate Embryos. Mol. Cell 2018, 70, 72–82.e7. [Google Scholar] [CrossRef] [Green Version]
- Morgan, M.; Much, C.; DiGiacomo, M.; Azzi, C.; Ivanova, I.; Vitsios, D.M.; Pistolic, J.; Collier, P.; Moreira, P.N.; Benes, V.; et al. mRNA 3′ uridylation and poly(A) tail length sculpt the mammalian maternal transcriptome. Nature 2017, 548, 347–351. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.; Ha, M.; Loeff, L.; Chang, H.; Simanshu, D.K.; Li, S.; Fareh, M.; Patel, D.J.; Joo, C.; Kim, V.N. Tut7 Controls the Fate of Precursor MicroRNAs by Using Three Different Uridylation Mechanisms. EMBO J. 2015, 34, 1801–1815. [Google Scholar] [CrossRef]
- Medhi, R.; Price, J.; Furlan, G.; Gorges, B.; Sapetschnig, A.; Miska, E.A. RNA Uridyl Transferases Tut4/7 Differentially Regulate MiRNA Variants Depending on the Cancer Cell Type. RNA 2022, 28, 353–370. [Google Scholar] [CrossRef]
- Chang, H.-M.; Triboulet, R.; Thornton, J.E.; Gregory, R.I. A role for the Perlman syndrome exonuclease Dis3l2 in the Lin28–let-7 pathway. Nature 2013, 497, 244–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ustianenko, D.; Hrossova, D.; Potesil, D.; Chalupnikova, K.; Hrazdilova, K.; Pachernik, J.; Cetkovska, K.; Uldrijan, S.; Zdrahal, Z.; Vanacova, S. Mammalian DIS3L2 exoribonuclease targets the uridylated precursors of let-7 miRNAs. RNA 2013, 19, 1632–1638. [Google Scholar] [CrossRef] [Green Version]
- Yang, A.; Shao, T.-J.; Ros, X.B.-D.; Lian, C.; Villanueva, P.; Dai, L.; Gu, S. AGO-bound mature miRNAs are oligouridylated by TUTs and subsequently degraded by DIS3L2. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Heo, I.; Ha, M.; Lim, J.; Yoon, M.-J.; Park, J.-E.; Kwon, S.C.; Chang, H.; Kim, V.N. Mono-Uridylation of Pre-MicroRNA as a Key Step in the Biogenesis of Group II let-7 MicroRNAs. Cell 2012, 151, 521–532. [Google Scholar] [CrossRef] [Green Version]
- Thornton, J.E.; Chang, H.-M.; Piskounova, E.; Gregory, R.I. Lin28-mediated control of let-7 microRNA expression by alternative TUTases Zcchc11 (TUT4) and Zcchc6 (TUT7). RNA 2012, 18, 1875–1885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Zheng, Q.; Vrettos, N.; Maragkakis, M.; Alexiou, P.; Gregory, B.D.; Mourelatos, Z. A MicroRNA Precursor Surveillance System in Quality Control of MicroRNA Synthesis. Mol. Cell 2014, 55, 868–879. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, S.; Nagaike, T.; Tomita, K. Crystal structure of the Lin28-interacting module of human terminal uridylyltransferase that regulates let-7 expression. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Chung, C.Z.; Balasuriya, N.; Siddika, T.; Frederick, M.I.; Heinemann, I.U. Gld2 activity and RNA specificity is dynamically regulated by phosphorylation and interaction with QKI-7. RNA Biol. 2021, 18, 397–408. [Google Scholar] [CrossRef]
- Yang, A.; Bofill-De Ros, X.; Shao, T.-J.; Jiang, M.; Li, K.; Villanueva, P.; Dai, L.; Gu, S. 3′ Uridylation Confers miRNAs with Non-canonical Target Repertoires. Mol. Cell 2019, 75, 511–522.e4. [Google Scholar] [CrossRef] [PubMed]
- Frederick, M.I.; Siddika, T.; Zhang, P.; Balasuriya, N.; Turk, M.A.; O’Donoghue, P.; Heinemann, I.U. MiRNA-Dependent Regulation of Akt1 Phosphorylation. Cells 2022, 11, 821. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.J.; Jung, G.S.; Jeon, W.B.; Lee, K.M. Arg-Gly-Asp-Modified Elastin-Like Polypeptide Regulates Cell Proliferation and Cell Cycle Proteins Via the Phosphorylation of Erk and Akt in Pancreatic Beta-Cell. Heliyon 2020, 6, e04918. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The String Database in 2017: Quality-Controlled Protein-Protein Association Networks, Made Broadly Accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef]
- Chang, L.; Zhou, G.; Soufan, O.; Xia, J. miRNet 2.0: Network-based visual analytics for miRNA functional analysis and systems biology. Nucleic Acids Res. 2020, 48, W244–W251. [Google Scholar] [CrossRef]
- Huang, H.Y.; Lin, Y.C.; Li, J.; Huang, K.Y.; Shrestha, S.; Hong, H.C.; Tang, Y.; Chen, Y.G.; Jin, C.N.; Yu, Y.; et al. Mirtarbase 2020: Updates to the Experimentally Validated MicroRNA-Target Interaction Database. Nucleic Acids Res. 2020, 48, D148–D154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fromm, B.; Domanska, D.; Høye, E.; Ovchinnikov, V.; Kang, W.; Aparicio-Puerta, E.; Johansen, M.; Flatmark, K.; Mathelier, A.; Hovig, E.; et al. Mirgenedb 2.0: The Metazoan MicroRNA Complement. Nucleic Acids Res. 2020, 48, D1172. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Han, X.; Wan, Y.; Zhang, S.; Zhao, Y.; Fan, R.; Cui, Q.; Zhou, Y. TAM 2.0: Tool for MicroRNA set analysis. Nucleic Acids Res. 2018, 46, W180–W185. [Google Scholar] [CrossRef] [Green Version]
- Letafati, A.; Najafi, S.; Mottahedi, M.; Karimzadeh, M.; Shahini, A.; Garousi, S.; Abbasi-Kolli, M.; Nahand, J.S.; Zadeh, S.S.T.; Hamblin, M.R.; et al. MicroRNA let-7 and viral infections: Focus on mechanisms of action. Cell. Mol. Biol. Lett. 2022, 27, 1–47. [Google Scholar] [CrossRef]
- Yang, A.; Ros, X.B.-D.; Stanton, R.; Shao, T.-J.; Villanueva, P.; Gu, S. TENT2, TUT4, and TUT7 selectively regulate miRNA sequence and abundance. Nat. Commun. 2022, 13, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; He, L.; Fominykh, K.; Yan, Z.; Guo, S.; Zhang, X.; Taylor, M.; Tang, L.; Li, J.; Liu, J.; et al. Evolution of the human-specific microRNA miR-941. Nat. Commun. 2012, 3, 1145. [Google Scholar] [CrossRef] [Green Version]
- Miao, J.; Wang, W.; Wu, S.; Zang, X.; Li, Y.; Wang, J.; Zhan, R.; Gao, M.; Hu, M.; Li, J.; et al. miR-194 Suppresses Proliferation and Migration and Promotes Apoptosis of Osteosarcoma Cells by Targeting CDH2. Cell. Physiol. Biochem. 2018, 45, 1966–1974. [Google Scholar] [CrossRef]
- Zhao, J.; Tao, Y.; Zhou, Y.; Qin, N.; Chen, C.; Tian, D.; Xu, L. MicroRNA-7: A promising new target in cancer therapy. Cancer Cell Int. 2015, 15, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Mazzelli, M.; Maj, C.; Mariani, N.; Mora, C.; Begni, V.; Pariante, C.M.; Riva, M.A.; Cattaneo, A.; Cattane, N. The Long-Term Effects of Early Life Stress on the Modulation of Mir-19 Levels. Front Psychiatry 2020, 11, 389. [Google Scholar] [CrossRef]
- Gao, Y.; Feng, B.; Han, S.; Lu, L.; Chen, Y.; Chu, X.; Wang, R.; Chen, L. MicroRNA-129 in Human Cancers: From Tumorigenesis to Clinical Treatment. Cell. Physiol. Biochem. 2016, 39, 2186–2202. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.W.; Kim, J.M.; Kim, J.E.; Cho, H.; Kim, D.; Kim, W.; Oh, J.-W.; Kwon, H.J. MiR-4435 is an UQCRB-related circulating miRNA in human colorectal cancer. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Liao, Y.; Tang, L. MicroRNA-34 family: A potential tumor suppressor and therapeutic candidate in cancer. J. Exp. Clin. Cancer Res. 2019, 38, 53. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Shi, Y.; Liu, Z.; Li, Z.; Xu, W. The emerging role of miR-10 family in gastric cancer. Cell Cycle 2021, 20, 1468–1476. [Google Scholar] [CrossRef]
- Yang, S.-J.; Wang, D.-D.; Chen, X.; Shen, H.-Y.; Zhang, X.-H.; Zhong, S.-L.; Tang, J.-H.; Zhao, J.-H. The miR-30 family: Versatile players in breast cancer. Tumor Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, C.Z.; Jo, D.H.S.; Heinemann, I.U. Nucleotide specificity of the human terminal nucleotidyltransferase Gld2 (TUT2). RNA 2016, 22, 1239–1249. [Google Scholar] [CrossRef] [Green Version]
- Chung, C.Z.; Seidl, L.E.; Mann, M.R.; Heinemann, I.U. Tipping the balance of RNA stability by 3′ editing of the transcriptome. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 2971–2979. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gene Ontology Consortium. The Gene Ontology Resource: Enriching a Gold Mine. Nucleic Acids Res. 2021, 49, D325–D334. [Google Scholar] [CrossRef]
- Chen, B.; Piel, W.H.; Gui, L.; Bruford, E.; Monteiro, A. The HSP90 family of genes in the human genome: Insights into their divergence and evolution. Genomics 2005, 86, 627–637. [Google Scholar] [CrossRef]
- Tsai, A.-C.; Liu, Y.; Yuan, X.; Ma, T. Compaction, Fusion, and Functional Activation of Three-Dimensional Human Mesenchymal Stem Cell Aggregate. Tissue Eng. Part A 2015, 21, 1705–1719. [Google Scholar] [CrossRef]
- Lin, R.Z.; Chang, H.Y. Recent Advances in Three-Dimensional Multicellular Spheroid Culture for Biomedical Research. Biotechnol. J. 2008, 3, 1172–1184. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, T.; Tomari, Y. Making Risc. Trends Biochem. Sci. 2010, 35, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Tooley, A.S.; Kazyken, D.; Bodur, C.; Gonzalez, I.E.; Fingar, D.C. The innate immune kinase TBK1 directly increases mTORC2 activity and downstream signaling to Akt. J. Biol. Chem. 2021, 297, 100942. [Google Scholar] [CrossRef]
- Pirkmajer, S.; Chibalin, A.V. Serum Starvation: Caveat Emptor. Am. J. Physiol. Cell Physiol. 2011, 301, C272–C279. [Google Scholar] [CrossRef] [Green Version]
- Hoefig, K.P.; Heissmeyer, V. Degradation of oligouridylated histone mRNAs: See UUUUU and goodbye. Wiley Interdiscip. Rev. RNA 2014, 5, 577–589. [Google Scholar] [CrossRef]
- Kim, Y.K.; Kim, B.; Kim, V.N. Re-Evaluation of the Roles of Drosha, Export in 5, and Dicer in MicroRNA Biogenesis. Proc. Natl. Acad. Sci. USA 2016, 113, E1881–E1889. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; An, D.; Liu, X.; Wang, X.; Li, B. MicroRNA-27a downregulates the expression of Hsp90 and enhances the radiosensitivity in esophageal squamous cell carcinoma. OncoTargets Ther. 2019, 12, 5967–5977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kariya, A.; Furusawa, Y.; Yunoki, T.; Kondo, T.; Tabuchi, Y. A microRNA-27a mimic sensitizes human oral squamous cell carcinoma HSC-4 cells to hyperthermia through downregulation of Hsp110 and Hsp90. Int. J. Mol. Med. 2014, 34, 334–340. [Google Scholar] [CrossRef] [Green Version]
- Towler, B.P.; Pashler, A.L.; Haime, H.J.; Przybyl, K.M.; Viegas, S.C.; Matos, R.G.; Morley, S.J.; Arraiano, C.M.; Newbury, S.F. Dis3L2 regulates cell proliferation and tissue growth through a conserved mechanism. PLoS Genet. 2020, 16, e1009297. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, P.; Frederick, M.I.; Heinemann, I.U. Terminal Uridylyltransferases TUT4/7 Regulate microRNA and mRNA Homeostasis. Cells 2022, 11, 3742. https://doi.org/10.3390/cells11233742
Zhang P, Frederick MI, Heinemann IU. Terminal Uridylyltransferases TUT4/7 Regulate microRNA and mRNA Homeostasis. Cells. 2022; 11(23):3742. https://doi.org/10.3390/cells11233742
Chicago/Turabian StyleZhang, Pengcheng, Mallory I. Frederick, and Ilka U. Heinemann. 2022. "Terminal Uridylyltransferases TUT4/7 Regulate microRNA and mRNA Homeostasis" Cells 11, no. 23: 3742. https://doi.org/10.3390/cells11233742