
Ferroptosis—A New Dawn in the Treatment of Organ Ischemia–Reperfusion Injury
 ,
,
Abstract
:1. Introduction
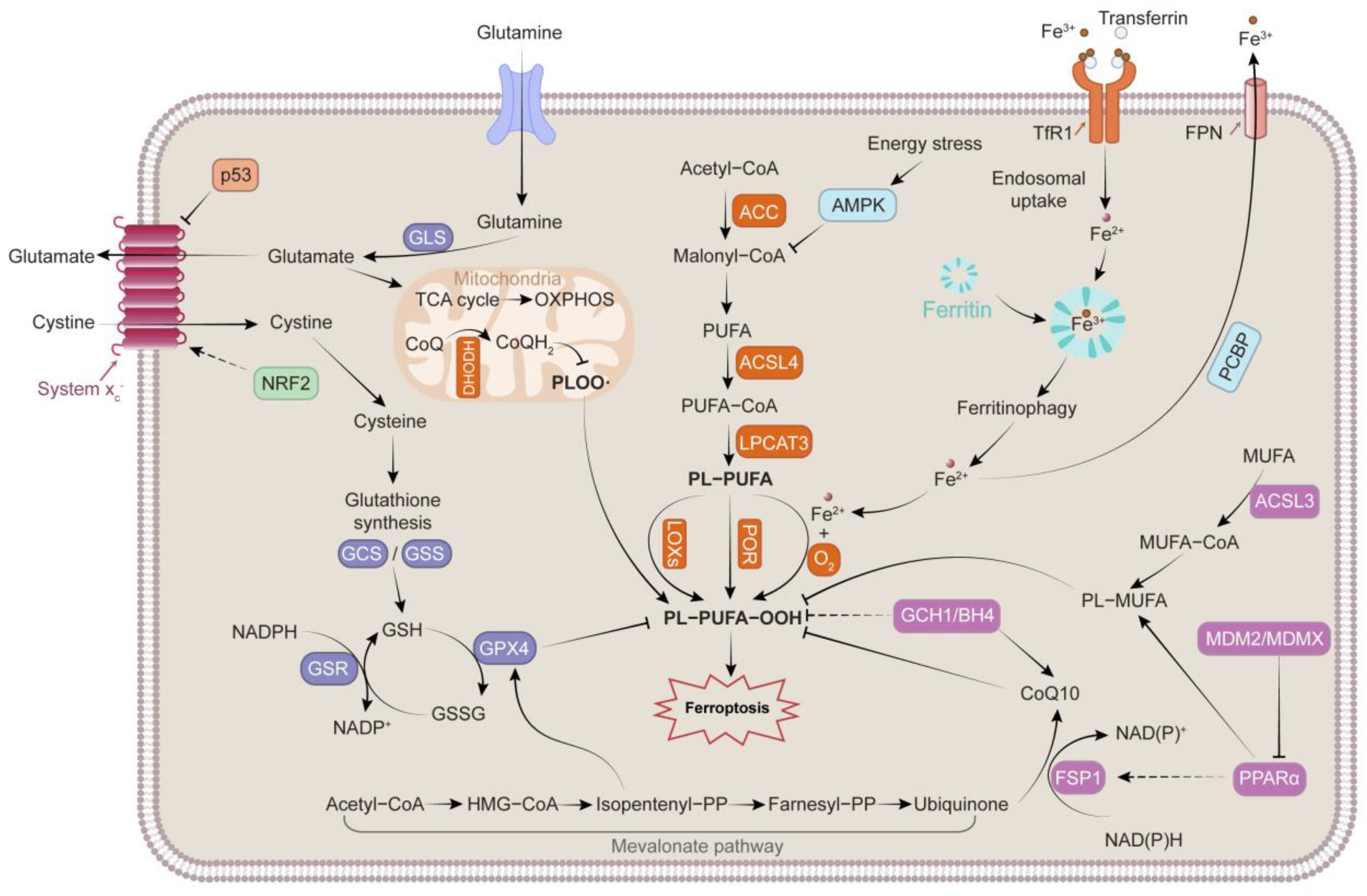
2. Mechanisms Governing Ferroptosis
2.1. Iron Metabolism
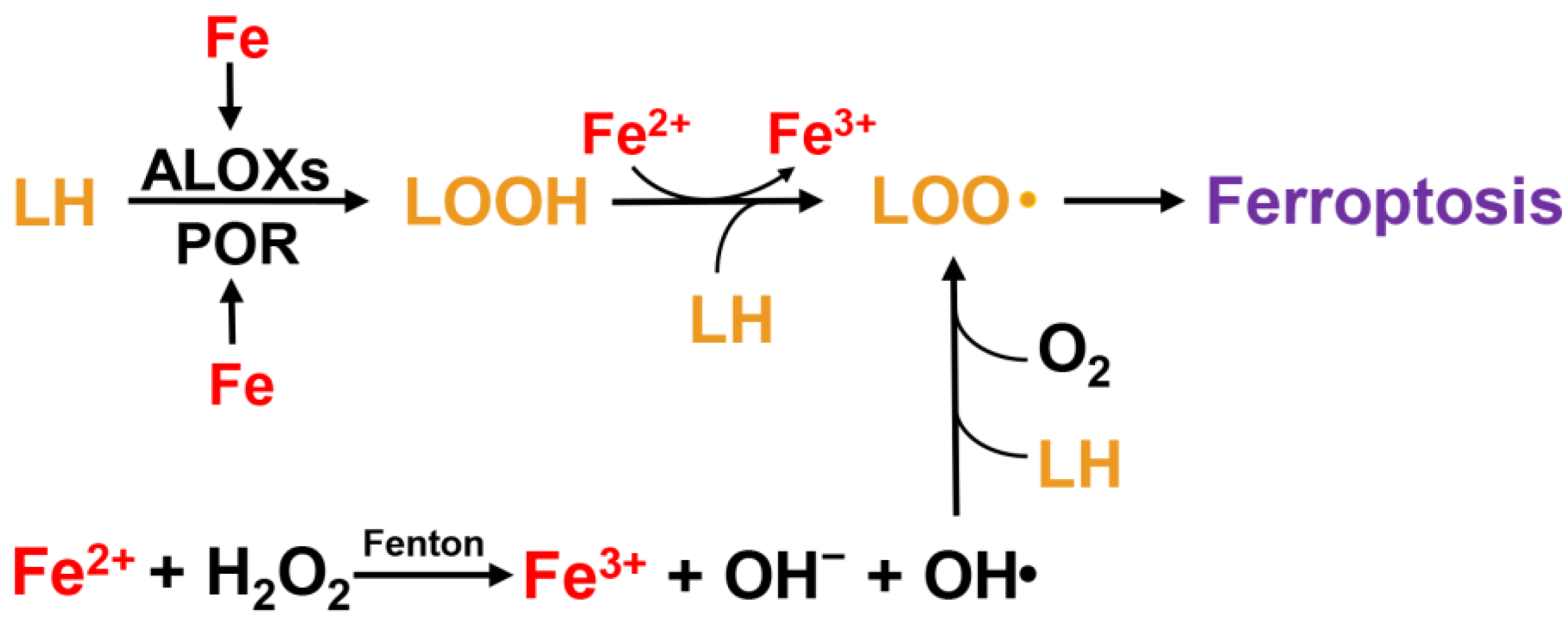
2.2. Lipid Peroxidation
2.3. Antioxidant System
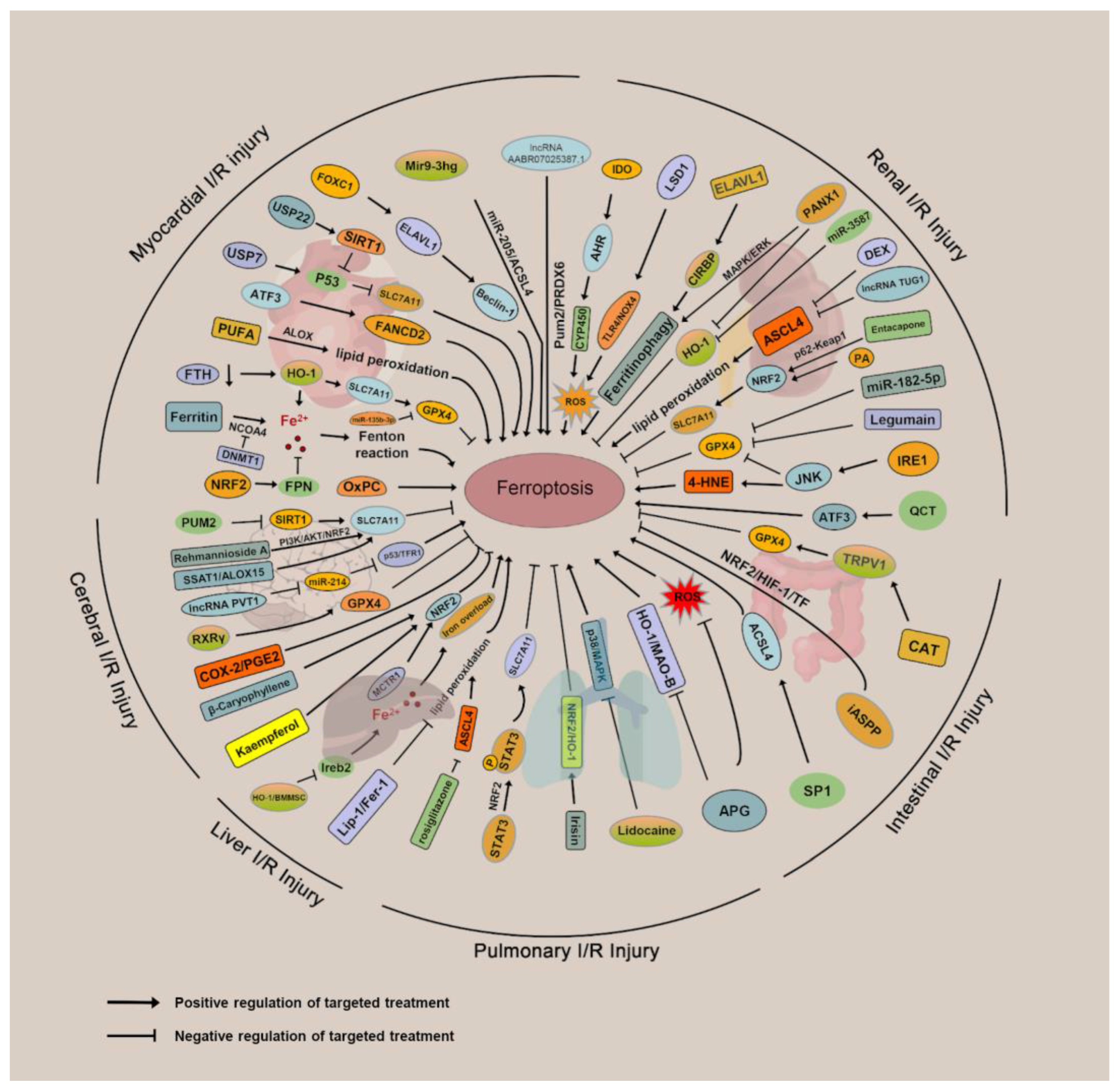
3. Mechanisms and Targeted Therapies for Ferroptosis in IRI
3.1. Ferroptosis and Myocardial IRI
3.2. Ferroptosis and Renal IRI
3.3. Ferroptosis and Cerebral IRI
3.4. Ferroptosis and Intestinal IRI
3.5. Ferroptosis and Hepatic IRI
3.6. Ferroptosis and Pulmonary IRI
4. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 4-HNE | 4-hydroxynonenal |
| AA | arachidonic acid |
| ACSL4 | acyl-CoA synthetase long-chain family member 4 |
| AdA | adrenic acid |
| AKI | acute kidney injury |
| ALI | acute lung injury |
| ALOX | arachidonate lipoxygenases |
| AMPK | AMP-activated protein kinase |
| APG | apigenin-7-O-β-D-(-6′′-p-coumaroyl)-glucopyranoside |
| ATF3 | activating transcription factor 3 |
| BH4/BH2 | tetrahydrobiopterin/dihydrobiopterin |
| BMSC | bone marrow mesenchymal stem cells |
| C3G | cyanidin-3-glucoside |
| CAT | capsiate |
| CIRBP | cold inducible RNA binding protein |
| CISD1 | CDGSH iron sulfur domain 1 |
| CISD2 | CDGSH iron sulfur domain 1 |
| CoQ | coenzyme Q |
| CoQ10 | coenzyme Q10 |
| Dex | dexmedetomidine |
| DHODH | dihydroorotate dehydrogenase |
| DMT1 | divalent metal transporter 1 |
| ELAVL1 | ELAV like RNA binding protein 1 |
| ER | endoplasmic reticulum |
| Fer-1 | ferrostatin-1 |
| FSP1 | ferroptosis suppressor protein 1 |
| FTH | ferritin heavy chain |
| GAA | gossypol acetic acid |
| GCH1 | GTP cyclohydrolase 1 |
| GLS | glutaminases |
| GPX4 | glutathione peroxidase 4 |
| GSK3β | glycogen synthase kinase 3 beta |
| HC | histochrome |
| HO-1 | heme oxygenase 1 |
| I/R | ischemia-reperfusion |
| IDO | indoleamine 2,3-dioxygenase 1 |
| IRE1 | inositol requiring enzyme 1 |
| IRI | ischemia-reperfusion injury |
| JNK | Jun N-terminal kinase |
| Lgmn | legumain |
| LH | polyunsaturated lipid |
| LIP | labile iron pool |
| LOO· | lipid peroxyl radical |
| LOOH | hydroperoxide |
| LPCAT3 | lysophosphatidylcholine acyltransferase 3 |
| LSD1 | lysine-specific demethylase 1 |
| MAPK | mitogen-activated protein kinase |
| MCAO | middle cerebral artery occlusion |
| MCTR1 | maresin conjugate in tissue regeneration 1 |
| MDA | malondialdehyde |
| MDM2 | murine double minute 2 |
| METs | macrophage extracellular traps |
| MUFA | monounsaturated fatty acid |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NAR | naringenin |
| NCOA4 | nuclear receptor coactivator 4 |
| NOX4 | NADPH oxidase 4 |
| NRF2 | nuclear factor erythroid 2-related factor 2 |
| NTBI | non-TF-bound iron |
| OGD/R | oxygen-glucose deprivation/reoxygenation |
| OH· | hydroxyl radical |
| OxPCs | oxidized phosphatidylcholines |
| OXPHOS | oxidative phosphorylation |
| Panx1 | pannexin 1 |
| PCBP | poly(rC)-binding protein |
| PGE2 | prostaglandin E2 |
| PLOO· | phospholipid hydroperoxyl radical |
| POR | cytochrome P450 oxidoreductase |
| PTGS2 | prostaglandin-endoperoxide synthase 2 |
| PUFAs | polyunsaturated fatty acids |
| PUM2 | pumilio 2 |
| QCT | quercetin |
| Res | resveratrol |
| ROS | reactive oxygen species |
| SIRT1 | sirtuin 1 |
| SLC7A11 | solute carrier family 7 member 11 |
| SLC25A28 | solute carrier family 25 member 28 |
| SLC25A37 | solute carrier family 25 member 37 |
| SLC40A1/FPN | solute carrier family 40 member 1 |
| Sp1 | special protein 1 |
| TF | transferrin |
| TFR1 | transferrin receptor 1 |
| TLR4 | toll like receptor 4 |
| USC | urine-derived stem cells |
| VDAC1 | voltage dependent anion channel 1 |
References
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion—From mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jassem, W.; Heaton, N.D. The role of mitochondria in ischemia/reperfusion injury in organ transplantation. Kidney Int. 2004, 66, 514–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Ma, N.; Xu, J.; Zhang, Y.; Yang, P.; Su, X.; Xing, Y.; An, N.; Yang, F.; Zhang, G.; et al. Targeting Ferroptosis: Pathological Mechanism and Treatment of Ischemia-Reperfusion Injury. Oxid. Med. Cell Longev. 2021, 2021, 1587922. [Google Scholar] [CrossRef]
- Chen, Y.; Fan, H.; Wang, S.; Tang, G.; Zhai, C.; Shen, L. Ferroptosis: A Novel Therapeutic Target for Ischemia-Reperfusion Injury. Front. Cell Dev. Biol. 2021, 9, 688605. [Google Scholar] [CrossRef]
- Chen, X.; Li, J.; Kang, R.; Klionsky, D.J.; Tang, D. Ferroptosis: Machinery and regulation. Autophagy 2021, 17, 2054–2081. [Google Scholar] [CrossRef]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef]
- Belcher, J.D.; Beckman, J.D.; Balla, G.; Balla, J.; Vercellotti, G. Heme degradation and vascular injury. Antioxid. Redox Signal. 2010, 12, 233–248. [Google Scholar] [CrossRef] [Green Version]
- Higashimoto, Y.; Sato, H.; Sakamoto, H.; Takahashi, K.; Palmer, G.; Noguchi, M. The reactions of heme- and verdoheme-heme oxygenase-1 complexes with FMN-depleted NADPH-cytochrome P450 reductase. Electrons required for verdoheme oxidation can be transferred through a pathway not involving FMN. J. Biol. Chem. 2006, 281, 31659–31667. [Google Scholar] [CrossRef]
- Atamna, H. Heme, iron, and the mitochondrial decay of ageing. Ageing Res. Rev. 2004, 3, 303–318. [Google Scholar] [CrossRef] [PubMed]
- Shaw, G.C.; Cope, J.J.; Li, L.; Corson, K.; Hersey, C.; Ackermann, G.E.; Gwynn, B.; Lambert, A.J.; Wingert, R.A.; Traver, D.; et al. Mitoferrin is essential for erythroid iron assimilation. Nature 2006, 440, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Paradkar, P.N.; Zumbrennen, K.B.; Paw, B.H.; Ward, D.M.; Kaplan, J. Regulation of mitochondrial iron import through differential turnover of mitoferrin 1 and mitoferrin 2. Mol. Cell Biol. 2009, 29, 1007–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation. Biochem. Biophys. Res. Commun. 2016, 478, 838–844. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Lin, F.; Qi, Y.; Liu, C.; Zhou, L.; Xia, Y.; Chen, K.; Xing, J.; Liu, Z.; Yu, W.; et al. HO-1 Contributes to Luteolin-Triggered Ferroptosis in Clear Cell Renal Cell Carcinoma via Increasing the Labile Iron Pool and Promoting Lipid Peroxidation. Oxid. Med. Cell Longev. 2022, 2022, 3846217. [Google Scholar] [CrossRef]
- Shintoku, R.; Takigawa, Y.; Yamada, K.; Kubota, C.; Yoshimoto, Y.; Takeuchi, T.; Koshiishi, I.; Torii, S. Lipoxygenase-mediated generation of lipid peroxides enhances ferroptosis induced by erastin and RSL3. Cancer Sci. 2017, 108, 2187–2194. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef] [Green Version]
- De Domenico, I.; Vaughn, M.B.; Li, L.; Bagley, D.; Musci, G.; Ward, D.M.; Kaplan, J. Ferroportin-mediated mobilization of ferritin iron precedes ferritin degradation by the proteasome. EMBO J. 2006, 25, 5396–5404. [Google Scholar] [CrossRef] [Green Version]
- Ganz, T. Cellular iron: Ferroportin is the only way out. Cell Metab. 2005, 1, 155–157. [Google Scholar] [CrossRef]
- Ma, S.; Henson, E.S.; Chen, Y.; Gibson, S.B. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. 2016, 7, e2307. [Google Scholar] [CrossRef] [Green Version]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, N.A.; Wolf, R.A.; Yarbro, E.M.; Weenen, H. The autoxidation of arachidonic acid: Formation of the proposed SRS-A intermediate. Biochem. Biophys. Res. Commun. 1979, 89, 1058–1064. [Google Scholar] [CrossRef]
- Yin, H.; Xu, L.; Porter, N.A. Free radical lipid peroxidation: Mechanisms and analysis. Chem. Rev. 2011, 111, 5944–5972. [Google Scholar] [CrossRef] [PubMed]
- Frank, C.E. Hydrocarbon autoxidation. Chem. Rev. 1950, 46, 155–169. [Google Scholar] [CrossRef]
- Yan, H.-F.; Zou, T.; Tuo, Q.-Z.; Xu, S.; Li, H.; Belaidi, A.A.; Lei, P. Ferroptosis: Mechanisms and links with diseases. Signal Transduct. Target Ther. 2021, 6, 49. [Google Scholar] [CrossRef]
- Kuhn, H.; Banthiya, S.; van Leyen, K. Mammalian lipoxygenases and their biological relevance. Biochim. Biophys. Acta 2015, 1851, 308–330. [Google Scholar] [CrossRef] [Green Version]
- Wenzel, S.E.; Tyurina, Y.Y.; Zhao, J.; St Croix, C.M.; Dar, H.H.; Mao, G.; Tyurin, V.A.; Anthonymuthu, T.S.; Kapralov, A.A.; Amoscato, A.A.; et al. PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals. Cell 2017, 171, 628–641.E26. [Google Scholar] [CrossRef] [Green Version]
- Seiler, A.; Schneider, M.; Förster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Rådmark, O.; Wurst, W.; et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008, 8, 237–248. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Maher, P.; Schubert, D. A role for 12-lipoxygenase in nerve cell death caused by glutathione depletion. Neuron 1997, 19, 453–463. [Google Scholar] [CrossRef]
- Jin, G.; Arai, K.; Murata, Y.; Wang, S.; Stins, M.F.; Lo, E.H.; van Leyen, K. Protecting against cerebrovascular injury: Contributions of 12/15-lipoxygenase to edema formation after transient focal ischemia. Stroke 2008, 39, 2538–2543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, R.; Shchepinov, M.S.; Pratt, D.A. Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS Cent. Sci. 2018, 4, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Brütsch, S.H.; Wang, C.C.; Li, L.; Stender, H.; Neziroglu, N.; Richter, C.; Kuhn, H.; Borchert, A. Expression of inactive glutathione peroxidase 4 leads to embryonic lethality, and inactivation of the Alox15 gene does not rescue such knock-in mice. Antioxid. Redox Signal. 2015, 22, 281–293. [Google Scholar] [CrossRef]
- Zou, Y.; Li, H.; Graham, E.T.; Deik, A.A.; Eaton, J.K.; Wang, W.; Sandoval-Gomez, G.; Clish, C.B.; Doench, J.G.; Schreiber, S.L. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat. Chem. Biol. 2020, 16, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [Green Version]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxidative Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong-Ekkabut, J.; Xu, Z.; Triampo, W.; Tang, I.M.; Tieleman, D.P.; Monticelli, L. Effect of lipid peroxidation on the properties of lipid bilayers: A molecular dynamics study. Biophys. J. 2007, 93, 4225–4236. [Google Scholar] [CrossRef] [Green Version]
- Kuang, F.; Liu, J.; Tang, D.; Kang, R. Oxidative Damage and Antioxidant Defense in Ferroptosis. Front. Cell Dev. Biol. 2020, 8, 586578. [Google Scholar] [CrossRef]
- Chen, J.J.; Galluzzi, L. Fighting Resilient Cancers with Iron. Trends Cell Biol. 2018, 28, 77–78. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef]
- Liang, C.; Zhang, X.; Yang, M.; Dong, X. Recent Progress in Ferroptosis Inducers for Cancer Therapy. Adv. Mater. 2019, 31, e1904197. [Google Scholar] [CrossRef] [PubMed]
- Forcina, G.C.; Dixon, S.J. GPX4 at the Crossroads of Lipid Homeostasis and Ferroptosis. Proteomics 2019, 19, e1800311. [Google Scholar] [CrossRef] [PubMed]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Kraft, V.A.N.; Bezjian, C.T.; Pfeiffer, S.; Ringelstetter, L.; Müller, C.; Zandkarimi, F.; Merl-Pham, J.; Bao, X.; Anastasov, N.; Kössl, J.; et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent. Sci. 2020, 6, 41–53. [Google Scholar] [CrossRef] [Green Version]
- Soula, M.; Weber, R.A.; Zilka, O.; Alwaseem, H.; La, K.; Yen, F.; Molina, H.; Garcia-Bermudez, J.; Pratt, D.A.; Birsoy, K. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat. Chem. Biol. 2020, 16, 1351–1360. [Google Scholar] [CrossRef]
- Mao, C.; Liu, X.; Zhang, Y.; Lei, G.; Yan, Y.; Lee, H.; Koppula, P.; Wu, S.; Zhuang, L.; Fang, B.; et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 2021, 593, 586–590. [Google Scholar] [CrossRef]
- Lee, H.; Zandkarimi, F.; Zhang, Y.; Meena, J.K.; Kim, J.; Zhuang, L.; Tyagi, S.; Ma, L.; Westbrook, T.F.; Steinberg, G.R.; et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat. Cell Biol. 2020, 22, 225–234. [Google Scholar] [CrossRef]
- Song, X.; Zhu, S.; Chen, P.; Hou, W.; Wen, Q.; Liu, J.; Xie, Y.; Liu, J.; Klionsky, D.J.; Kroemer, G.; et al. AMPK-Mediated BECN1 Phosphorylation Promotes Ferroptosis by Directly Blocking System X Activity. Curr. Biol. 2018, 28, 2388–2399.E5. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-M.; Calkins, M.J.; Chan, K.; Kan, Y.W.; Johnson, J.A. Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J. Biol. Chem. 2003, 278, 12029–12038. [Google Scholar] [CrossRef]
- Kerins, M.J.; Ooi, A. The Roles of NRF2 in Modulating Cellular Iron Homeostasis. Antioxid. Redox Signal. 2018, 29, 1756–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, K.C.; Cui, J.Y.; Klaassen, C.D. Beneficial role of Nrf2 in regulating NADPH generation and consumption. Toxicol. Sci. 2011, 123, 590–600. [Google Scholar] [CrossRef] [Green Version]
- Lillo-Moya, J.; Rojas-Solé, C.; Muñoz-Salamanca, D.; Panieri, E.; Saso, L.; Rodrigo, R. Targeting Ferroptosis against Ischemia/Reperfusion Cardiac Injury. Antioxidants 2021, 10, 667. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Jiang, W.; Wang, W.; Xiong, R.; Wu, X.; Geng, Q. Ferroptosis and its emerging roles in cardiovascular diseases. Pharmacol. Res. 2021, 166, 105466. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Toan, S.; Mui, D.; Zhou, H. Mitochondrial quality surveillance as a therapeutic target in myocardial infarction. Acta Physiol. (Oxf.) 2021, 231, e13590. [Google Scholar] [CrossRef]
- Zhou, H.; Ren, J.; Toan, S.; Mui, D. Role of mitochondrial quality surveillance in myocardial infarction: From bench to bedside. Ageing Res. Rev. 2021, 66, 101250. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.; Lochner, A.; Wang, H.H.; Wang, S.; Zhu, H.; Ren, J.; Zhou, H. Coronary microvascular injury in myocardial infarction: Perception and knowledge for mitochondrial quality control. Theranostics 2021, 11, 6766–6785. [Google Scholar] [CrossRef]
- Zhu, H.; Tan, Y.; Du, W.; Li, Y.; Toan, S.; Mui, D.; Tian, F.; Zhou, H. Phosphoglycerate mutase 5 exacerbates cardiac ischemia-reperfusion injury through disrupting mitochondrial quality control. Redox Biol. 2021, 38, 101777. [Google Scholar] [CrossRef]
- Tang, L.-J.; Luo, X.-J.; Tu, H.; Chen, H.; Xiong, X.-M.; Li, N.-S.; Peng, J. Ferroptosis occurs in phase of reperfusion but not ischemia in rat heart following ischemia or ischemia/reperfusion. Naunyn Schmiedebergs Arch. Pharm. 2021, 394, 401–410. [Google Scholar] [CrossRef]
- Ma, X.-H.; Liu, J.-H.-Z.; Liu, C.-Y.; Sun, W.-Y.; Duan, W.-J.; Wang, G.; Kurihara, H.; He, R.-R.; Li, Y.-F.; Chen, Y.; et al. ALOX15-launched PUFA-phospholipids peroxidation increases the susceptibility of ferroptosis in ischemia-induced myocardial damage. Signal Transduct. Target. Ther. 2022, 7, 288. [Google Scholar] [CrossRef]
- Liu, H.; Mo, H.; Yang, C.; Mei, X.; Song, X.; Lu, W.; Xiao, H.; Yan, J.; Wang, X.; Yan, J.; et al. A novel function of ATF3 in suppression of ferroptosis in mouse heart suffered ischemia/reperfusion. Free Radic. Biol. Med. 2022, 189, 122–135. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, W.; Wang, Y.; Leng, Y.; Xia, Z. Inhibition of DNMT-1 alleviates ferroptosis through NCOA4 mediated ferritinophagy during diabetes myocardial ischemia/reperfusion injury. Cell Death Discov. 2021, 7, 267. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Xiong, Y.; Zhang, Y.; Leng, Y.; Tao, J.; Li, L.; Qiu, Z.; Xia, Z. Activation of NRF2/FPN1 pathway attenuates myocardial ischemia-reperfusion injury in diabetic rats by regulating iron homeostasis and ferroptosis. Cell Stress Chaperones 2021, 27, 149–164. [Google Scholar] [CrossRef]
- Machado, S.E.; Spangler, D.; Stacks, D.A.; Darley-Usmar, V.; Benavides, G.A.; Xie, M.; Balla, J.; Zarjou, A. Counteraction of Myocardial Ferritin Heavy Chain Deficiency by Heme Oxygenase-1. Int. J. Mol. Sci. 2022, 23, 8300. [Google Scholar] [CrossRef]
- Ryter, S.W.; Alam, J.; Choi, A.M.K. Heme oxygenase-1/carbon monoxide: From basic science to therapeutic applications. Physiol. Rev. 2006, 86, 583–650. [Google Scholar] [CrossRef]
- Jang, S.; Chapa-Dubocq, X.R.; Tyurina, Y.Y.; St Croix, C.M.; Kapralov, A.A.; Tyurin, V.A.; Bayır, H.; Kagan, V.E.; Javadov, S. Elucidating the contribution of mitochondrial glutathione to ferroptosis in cardiomyocytes. Redox Biol. 2021, 45, 102021. [Google Scholar] [CrossRef] [PubMed]
- Stamenkovic, A.; O’Hara, K.A.; Nelson, D.C.; Maddaford, T.G.; Edel, A.L.; Maddaford, G.; Dibrov, E.; Aghanoori, M.; Kirshenbaum, L.A.; Fernyhough, P.; et al. Oxidized phosphatidylcholines trigger ferroptosis in cardiomyocytes during ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H1170–H1184. [Google Scholar] [CrossRef]
- Tang, L.-J.; Zhou, Y.-J.; Xiong, X.-M.; Li, N.-S.; Zhang, J.-J.; Luo, X.-J.; Peng, J. Ubiquitin-specific protease 7 promotes ferroptosis via activation of the p53/TfR1 pathway in the rat hearts after ischemia/reperfusion. Free Radic. Biol. Med. 2021, 162, 339–352. [Google Scholar] [CrossRef]
- Ma, S.; Sun, L.; Wu, W.; Wu, J.; Sun, Z.; Ren, J. USP22 Protects Against Myocardial Ischemia-Reperfusion Injury via the SIRT1-p53/SLC7A11-Dependent Inhibition of Ferroptosis-Induced Cardiomyocyte Death. Front. Physiol. 2020, 11, 551318. [Google Scholar] [CrossRef]
- Sun, W.; Wu, X.; Yu, P.; Zhang, Q.; Shen, L.; Chen, J.; Tong, H.; Fan, M.; Shi, H.; Chen, X. LncAABR07025387.1 Enhances Myocardial Ischemia/Reperfusion Injury miR-205/ACSL4-Mediated Ferroptosis. Front. Cell Dev. Biol. 2022, 10, 672391. [Google Scholar] [CrossRef]
- Sun, W.; Shi, R.; Guo, J.; Wang, H.; Shen, L.; Shi, H.; Yu, P.; Chen, X. miR-135b-3p Promotes Cardiomyocyte Ferroptosis by Targeting GPX4 and Aggravates Myocardial Ischemia/Reperfusion Injury. Front. Cardiovasc. Med. 2021, 8, 663832. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-K.; Zhang, Z.; Guo, Z.-A.; Fu, Y.; Chen, X.-J.; Chen, W.-J.; Wu, H.-F.; Cui, X.-J. The BMSC-derived exosomal lncRNA Mir9-3hg suppresses cardiomyocyte ferroptosis in ischemia-reperfusion mice via the Pum2/PRDX6 axis. Nutr. Metab. Cardiovasc. Dis. 2022, 32, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Feng, G.; Gauthier, J.M.; Lokshina, I.; Higashikubo, R.; Evans, S.; Liu, X.; Hassan, A.; Tanaka, S.; Cicka, M.; et al. Ferroptotic cell death and TLR4/Trif signaling initiate neutrophil recruitment after heart transplantation. J. Clin. Investig. 2019, 129, 2293–2304. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Madungwe, N.B.; Imam Aliagan, A.D.; Tombo, N.; Bopassa, J.C. Liproxstatin-1 protects the mouse myocardium against ischemia/reperfusion injury by decreasing VDAC1 levels and restoring GPX4 levels. Biochem. Biophys. Res. Commun. 2019, 520, 606–611. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.-W.; Park, J.-H.; Park, B.-W.; Kim, H.; Kim, J.-J.; Sim, W.-S.; Mishchenko, N.P.; Fedoreyev, S.A.; Vasileva, E.A.; Ban, K.; et al. Histochrome Attenuates Myocardial Ischemia-Reperfusion Injury by Inhibiting Ferroptosis-Induced Cardiomyocyte Death. Antioxidants 2021, 10, 1624. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yao, M.; Jiang, L.; Wang, L.; Yang, Y.; Wang, Q.; Qian, X.; Zhao, Y.; Qian, J. Dexmedetomidine attenuates myocardial ischemia/reperfusion-induced ferroptosis via AMPK/GSK-3β/Nrf2 axis. Biomed. Pharmacother. 2022, 154, 113572. [Google Scholar] [CrossRef]
- Lu, H.; Xiao, H.; Dai, M.; Xue, Y.; Zhao, R. Britanin relieves ferroptosis-mediated myocardial ischaemia/reperfusion damage by upregulating GPX4 through activation of AMPK/GSK3β/Nrf2 signalling. Pharm. Biol. 2022, 60, 38–45. [Google Scholar] [CrossRef]
- Lin, J.-H.; Yang, K.-T.; Ting, P.-C.; Luo, Y.-P.; Lin, D.-J.; Wang, Y.-S.; Chang, J.-C. Gossypol Acetic Acid Attenuates Cardiac Ischemia/Reperfusion Injury in Rats via an Antiferroptotic Mechanism. Biomolecules 2021, 11, 1667. [Google Scholar] [CrossRef]
- Lv, Z.; Wang, F.E.; Zhang, X.; Zhang, X.; Zhang, J.; Liu, R. Etomidate Attenuates the Ferroptosis in Myocardial Ischemia/Reperfusion Rat Model via Nrf2/HO-1 Pathway. Shock 2021, 56, 440–449. [Google Scholar] [CrossRef]
- Xu, S.; Wu, B.; Zhong, B.; Lin, L.; Ding, Y.; Jin, X.; Huang, Z.; Lin, M.; Wu, H.; Xu, D. Naringenin alleviates myocardial ischemia/reperfusion injury by regulating the nuclear factor-erythroid factor 2-related factor 2 (Nrf2)/System xc-/glutathione peroxidase 4 (GPX4) axis to inhibit ferroptosis. Bioengineered 2021, 12, 10924–10934. [Google Scholar] [CrossRef]
- Lin, J.-H.; Yang, K.-T.; Lee, W.-S.; Ting, P.-C.; Luo, Y.-P.; Lin, D.-J.; Wang, Y.-S.; Chang, J.-C. Xanthohumol Protects the Rat Myocardium against Ischemia/Reperfusion Injury-Induced Ferroptosis. Oxidative Med. Cell. Longev. 2022, 2022, 9523491. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Qi, K.; Gong, Y.; Long, X.; Zhu, S.; Lu, F.; Lin, K.; Xu, J. Ferulic Acid Alleviates Myocardial Ischemia Reperfusion Injury Via Upregulating AMPKα2 Expression-Mediated Ferroptosis Depression. J. Cardiovasc. Pharmacol. 2021, 79, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Tan, Y.; Ouyang, S.; He, J.; Liu, L. Resveratrol protects against myocardial ischemia-reperfusion injury via attenuating ferroptosis. Gene 2022, 808, 145968. [Google Scholar] [CrossRef] [PubMed]
- Shan, X.; Lv, Z.-Y.; Yin, M.-J.; Chen, J.; Wang, J.; Wu, Q.-N. The Protective Effect of Cyanidin-3-Glucoside on Myocardial Ischemia-Reperfusion Injury through Ferroptosis. Oxidative Med. Cell. Longev. 2021, 2021, 8880141. [Google Scholar] [CrossRef]
- Li, Y.M.; Sun, J.G.; Hu, L.H.; Ma, X.C.; Zhou, G.; Huang, X.Z. Propofol-mediated cardioprotection dependent of microRNA-451/HMGB1 against myocardial ischemia-reperfusion injury. J. Cell Physiol. 2019, 234, 23289–23301. [Google Scholar] [CrossRef]
- Wang, W.; Qin, J.-J.; Rajaei, M.; Li, X.; Yu, X.; Hunt, C.; Zhang, R. Targeting MDM2 for novel molecular therapy: Beyond oncology. Med. Res. Rev. 2020, 40, 856–880. [Google Scholar] [CrossRef]
- Feng, J.; Tamaskovic, R.; Yang, Z.; Brazil, D.P.; Merlo, A.; Hess, D.; Hemmings, B.A. Stabilization of Mdm2 via decreased ubiquitination is mediated by protein kinase B/Akt-dependent phosphorylation. J. Biol. Chem. 2004, 279, 35510–35517. [Google Scholar] [CrossRef] [Green Version]
- Shravah, J.; Wang, B.; Pavlovic, M.; Kumar, U.; Chen, D.D.; Luo, H.; Ansley, D.M. Propofol mediates signal transducer and activator of transcription 3 activation and crosstalk with phosphoinositide 3-kinase/AKT. JAKSTAT 2014, 3, e29554. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.; Tian, L.; Zhao, X.; Lei, S.; Zhao, B.; Qiu, Z.; Xia, Z.-Y. Rev-erbs agonist SR9009 alleviates ischemia-reperfusion injury by heightening endogenous cardioprotection at onset of type-2 diabetes in rats: Down-regulating ferritinophagy/ferroptosis signaling. Biomed. Pharmacother. 2022, 154, 113595. [Google Scholar] [CrossRef]
- Cetin, N.; Suleyman, H.; Sener, E.; Demirci, E.; Gundogdu, C.; Akcay, F. The prevention of ischemia/reperfusion induced oxidative damage by venous blood in rabbit kidneys monitored with biochemical, histopatological and immunohistochemical analysis. J. Physiol. Pharmacol. 2014, 65, 383–392. [Google Scholar] [PubMed]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, X.; Zhang, X.; Liu, L.C.; Zhang, S.; Pelger, C.B.; Lughmani, H.Y.; Haller, S.T.; Gunning, W.T.; Cooper, C.J.; Gong, R.; et al. Hemopexin accumulates in kidneys and worsens acute kidney injury by causing hemoglobin deposition and exacerbation of iron toxicity in proximal tubules. Kidney Int. 2022, in press. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Jiang, X.; Yang, C.; Zhang, J.; Chen, B.; Li, Y.; Yao, S.; Xie, Q.; Gomez, H.; Murugan, R.; et al. Pannexin 1 mediates ferroptosis that contributes to renal ischemia/reperfusion injury. J. Biol. Chem. 2019, 294, 19395–19404. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Wu, J.; Bi, Q.; Wang, W. Exosomal lncRNA TUG1 derived from human urine-derived stem cells attenuates renal ischemia/reperfusion injury by interacting with SRSF1 to regulate ASCL4-mediated ferroptosis. Stem. Cell Res. Ther. 2022, 13, 297. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Ding, X.; Zheng, J.; Wang, B.; Li, Y.; Xiang, H.; Dou, M.; Qiao, Y.; Tian, P.; Xue, W. miR-182-5p and miR-378a-3p regulate ferroptosis in I/R-induced renal injury. Cell Death Dis. 2020, 11, 929. [Google Scholar] [CrossRef]
- Chen, C.A.; Wang, D.; Yu, Y.; Zhao, T.; Min, N.; Wu, Y.; Kang, L.; Zhao, Y.; Du, L.; Zhang, M.; et al. Legumain promotes tubular ferroptosis by facilitating chaperone-mediated autophagy of GPX4 in AKI. Cell Death Dis. 2021, 12, 65. [Google Scholar] [CrossRef]
- Tao, W.; Liu, F.; Zhang, J.; Fu, S.; Zhan, H.; Qian, K. miR-3587 Inhibitor Attenuates Ferroptosis Following Renal Ischemia-Reperfusion Through HO-1. Front. Mol. Biosci. 2021, 8, 789927. [Google Scholar] [CrossRef]
- Eleftheriadis, T.; Pissas, G.; Golfinopoulos, S.; Liakopoulos, V.; Stefanidis, I. Role of indoleamine 2,3-dioxygenase in ischemia-reperfusion injury of renal tubular epithelial cells. Mol. Med. Rep. 2021, 23, 12111. [Google Scholar] [CrossRef]
- Liang, Y.; Liu, Z.; Qu, L.; Wang, Y.; Zhou, Y.; Liang, L.; Guo, Y.; Tang, L. Inhibition of the IRE1/JNK pathway in renal tubular epithelial cells attenuates ferroptosis in acute kidney injury. Front. Pharmacol. 2022, 13, 927641. [Google Scholar] [CrossRef]
- Feng, R.; Xiong, Y.; Lei, Y.; Huang, Q.; Liu, H.; Zhao, X.; Chen, Z.; Chen, H.; Liu, X.; Wang, L.; et al. Lysine-specific demethylase 1 aggravated oxidative stress and ferroptosis induced by renal ischemia and reperfusion injury through activation of TLR4/NOX4 pathway in mice. J. Cell Mol. Med. 2022, 26, 4254–4267. [Google Scholar] [CrossRef] [PubMed]
- Sui, M.; Xu, D.; Zhao, W.; Lu, H.; Chen, R.; Duan, Y.; Li, Y.; Zhu, Y.; Zhang, L.; Zeng, L. CIRBP promotes ferroptosis by interacting with ELAVL1 and activating ferritinophagy during renal ischaemia-reperfusion injury. J. Cell Mol. Med. 2021, 25, 6203–6216. [Google Scholar] [CrossRef] [PubMed]
- Linkermann, A.; Skouta, R.; Himmerkus, N.; Mulay, S.R.; Dewitz, C.; De Zen, F.; Prokai, A.; Zuchtriegel, G.; Krombach, F.; Welz, P.-S.; et al. Synchronized renal tubular cell death involves ferroptosis. Proc. Natl. Acad. Sci. USA 2014, 111, 16836–16841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Bi, J.; Ren, Y.; Du, Z.; Li, T.; Wang, T.; Zhang, L.; Wang, M.; Wei, S.; Lv, Y.; et al. Involvement of GPX4 in irisin’s protection against ischemia reperfusion-induced acute kidney injury. J. Cell Physiol. 2021, 236, 931–945. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Wu, J.; Xu, H.; Zhou, C.; Han, B.; Zhu, H.; Hu, Z.; Ma, Z.; Ming, Z.; Yao, Y.; et al. XJB-5-131 inhibited ferroptosis in tubular epithelial cells after ischemia-reperfusion injury. Cell Death Dis. 2020, 11, 629. [Google Scholar] [CrossRef]
- Tao, W.-H.; Shan, X.-S.; Zhang, J.-X.; Liu, H.-Y.; Wang, B.-Y.; Wei, X.; Zhang, M.; Peng, K.; Ding, J.; Xu, S.-X.; et al. Dexmedetomidine Attenuates Ferroptosis-Mediated Renal Ischemia/Reperfusion Injury and Inflammation by Inhibiting ACSL4 α2-AR. Front. Pharmacol. 2022, 13, 782466. [Google Scholar] [CrossRef]
- Wang, Y.; Quan, F.; Cao, Q.; Lin, Y.; Yue, C.; Bi, R.; Cui, X.; Yang, H.; Yang, Y.; Birnbaumer, L.; et al. Quercetin alleviates acute kidney injury by inhibiting ferroptosis. J. Adv. Res. 2021, 28, 231–243. [Google Scholar] [CrossRef]
- Jiang, G.-P.; Liao, Y.-J.; Huang, L.-L.; Zeng, X.-J.; Liao, X.-H. Effects and molecular mechanism of pachymic acid on ferroptosis in renal ischemia reperfusion injury. Mol. Med. Rep. 2021, 23, 11704. [Google Scholar] [CrossRef]
- Huang, Y.-B.; Jiang, L.; Liu, X.-Q.; Wang, X.; Gao, L.; Zeng, H.-X.; Zhu, W.; Hu, X.-R.; Wu, Y.-G. Melatonin Alleviates Acute Kidney Injury by Inhibiting NRF2/Slc7a11 Axis-Mediated Ferroptosis. Oxidative Med. Cell. Longev. 2022, 2022, 4776243. [Google Scholar] [CrossRef]
- Yang, J.; Sun, X.; Huang, N.; Li, P.; He, J.; Jiang, L.; Zhang, X.; Han, S.; Xin, H. Entacapone alleviates acute kidney injury by inhibiting ferroptosis. FASEB J. 2022, 36, e22399. [Google Scholar] [CrossRef]
- Cetin, N.; Dasdelen, D.; Mogulkoc, R.; Menevse, E.; Baltaci, A.K. Role of exogenous putrescine in the status of energy, DNA damage, inflammation, and spermidine/spermine-n(1)- acetyltransferase in brain ischemia-reperfusion in rats. Iran J. Basic Med. Sci. 2022, 25, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Bu, Z.-Q.; Yu, H.-Y.; Wang, J.; He, X.; Cui, Y.-R.; Feng, J.-C.; Feng, J. Emerging Role of Ferroptosis in the Pathogenesis of Ischemic Stroke: A New Therapeutic Target? ASN Neuro 2021, 13, 17590914211037505. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Jiang, L.; Hu, Y.; Tang, N.; Liang, N.; Li, X.-F.; Chen, Y.-W.; Qin, H.; Wu, L. Ferritin reduction is essential for cerebral ischemia-induced hippocampal neuronal death through p53/SLC7A11-mediated ferroptosis. Brain Res. 2021, 1752, 147216. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Liu, Y.; Li, Y.; Hong, Z.; Li, S.; Liu, C. PUM2 aggravates the neuroinflammation and brain damage induced by ischemia-reperfusion through the SLC7A11-dependent inhibition of ferroptosis via suppressing the SIRT1. Mol. Cell Biochem. 2022. [CrossRef] [PubMed]
- Tuo, Q.Z.; Lei, P.; Jackman, K.A.; Li, X.L.; Xiong, H.; Li, X.L.; Liuyang, Z.Y.; Roisman, L.; Zhang, S.T.; Ayton, S.; et al. Tau-mediated iron export prevents ferroptotic damage after ischemic stroke. Mol. Psychiatry 2017, 22, 1520–1530. [Google Scholar] [CrossRef]
- Li, C.; Sun, G.; Chen, B.; Xu, L.; Ye, Y.; He, J.; Bao, Z.; Zhao, P.; Miao, Z.; Zhao, L.; et al. Nuclear receptor coactivator 4-mediated ferritinophagy contributes to cerebral ischemia-induced ferroptosis in ischemic stroke. Pharmacol. Res. 2021, 174, 105933. [Google Scholar] [CrossRef]
- Yang, L.; Du, B.; Zhang, S.; Wang, M. RXRγ attenuates cerebral ischemia-reperfusion induced ferroptosis in neurons in mice through transcriptionally promoting the expression of GPX4. Metab. Brain Dis. 2022, 37, 1351–1363. [Google Scholar] [CrossRef]
- Zhao, J.; Wu, Y.; Liang, S.; Piao, X. Activation of SSAT1/ALOX15 Axis Aggravates Cerebral Ischemia/Reperfusion Injury via Triggering Neuronal Ferroptosis. Neuroscience 2022, 485, 78–90. [Google Scholar] [CrossRef]
- Zhai, Q.-Y.; Ren, Y.-Q.; Ni, Q.-S.; Song, Z.-H.; Ge, K.-L.; Guo, Y.-L. Transplantation of Human Umbilical Cord Mesenchymal Stem Cells-Derived Neural Stem Cells Pretreated with Neuregulin1β Ameliorate Cerebral Ischemic Reperfusion Injury in Rats. Biomolecules 2022, 12, 428. [Google Scholar] [CrossRef]
- Li, M.; Meng, Z.; Yu, S.; Li, J.; Wang, Y.; Yang, W.; Wu, H. Baicalein ameliorates cerebral ischemia-reperfusion injury by inhibiting ferroptosis via regulating GPX4/ACSL4/ACSL3 axis. Chem. Biol. Interact. 2022, 366, 110137. [Google Scholar] [CrossRef]
- Fu, C.; Wu, Y.; Liu, S.; Luo, C.; Lu, Y.; Liu, M.; Wang, L.; Zhang, Y.; Liu, X. Rehmannioside A improves cognitive impairment and alleviates ferroptosis via activating PI3K/AKT/Nrf2 and SLC7A11/GPX4 signaling pathway after ischemia. J. Ethnopharmacol. 2022, 289, 115021. [Google Scholar] [CrossRef]
- Hu, Q.; Zuo, T.; Deng, L.; Chen, S.; Yu, W.; Liu, S.; Liu, J.; Wang, X.; Fan, X.; Dong, Z. β-Caryophyllene suppresses ferroptosis induced by cerebral ischemia reperfusion via activation of the NRF2/HO-1 signaling pathway in MCAO/R rats. Phytomedicine 2022, 102, 154112. [Google Scholar] [CrossRef]
- Yuan, Y.; Zhai, Y.; Chen, J.; Xu, X.; Wang, H. Kaempferol Ameliorates Oxygen-Glucose Deprivation/Reoxygenation-Induced Neuronal Ferroptosis by Activating Nrf2/SLC7A11/GPX4 Axis. Biomolecules 2021, 11, 923. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Li, Z.; Zhu, S.; Cheng, M.; Ju, Y.; Ren, L.; Yang, G.; Min, D. Galangin attenuated cerebral ischemia-reperfusion injury by inhibition of ferroptosis through activating the SLC7A11/GPX4 axis in gerbils. Life Sci. 2021, 264, 118660. [Google Scholar] [CrossRef]
- Guan, X.; Li, X.; Yang, X.; Yan, J.; Shi, P.; Ba, L.; Cao, Y.; Wang, P. The neuroprotective effects of carvacrol on ischemia/reperfusion-induced hippocampal neuronal impairment by ferroptosis mitigation. Life Sci. 2019, 235, 116795. [Google Scholar] [CrossRef]
- Guo, H.; Zhu, L.; Tang, P.; Chen, D.; Li, Y.; Li, J.; Bao, C. Carthamin yellow improves cerebral ischemia-reperfusion injury by attenuating inflammation and ferroptosis in rats. Int. J. Mol. Med. 2021, 47, 4885. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Huang, J.; Chen, Y.; Li, X.; Wen, J.; Tian, M.; Ren, J.; Zhou, L.; Yang, Q. Resveratrol pretreatment protects neurons from oxygen-glucose deprivation/reoxygenation and ischemic injury through inhibiting ferroptosis. Biosci. Biotechnol. Biochem. 2022, 86, 704–716. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Liu, J.; He, J.; Hu, Z.; Tan, F.; Zhu, X.; Yuan, F.; Jiang, Z. UBIAD1 alleviates ferroptotic neuronal death by enhancing antioxidative capacity by cooperatively restoring impaired mitochondria and Golgi apparatus upon cerebral ischemic/reperfusion insult. Cell Biosci. 2022, 12, 42. [Google Scholar] [CrossRef]
- Wang, P.; Cui, Y.; Ren, Q.; Yan, B.; Zhao, Y.; Yu, P.; Gao, G.; Shi, H.; Chang, S.; Chang, Y.-Z. Mitochondrial ferritin attenuates cerebral ischaemia/reperfusion injury by inhibiting ferroptosis. Cell Death Dis. 2021, 12, 447. [Google Scholar] [CrossRef]
- Wang, P.; Ren, Q.; Shi, M.; Liu, Y.; Bai, H.; Chang, Y.-Z. Overexpression of Mitochondrial Ferritin Enhances Blood-Brain Barrier Integrity Following Ischemic Stroke in Mice by Maintaining Iron Homeostasis in Endothelial Cells. Antioxidants 2022, 11, 1257. [Google Scholar] [CrossRef]
- Tuo, Q.-Z.; Liu, Y.; Xiang, Z.; Yan, H.-F.; Zou, T.; Shu, Y.; Ding, X.-L.; Zou, J.-J.; Xu, S.; Tang, F.; et al. Thrombin induces ACSL4-dependent ferroptosis during cerebral ischemia/reperfusion. Signal Transduct. Target. Ther. 2022, 7, 59. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Liu, Y.; Li, K.; Yuan, D.; Yang, S.; Zhou, L.; Zhao, Y.; Miao, S.; Lv, C.; Zhao, J. COX-2/PGE2 Pathway Inhibits the Ferroptosis Induced by Cerebral Ischemia Reperfusion. Mol. Neurobiol. 2022, 59, 1619–1631. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Xu, F.; Lu, H. LncRNA PVT1 regulates ferroptosis through miR-214-mediated TFR1 and p53. Life Sci. 2020, 260, 118305. [Google Scholar] [CrossRef] [PubMed]
- Akbari, G. Emerging roles of microRNAs in intestinal ischemia/reperfusion-induced injury: A review. J. Physiol. Biochem. 2020, 76, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wen, S.; Yao, X.; Liu, W.; Shen, J.; Deng, W.; Tang, J.; Li, C.; Liu, K. MicroRNA-378 protects against intestinal ischemia/reperfusion injury via a mechanism involving the inhibition of intestinal mucosal cell apoptosis. Cell Death Dis. 2017, 8, e3127. [Google Scholar] [CrossRef] [Green Version]
- Mallick, I.H.; Yang, W.; Winslet, M.C.; Seifalian, A.M. Ischemia-reperfusion injury of the intestine and protective strategies against injury. Dig. Dis. Sci. 2004, 49, 1359–1377. [Google Scholar] [CrossRef]
- Diepenhorst, G.M.P.; van Gulik, T.M.; Hack, C.E. Complement-mediated ischemia-reperfusion injury: Lessons learned from animal and clinical studies. Ann. Surg. 2009, 249, 889–899. [Google Scholar] [CrossRef]
- Zhang, X.-Y.; Liu, Z.-M.; Zhang, H.-F.; Li, Y.-S.; Wen, S.-H.; Shen, J.-T.; Huang, W.-Q.; Liu, K.-X. TGF-β1 improves mucosal IgA dysfunction and dysbiosis following intestinal ischaemia-reperfusion in mice. J. Cell Mol. Med. 2016, 20, 1014–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Mao, Z.; Xu, L.; Yin, L.; Tao, X.; Tang, Z.; Qi, Y.; Sun, P.; Peng, J. Protective effect of dioscin against intestinal ischemia/reperfusion injury via adjusting miR-351-5p-mediated oxidative stress. Pharmacol. Res. 2018, 137, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Stefanutti, G.; Pierro, A.; Parkinson, E.J.; Smith, V.V.; Eaton, S. Moderate hypothermia as a rescue therapy against intestinal ischemia and reperfusion injury in the rat. Crit. Care Med. 2008, 36, 1564–1572. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Qiang, Z.; Chai, D.; Peng, J.; Xia, Y.; Hu, R.; Jiang, H. Nrf2 inhibits ferroptosis and protects against acute lung injury due to intestinal ischemia reperfusion via regulating SLC7A11 and HO-1. Aging 2020, 12, 12943–12959. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.-D.; Ye, W.; Tian, W.; Meng, J.-R.; Zhang, M.; Sun, Y.; Zhang, H.-N.; Wang, S.-J.; Wu, K.-H.; Liu, C.-X.; et al. Old targets, new strategy: Apigenin-7-O-β-d-(-6″-p-coumaroyl)-glucopyranoside prevents endothelial ferroptosis and alleviates intestinal ischemia-reperfusion injury through HO-1 and MAO-B inhibition. Free Radic. Biol. Med. 2022, 184, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Deng, F.; Zhao, B.-C.; Yang, X.; Lin, Z.-B.; Sun, Q.-S.; Wang, Y.-F.; Yan, Z.-Z.; Liu, W.-F.; Li, C.; Hu, J.-J.; et al. The gut microbiota metabolite capsiate promotes Gpx4 expression by activating to inhibit intestinal ischemia reperfusion-induced ferroptosis. Gut Microbes 2021, 13, 1902719. [Google Scholar] [CrossRef]
- Li, Y.; Feng, D.; Wang, Z.; Zhao, Y.; Sun, R.; Tian, D.; Liu, D.; Zhang, F.; Ning, S.; Yao, J.; et al. Ischemia-induced ACSL4 activation contributes to ferroptosis-mediated tissue injury in intestinal ischemia/reperfusion. Cell Death Differ. 2019, 26, 2284–2299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Cao, Y.; Xiao, J.; Shang, J.; Tan, Q.; Ping, F.; Huang, W.; Wu, F.; Zhang, H.; Zhang, X. Inhibitor of apoptosis-stimulating protein of p53 inhibits ferroptosis and alleviates intestinal ischemia/reperfusion-induced acute lung injury. Cell Death Differ. 2020, 27, 2635–2650. [Google Scholar] [CrossRef]
- Macías-Rodríguez, R.U.; Inzaugarat, M.E.; Ruiz-Margáin, A.; Nelson, L.J.; Trautwein, C.; Cubero, F.J. Reclassifying Hepatic Cell Death during Liver Damage: Ferroptosis-A Novel Form of Non-Apoptotic Cell Death? Int. J. Mol. Sci. 2020, 21, 1651. [Google Scholar] [CrossRef] [Green Version]
- Yamada, N.; Karasawa, T.; Wakiya, T.; Sadatomo, A.; Ito, H.; Kamata, R.; Watanabe, S.; Komada, T.; Kimura, H.; Sanada, Y.; et al. Iron overload as a risk factor for hepatic ischemia-reperfusion injury in liver transplantation: Potential role of ferroptosis. Am. J. Transplant. 2020, 20, 1606–1618. [Google Scholar] [CrossRef]
- Decuypere, J.-P.; Ceulemans, L.J.; Agostinis, P.; Monbaliu, D.; Naesens, M.; Pirenne, J.; Jochmans, I. Autophagy and the Kidney: Implications for Ischemia-Reperfusion Injury and Therapy. Am. J. Kidney Dis. 2015, 66, 699–709. [Google Scholar] [CrossRef]
- Oliveira, T.H.C.D.; Marques, P.E.; Proost, P.; Teixeira, M.M.M. Neutrophils: A cornerstone of liver ischemia and reperfusion injury. Lab. Investig. 2018, 98, 51–62. [Google Scholar] [CrossRef] [Green Version]
- Dickson, K.B.; Zhou, J. Role of reactive oxygen species and iron in host defense against infection. Front. Biosci. 2020, 25, 1600–1616. [Google Scholar]
- Skaar, E.P. The battle for iron between bacterial pathogens and their vertebrate hosts. PLoS Pathog. 2010, 6, e1000949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, J.; Peng, J.; Liu, K.; Zhang, T.; Huang, W. MCTR1 inhibits ferroptosis by promoting NRF2 expression to attenuate hepatic ischemia-reperfusion injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2022, 323, G283–G293. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, L.; Tian, X.; Zheng, W.; Yuan, M.; Tian, X.; Zuo, H.; Song, H.; Shen, Z. miR-29a-3p in Exosomes from Heme Oxygenase-1 Modified Bone Marrow Mesenchymal Stem Cells Alleviates Steatotic Liver Ischemia-Reperfusion Injury in Rats by Suppressing Ferroptosis via Iron Responsive Element Binding Protein 2. Oxidative Med. Cell. Longev. 2022, 2022, 6520789. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Yang, J.; Sun, G.; Hu, J.; Zhang, Q.; Cai, J.; Yuan, D.; Li, H.; Hei, Z.; Yao, W. Macrophage extracellular traps aggravate iron overload-related liver ischaemia/reperfusion injury. Br. J. Pharmacol. 2021, 178, 3783–3796. [Google Scholar] [CrossRef]
- Xu, Y.; Li, X.; Cheng, Y.; Yang, M.; Wang, R. Inhibition of ACSL4 attenuates ferroptotic damage after pulmonary ischemia-reperfusion. FASEB J. 2020, 34, 16262–16275. [Google Scholar] [CrossRef]
- Qiang, Z.; Dong, H.; Xia, Y.; Chai, D.; Hu, R.; Jiang, H. Nrf2 and STAT3 Alleviates Ferroptosis-Mediated IIR-ALI by Regulating SLC7A11. Oxidative Med. Cell. Longev. 2020, 2020, 5146982. [Google Scholar] [CrossRef]
- Li, Y.; Yang, Y.; Yang, Y. Multifaceted Roles of Ferroptosis in Lung Diseases. Front. Mol. Biosci. 2022, 9, 919187. [Google Scholar] [CrossRef]
- Wang, Y.; Dong, Z.; Zhang, Z.; Wang, Y.; Yang, K.; Li, X. Postconditioning with Irisin Attenuates Lung Ischemia/Reperfusion Injury by Suppressing Ferroptosis via Induction of the Nrf2/HO-1 Signal Axis. Oxidative Med. Cell. Longev. 2022, 2022, 9911167. [Google Scholar] [CrossRef]
- Ma, X.; Yan, W.; He, N. Lidocaine attenuates hypoxia/reoxygenation-induced inflammation, apoptosis and ferroptosis in lung epithelial cells by regulating the p38 MAPK pathway. Mol. Med. Rep. 2022, 25, 12666. [Google Scholar] [CrossRef]
- Lu, F.; Zander, D.S.; Visner, G.A. Increased expression of heme oxygenase-1 in human lung transplantation. J. Heart Lung. Transplant. 2002, 21, 1120–1126. [Google Scholar] [CrossRef]
- Pugh, C.; Hathwar, V.; Richards, J.H.; Stonehuerner, J.; Ghio, A.J. Disruption of iron homeostasis in the lungs of transplant patients. J. Heart Lung Transplant. 2005, 24, 1821–1827. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Drew, P.; Cheng, Y.; Visner, G.A. Pirfenidone inhibits inflammatory responses and ameliorates allograft injury in a rat lung transplant model. J. Thorac. Cardiovasc. Surg. 2005, 130, 852–858. [Google Scholar] [CrossRef] [Green Version]
- Tu, H.; Zhou, Y.-J.; Tang, L.-J.; Xiong, X.-M.; Zhang, X.-J.; Ali Sheikh, M.S.; Zhang, J.-J.; Luo, X.-J.; Yuan, C.; Peng, J. Combination of ponatinib with deferoxamine synergistically mitigates ischemic heart injury via simultaneous prevention of necroptosis and ferroptosis. Eur. J. Pharmacol. 2021, 898, 173999. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Wang, H.; Han, D.; Xie, E.; Yang, X.; Wei, J.; Gu, S.; Gao, F.; Zhu, N.; Yin, X.; et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2672–2680. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Morphological Features | Biochemical Features | Core genes | Inducers | Inhibitors | |
|---|---|---|---|---|---|
| Ferroptosis | Mitochondrial shrinkage and morphological abnormalities (increased membrane density, diminished or vanished cristae, ruptured outer membrane), with normal nucleus | Iron accumulation, lipid peroxidation, inhibition of SLC7A11/GSH/GPX4 | GPX4, TFR1, SLC7A11, NRF2, NCOA4, P53, ALOXs, ACSL4, FSP1 | Erastin, RSL3 | Ferrostatin-1, liproxstatin-1, vitamin E, desferoxamine |
| Apoptosis | Cell shrinkage, plasma membrane blebbing, chromatin agglutination, nuclear fragmentation, apoptotic bodies | DNA fragmentation, activation of caspase pathway | Caspase, Bcl-2, Bax, P53, Fas | FASL, DCC, UNC5B | zVAD-FMK, XIAP, c-IAP1 |
| Necroptosis | Swelling and rupture of cells and organelles, leakage of cell contents, moderate condensation of chromatin | Drop in ATP levels | RIP1, RIP3, LEF1 | zVAD-fmk, TNF-α | Necrostatin-1, NSA |
| Pyroptosis | Cell swelling and plasma membrane bubbling, formation of inflammasome, release of cellular components | Activation of caspase-1 and GSDMD, release of pro-inflammatory cytokines | Caspase-1, IL-1β, IL-18, GSDMD | Lipopolysacc-haride, ivermectin | NAC, GSH |
| Autophagy | Formation of double-membraned autolysosomes | Increased lysosomal activity | ATG5, ATG7, LC3, BECN1, DRAM3, AMPK, mTOR | Rapamycin, valproate | Chloroquine, 3-methyladenine, wortmannin, Spautin-1, bafilomycin A1 |
| I/R Injury Model | Reagents | Target | References |
|---|---|---|---|
| Myocardial I/R injury | Ferrostatin-1 | Inhibit lipid peroxidation | [73] |
| Deferoxamine | Chelation of iron | [59,163] | |
| Dexrazoxane | Chelation of iron | [164] | |
| Liproxstatin-1 | Reduce ROS levels/VDAC1 | [74] | |
| Histochrome | Increase expression of NRF2 | [75] | |
| Dexmedetomidine | AMPK/GSK-3β/Nrf2 | [76] | |
| Britanin | AMPK/GSK3β/Nrf2 | [77] | |
| Gossypol acetic acid | Inhibit lipid peroxidation | [78] | |
| Etomidate | Nrf2/HO-1 | [79] | |
| Naringenin | Nrf2/System xc-/Gpx4 | [80] | |
| Xanthohumol | Inhibit lipid peroxidation /Chelation of iron | [81] | |
| Ferulic acid | Increase activity of antioxidant enzymes/AMPKα2 | [82] | |
| Compound 968 | Inhibit glutamine catabolism | [83] | |
| Resveratrol | Reduce oxidative stress/ USP19-Beclin1 | [84] | |
| Cyanidin-3-Glucoside | Inhibit oxidative stress | [85] | |
| Propofol | microRNA-451/HMGB1 | [86] | |
| SR9009 | Inhibit ferritinophagy | [87] | |
| Renal I/R injury | Liproxstatin-1 | Inhibit lipid peroxidation | [92] |
| Deferoxamine | Chelation of iron | [93] | |
| Ferrostatin-1 | Inhibit lipid peroxidation | [103] | |
| 16-86 | Inhibit lipid peroxidation | [103] | |
| XJB-5-131 | GPX4, ACSL4 | [105] | |
| Dexmedetomidine | Inhibit ACSL4 via α2-AR | [106] | |
| Quercetin | ATF3 | [107] | |
| Pachymic acid | NRF2/SLC7A11/GPX4 | [108] | |
| Melatonin | NRF2/Slc7a11 | [109] | |
| Entacapone | p62/KEAP1/NRF2/Slc7a11 | [110] | |
| Cerebral I/R injury | Baicalein | GPX4/ACSL4/ACSL3 | [120] |
| Rehmannioside A | PI3K/AKT/Nrf2 and SLC7A11/GPX4 | [121] | |
| β-Caryophyllene | NRF2/HO-1 | [122] | |
| Kaempferol | Nrf2/SLC7A11/GPX4 | [123] | |
| Ferrostatin-1 | Inhibit lipid peroxidation | [115] | |
| Galangin | SLC7A11/GPX4 | [124] | |
| Carvacrol | GPX4 | [125] | |
| Carthamin yellow | Fe2+/ROS/lipid peroxidation | [126] | |
| Resveratrol | Fe2+/ROS | [127] | |
| Liproxstatin-1 | Inhibit lipid peroxidation | [115] | |
| Hepatic I/R injury | Ferrostatin-1 | Inhibit lipid peroxidation | [92,147] |
| Deferoxamine | Reduce intracellular iron | [147] | |
| Liproxstatin-1 | Inhibit lipid peroxidation | [115] | |
| α-tocopherol | Inhibit lipid peroxidation | [147] | |
| Intestinal I/R injury | APG | HO-1 and MAO-B inhibition | [142] |
| Dioscin | miR-351-5p/oxidative stress | [139] | |
| Liproxstatin-1 | Inhibit lipid peroxidation | [144] | |
| Rosiglitazone | Inhibit ACSL4 | [144] | |
| Capsiate | GPX4, TRPV1 | [143] | |
| Lung I/R injury | Liproxstatin-1 | Inhibit lipid peroxidation | [155] |
| Lidocaine | p38 MAPK pathway | [159] | |
| Rosiglitazone ACSL4 | ACSL4 | [155] | |
| Irisin | Nrf2/HO-1 | [158] | |
| Pirfenidone | Reduce iron levels | [162] | |
| Ferrostatin-1 | Inhibit lipid peroxidation | [158] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, L.; Han, S.; Guo, J.; Qiu, T.; Zhou, J.; Shen, L. Ferroptosis—A New Dawn in the Treatment of Organ Ischemia–Reperfusion Injury. Cells 2022, 11, 3653. https://doi.org/10.3390/cells11223653
Zhou L, Han S, Guo J, Qiu T, Zhou J, Shen L. Ferroptosis—A New Dawn in the Treatment of Organ Ischemia–Reperfusion Injury. Cells. 2022; 11(22):3653. https://doi.org/10.3390/cells11223653
Chicago/Turabian StyleZhou, Linxiang, Shangting Han, Jiayu Guo, Tao Qiu, Jiangqiao Zhou, and Lei Shen. 2022. "Ferroptosis—A New Dawn in the Treatment of Organ Ischemia–Reperfusion Injury" Cells 11, no. 22: 3653. https://doi.org/10.3390/cells11223653