
Opposing Roles of IGFBP-3 and Heparanase in Regulating A549 Lung Cancer Cell Survival


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Solid Phase Peptide Synthesis and Purification
2.4. ELISA
2.5. MTT Assay
2.6. Quantitation of Heparanase Levels
2.7. Heparanase Activity Assay
2.8. HS Quantitation Assay
2.9. Quantitation of IGFBP-3 Levels
2.10. p53 Transcription Factor Activity Assay
2.11. Western Blotting
2.12. SiRNA Transfection
2.13. Statistical Analysis
3. Results
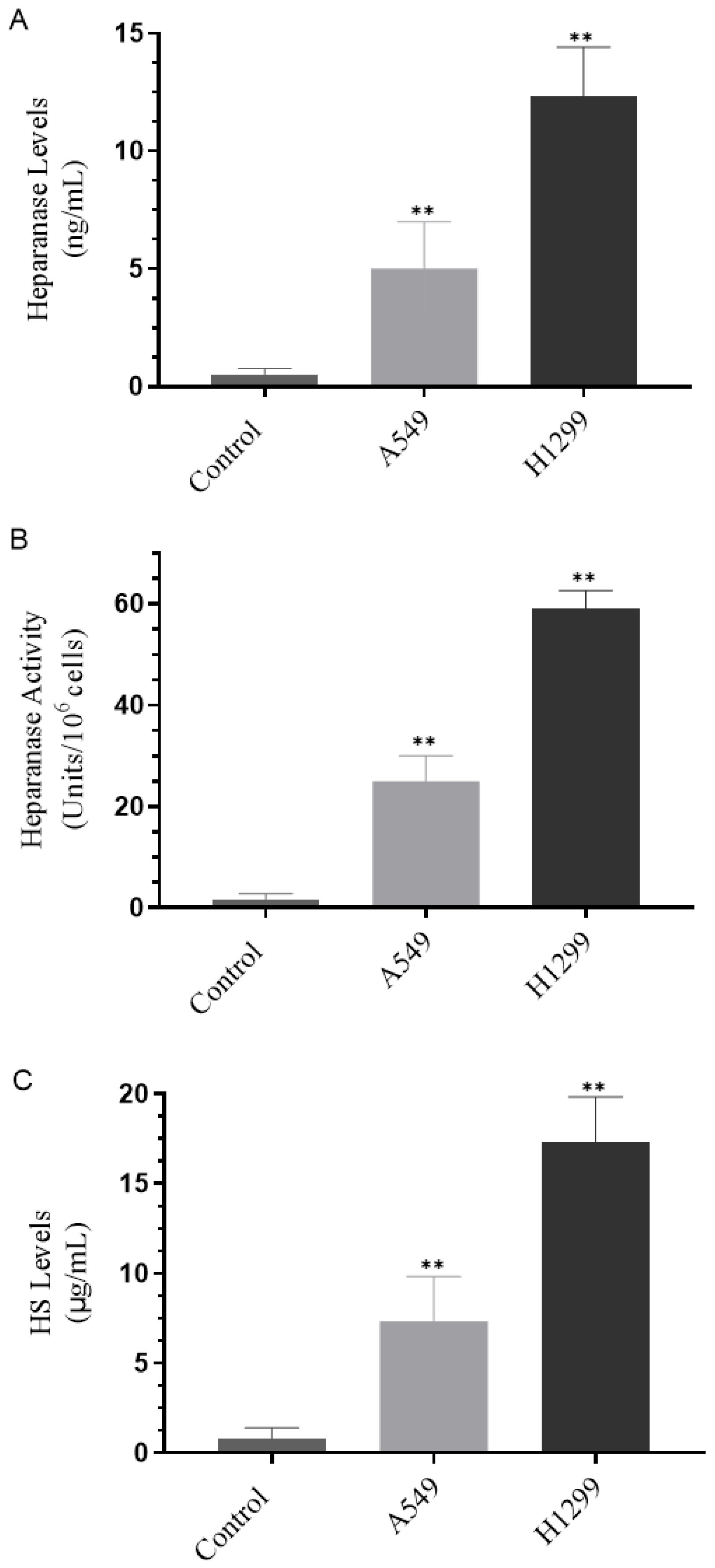
3.1. H1299 Cell Media Had Higher Heparanase Levels and Activity and Higher Levels of HS Than the Media of A549 Cells
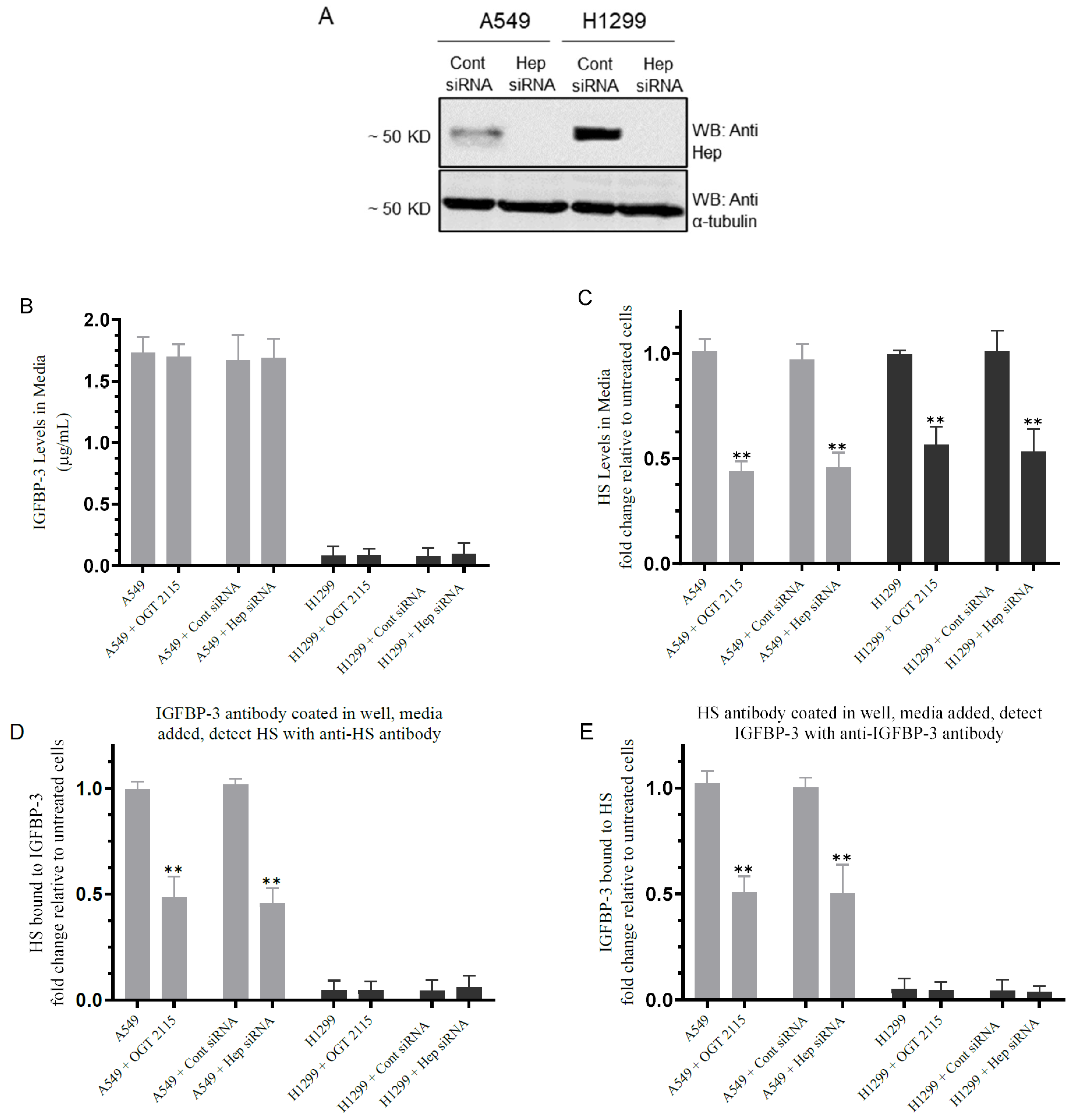
3.2. Inhibiting Heparanase Activity or Its Expression Using siRNA Led to Decreased HS Level in the Media and Reduced the Amount of IGFBP-3 Bound to HS
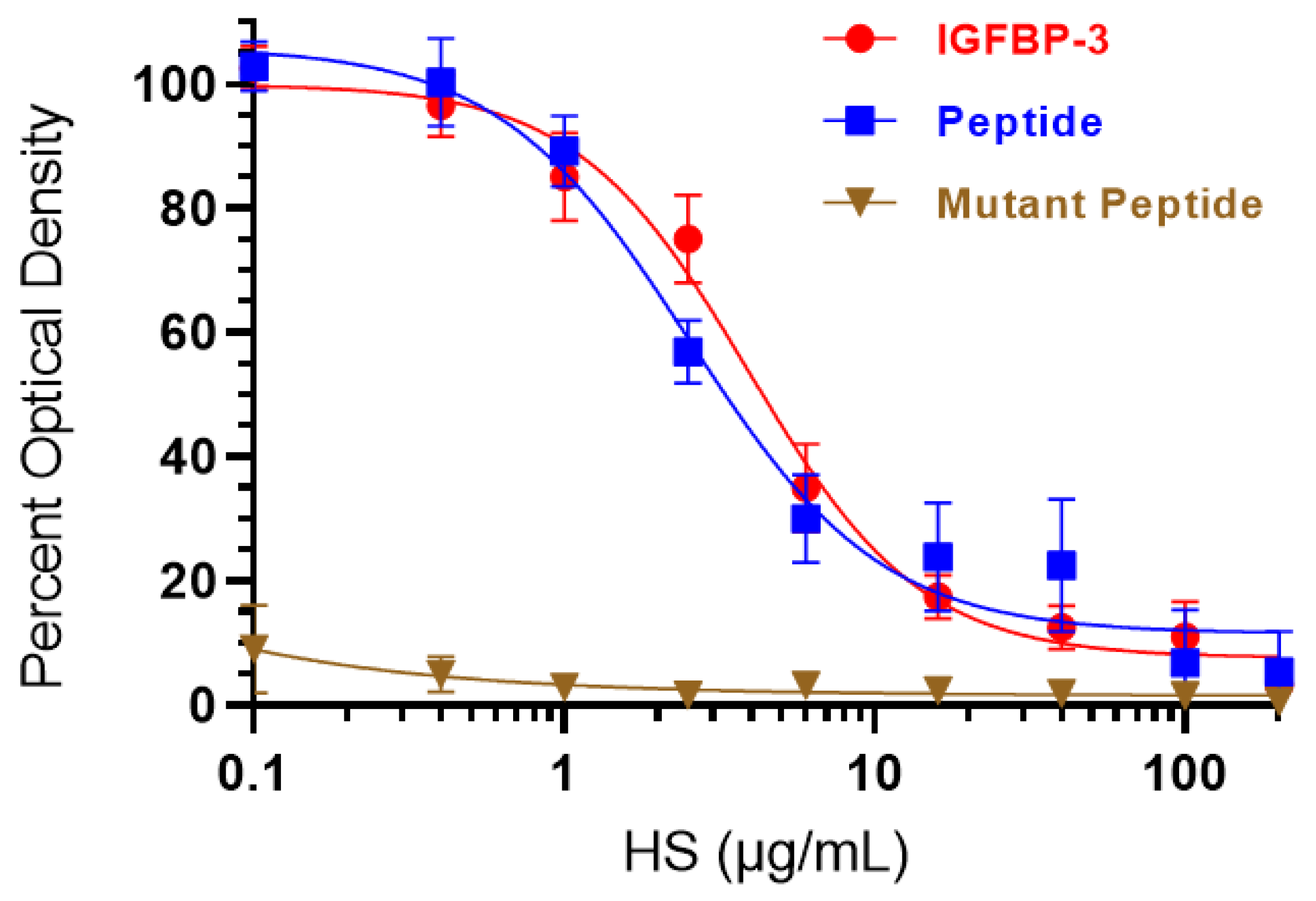
3.3. HS Competes with HA for Binding to IGFBP-3 or WT- but Not the Mutant-Peptide
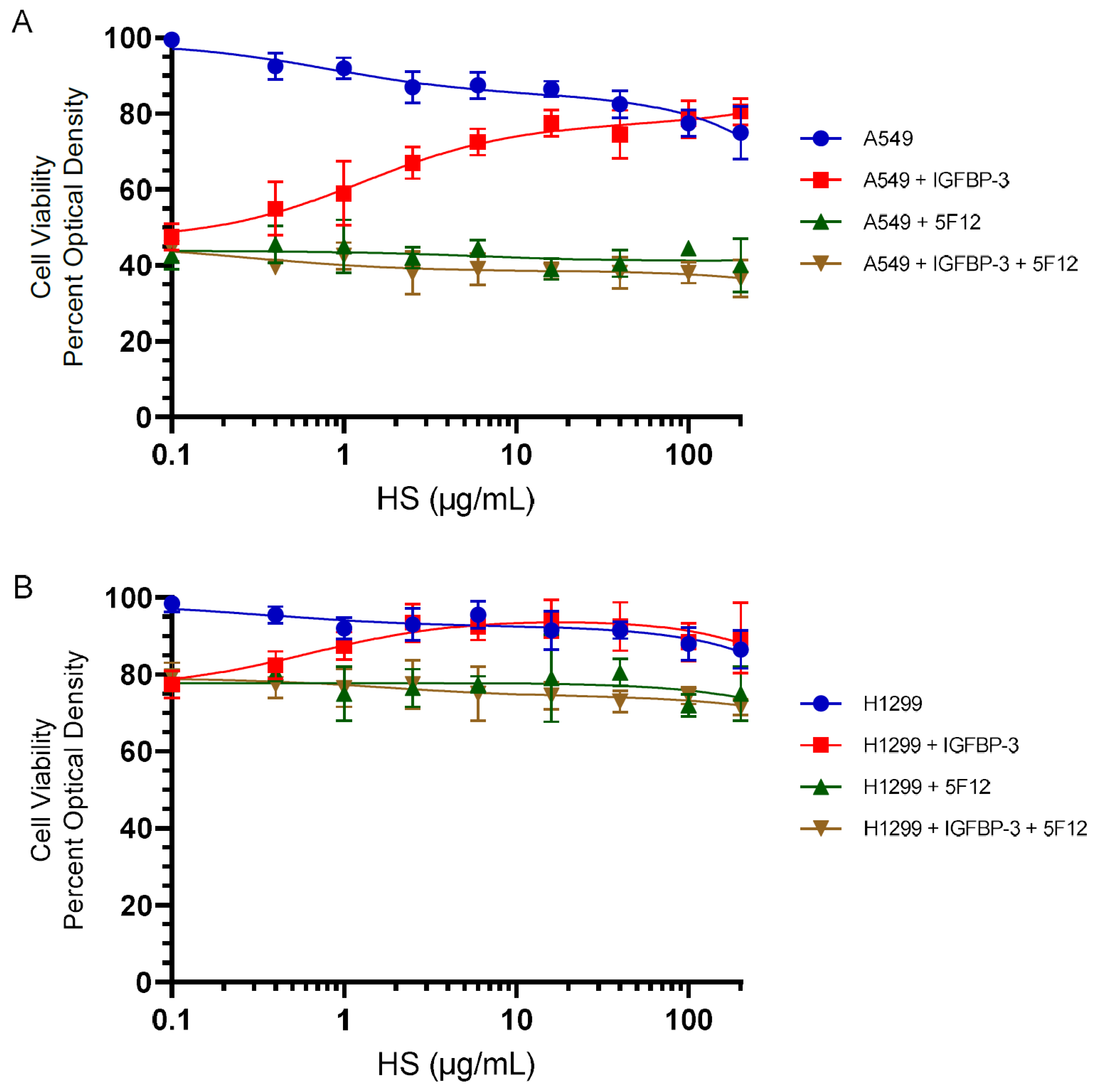
3.4. HS Abolished the Cytotoxic Effects of IGFBP-3 but Not upon Blocking HA–CD44 Signaling with the 5F12 Antibody
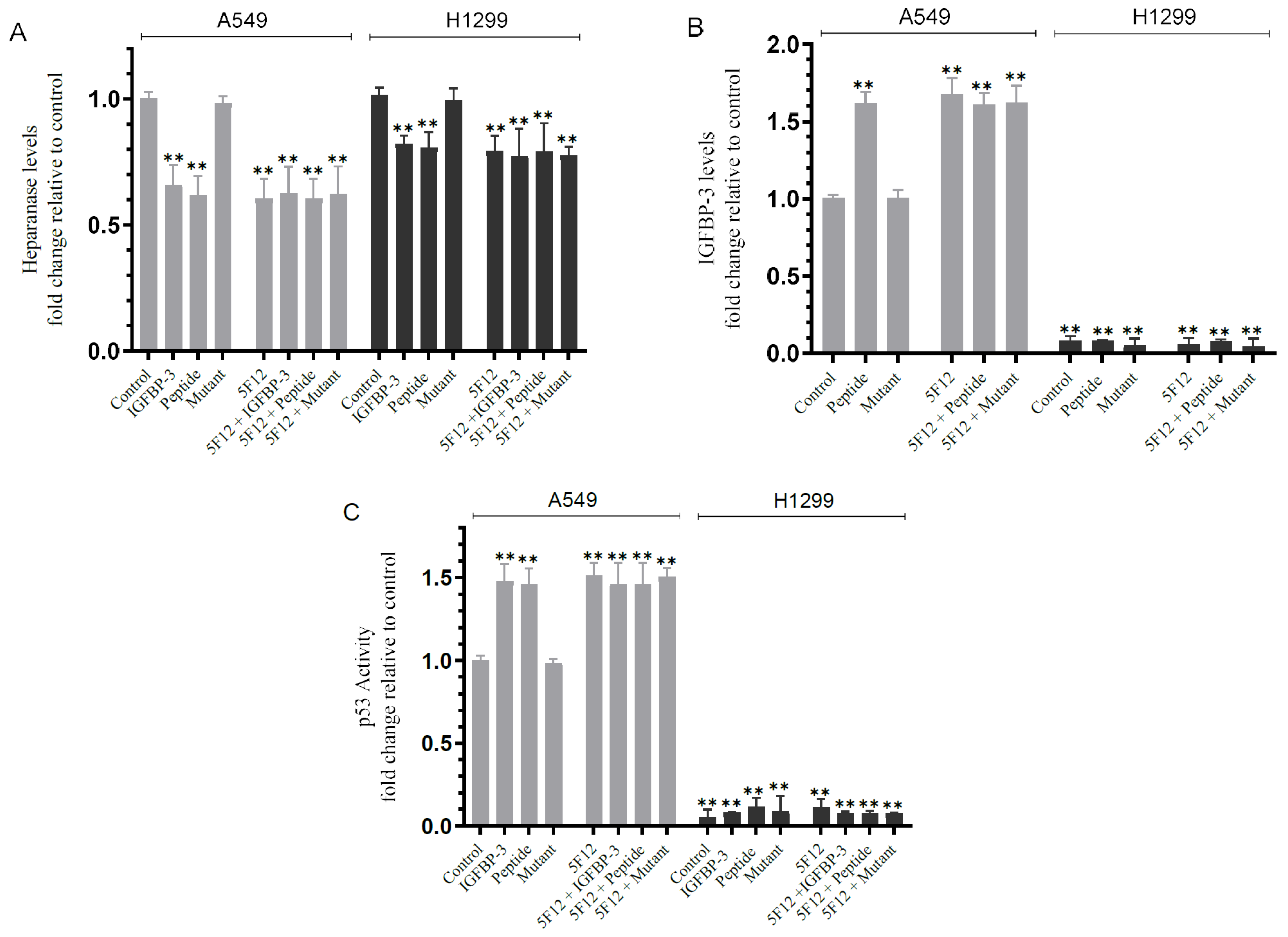
3.5. Blocking HA–CD44 Signaling Decreases the Levels of Heparanase in the Media of Both Cell Lines and Increases p53 Activity and the Levels of IGFBP-3 in A549 Cell Media
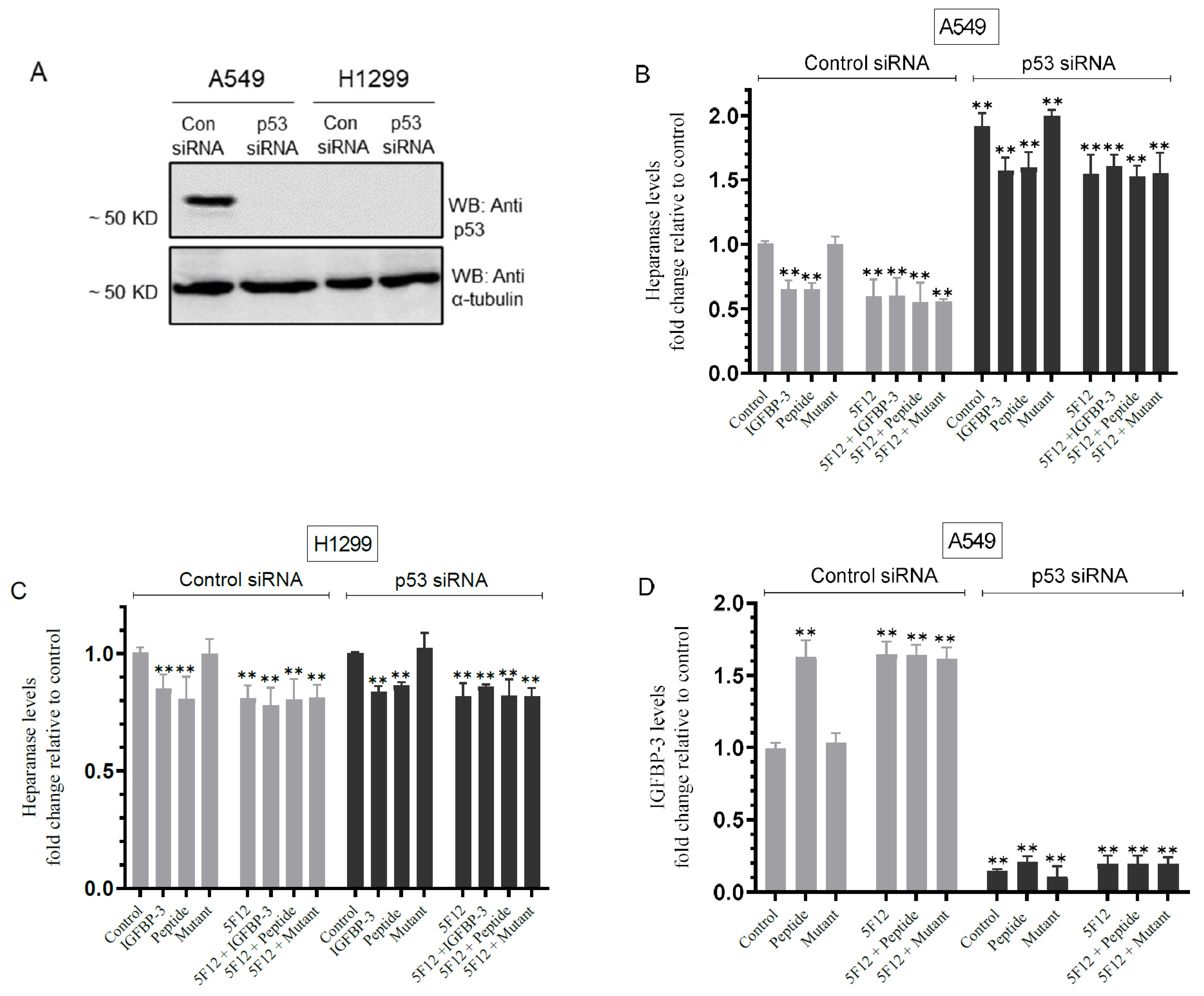
3.6. Knockdown of p53 Resulted in Increased Heparanase Levels and Reduced IGFBP-3 Levels in A549 Cell Media
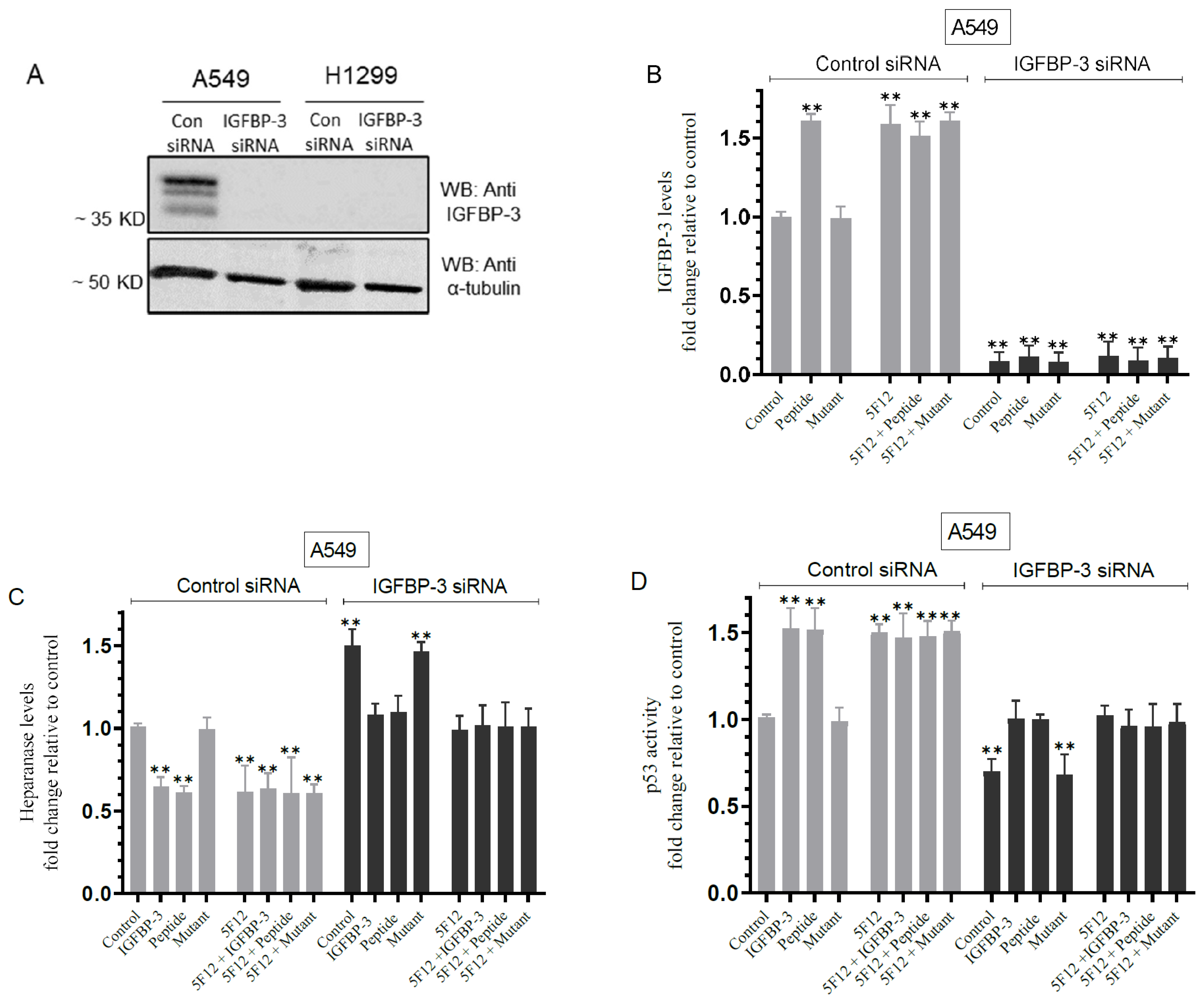
3.7. Knockdown of IGFBP-3 in A549 Cells Inhibited p53 Activity and Increased Heparanase Levels in the Media
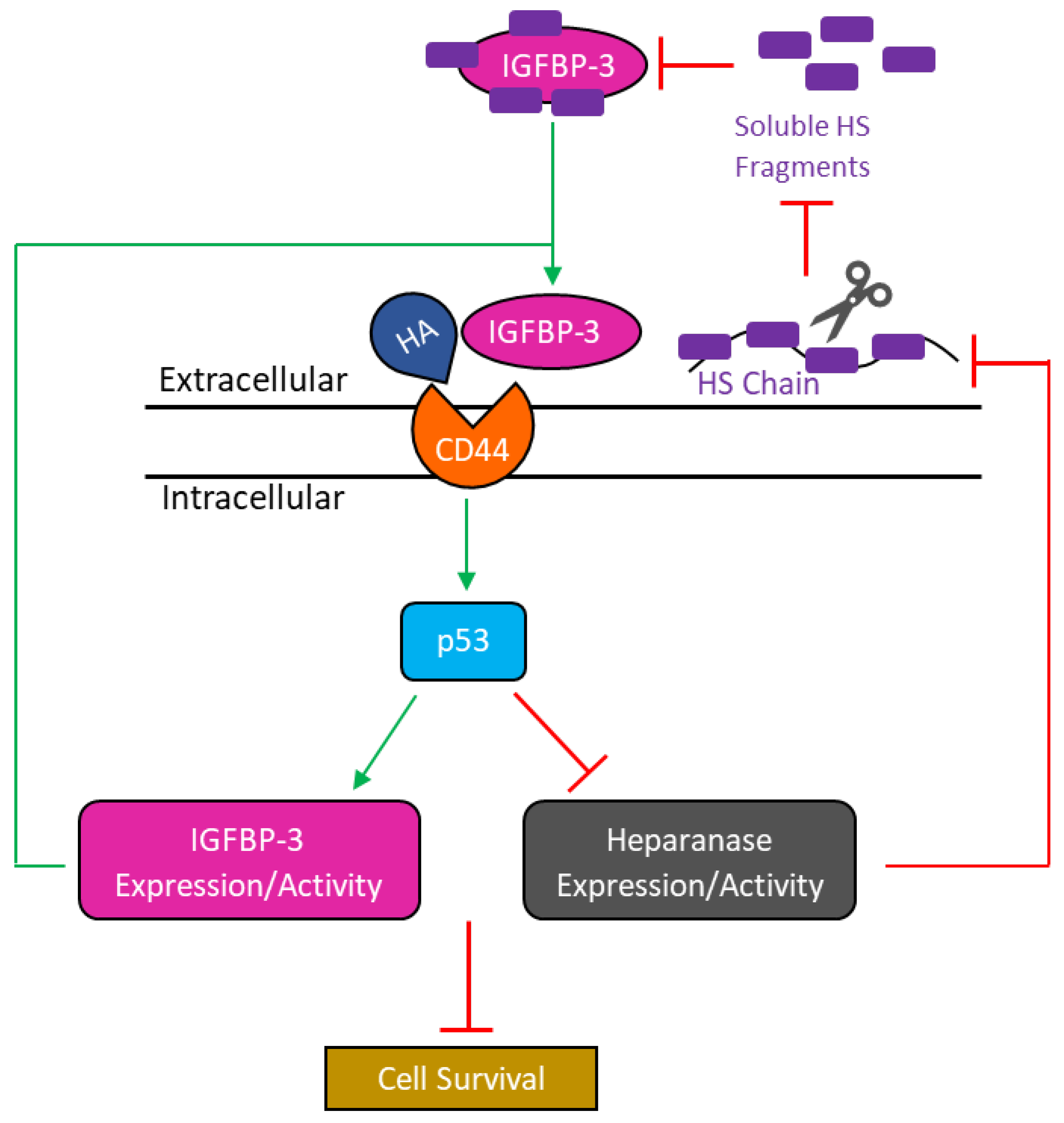
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The Biology and Management of Non-Small Cell Lung Cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, F.; Siddiqui, A.H. Lung Cancer. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Kang, H.; Wu, Q.; Sun, A.; Liu, X.; Fan, Y.; Deng, X. Cancer Cell Glycocalyx and Its Significance in Cancer Progression. Int. J. Mol. Sci. 2018, 19, 2484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knelson, E.H.; Nee, J.C.; Blobe, G.C. Heparan Sulfate Signaling in Cancer. Trends Biochem. Sci. 2014, 39, 277–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagarajan, A.; Malvi, P.; Wajapeyee, N. Heparan Sulfate and Heparan Sulfate Proteoglycans in Cancer Initiation and Progression. Front. Endocrinol. 2018, 9, 483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billings, P.C.; Pacifici, M. Interactions of Signaling Proteins, Growth Factors and Other Proteins with Heparan Sulfate: Mechanisms and Mysteries. Connect. Tissue Res. 2015, 56, 272–280. [Google Scholar] [CrossRef] [Green Version]
- Linker, A.; Hoffman, P.; Sampson, P.; Meyer, K. Heparitin Sulfate. Biochim. Biophys. Acta 1958, 29, 443–444. [Google Scholar] [CrossRef]
- Suhovskih, A.V.; Domanitskaya, N.V.; Tsidulko, A.Y.; Prudnikova, T.Y.; Kashuba, V.I.; Grigorieva, E.V. Tissue-Specificity of Heparan Sulfate Biosynthetic Machinery in Cancer. Cell Adh. Migr. 2015, 9, 452–459. [Google Scholar] [CrossRef] [Green Version]
- Friedmann, Y.; Vlodavsky, I.; Aingorn, H.; Aviv, A.; Peretz, T.; Pecker, I.; Pappo, O. Expression of Heparanase in Normal, Dysplastic, and Neoplastic Human Colonic Mucosa and Stroma. Evidence for Its Role in Colonic Tumorigenesis. Am. J. Pathol. 2000, 157, 1167–1175. [Google Scholar] [CrossRef]
- Heyman, B.; Yang, Y. Mechanisms of Heparanase Inhibitors in Cancer Therapy. Exp. Hematol. 2016, 44, 1002–1012. [Google Scholar] [CrossRef] [Green Version]
- Fux, L.; Ilan, N.; Sanderson, R.D.; Vlodavsky, I. Heparanase: Busy at the Cell Surface. Trends Biochem. Sci. 2009, 34, 511–519. [Google Scholar] [CrossRef]
- Vlodavsky, I.; Ilan, N.; Sanderson, R.D. Forty Years of Basic and Translational Heparanase Research. Adv. Exp. Med. Biol. 2020, 1221, 3–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misra, S.; Heldin, P.; Hascall, V.C.; Karamanos, N.K.; Skandalis, S.S.; Markwald, R.R.; Ghatak, S. HA/CD44 Interactions as Potential Targets for Cancer Therapy. FEBS J. 2011, 278, 1429–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, J.; Jiang, D.; Noble, P.W. Hyaluronan as a Therapeutic Target in Human Diseases. Adv. Drug Deliv. Rev. 2016, 97, 186–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cyphert, J.M.; Trempus, C.S.; Garantziotis, S. Size Matters: Molecular Weight Specificity of Hyaluronan Effects in Cell Biology. Int. J. Cell Biol. 2015, 2015, e563818. [Google Scholar] [CrossRef] [Green Version]
- Misra, S.; Hascall, V.C.; Markwald, R.R.; Ghatak, S. Interactions between Hyaluronan and Its Receptors (CD44, RHAMM) Regulate the Activities of Inflammation and Cancer. Front. Immunol. 2015, 6, 201. [Google Scholar] [CrossRef] [Green Version]
- Vigetti, D.; Karousou, E.; Viola, M.; Deleonibus, S.; De Luca, G.; Passi, A. Hyaluronan: Biosynthesis and Signaling. Biochim. Biophys. Acta 2014, 1840, 2452–2459. [Google Scholar] [CrossRef] [Green Version]
- Nam, E.J.; Park, P.W. Shedding of Cell Membrane-Bound Proteoglycans. Methods Mol. Biol. 2012, 836, 291–305. [Google Scholar] [CrossRef] [Green Version]
- Sherman, L.S.; Matsumoto, S.; Su, W.; Srivastava, T.; Back, S.A. Hyaluronan Synthesis, Catabolism, and Signaling in Neurodegenerative Diseases. Int. J. Cell Biol. 2015, 2015, e368584. [Google Scholar] [CrossRef] [Green Version]
- Toole, B.P. Hyaluronan: From Extracellular Glue to Pericellular Cue. Nat. Rev. Cancer 2004, 4, 528–539. [Google Scholar] [CrossRef]
- Nikitovic, D.; Kouvidi, K.; Kavasi, R.-M.; Berdiaki, A.; Tzanakakis, G.N. Hyaluronan/Hyaladherins—A Promising Axis for Targeted Drug Delivery in Cancer. Curr. Drug Deliv. 2016, 13, 500–511. [Google Scholar] [CrossRef]
- Chanmee, T.; Ontong, P.; Kimata, K.; Itano, N. Key Roles of Hyaluronan and Its CD44 Receptor in the Stemness and Survival of Cancer Stem Cells. Front. Oncol. 2015, 5, 180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kultti, A.; Li, X.; Jiang, P.; Thompson, C.B.; Frost, G.I.; Shepard, H.M. Therapeutic Targeting of Hyaluronan in the Tumor Stroma. Cancers 2012, 4, 873–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raso-Barnett, L.; Banky, B.; Barbai, T.; Becsagh, P.; Timar, J.; Raso, E. Demonstration of a Melanoma-Specific CD44 Alternative Splicing Pattern That Remains Qualitatively Stable, but Shows Quantitative Changes during Tumour Progression. PLoS ONE 2013, 8, e53883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aruffo, A.; Stamenkovic, I.; Melnick, M.; Underhill, C.B.; Seed, B. CD44 Is the Principal Cell Surface Receptor for Hyaluronate. Cell 1990, 61, 1303–1313. [Google Scholar] [CrossRef]
- Chen, C.; Zhao, S.; Karnad, A.; Freeman, J.W. The Biology and Role of CD44 in Cancer Progression: Therapeutic Implications. J. Hematol. Oncol. 2018, 11, 64. [Google Scholar] [CrossRef] [Green Version]
- Firth, S.M.; Baxter, R.C. Cellular Actions of the Insulin-like Growth Factor Binding Proteins. Endocr. Rev. 2002, 23, 824–854. [Google Scholar] [CrossRef]
- Jogie-Brahim, S.; Feldman, D.; Oh, Y. Unraveling Insulin-like Growth Factor Binding Protein-3 Actions in Human Disease. Endocr. Rev. 2009, 30, 417–437. [Google Scholar] [CrossRef] [Green Version]
- Johnson, M.A.; Firth, S.M. IGFBP-3: A Cell Fate Pivot in Cancer and Disease. Growth Horm. IGF Res. 2014, 24, 164–173. [Google Scholar] [CrossRef]
- Baxter, R.C. Insulin-like Growth Factor Binding Protein-3 (IGFBP-3): Novel Ligands Mediate Unexpected Functions. J. Cell Commun. Signal 2013, 7, 179–189. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.S.; Wang, L.; Liu, D.; Mao, L.; Hong, W.K.; Khuri, F.R.; Lee, H.-Y. Correlation between Insulin-like Growth Factor-Binding Protein-3 Promoter Methylation and Prognosis of Patients with Stage I Non-Small Cell Lung Cancer. Clin. Cancer Res. 2002, 8, 3669–3675. [Google Scholar]
- Ibanez de Caceres, I.; Cortes-Sempere, M.; Moratilla, C.; Machado-Pinilla, R.; Rodriguez-Fanjul, V.; Manguán-García, C.; Cejas, P.; López-Ríos, F.; Paz-Ares, L.; de CastroCarpeño, J.; et al. IGFBP-3 Hypermethylation-Derived Deficiency Mediates Cisplatin Resistance in Non-Small-Cell Lung Cancer. Oncogene 2010, 29, 1681–1690. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-Y.; Chun, K.-H.; Liu, B.; Wiehle, S.A.; Cristiano, R.J.; Hong, W.K.; Cohen, P.; Kurie, J.M. Insulin-like Growth Factor Binding Protein-3 Inhibits the Growth of Non-Small Cell Lung Cancer. Cancer Res. 2002, 62, 3530–3537. [Google Scholar] [PubMed]
- Chang, Y.S.; Wang, L.; Suh, Y.-A.; Mao, L.; Karpen, S.J.; Khuri, F.R.; Hong, W.K.; Lee, H.-Y. Mechanisms Underlying Lack of Insulin-like Growth Factor-Binding Protein-3 Expression in Non-Small-Cell Lung Cancer. Oncogene 2004, 23, 6569–6580. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-Y.; Moon, H.; Chun, K.-H.; Chang, Y.-S.; Hassan, K.; Ji, L.; Lotan, R.; Khuri, F.R.; Hong, W.K. Effects of Insulin-like Growth Factor Binding Protein-3 and Farnesyltransferase Inhibitor SCH66336 on Akt Expression and Apoptosis in Non–Small-Cell Lung Cancer Cells. J. Natl. Cancer Inst. 2004, 96, 1536–1548. [Google Scholar] [CrossRef] [Green Version]
- Põld, M.; Krysan, K.; Põld, A.; Dohadwala, M.; Heuze-Vourc’h, N.; Mao, J.T.; Riedl, K.L.; Sharma, S.; Dubinett, S.M. Cyclooxygenase-2 Modulates the Insulin-Like Growth Factor Axis in Non–Small-Cell Lung Cancer. Cancer Res. 2004, 64, 6549–6555. [Google Scholar] [CrossRef] [Green Version]
- Ho, G.Y.F.; Zheng, S.L.; Cushman, M.; Perez-Soler, R.; Kim, M.; Xue, X.; Wang, T.; Schlecht, N.F.; Tinker, L.; Rohan, T.E.; et al. Associations of Insulin and IGFBP-3 with Lung Cancer Susceptibility in Current Smokers. J. Natl. Cancer Inst. 2016, 108, djw012. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, K.; Laban, C.; McVittie, C.J.; Ogunkolade, W.; Khalaf, S.; Bustin, S.; Carpenter, R.; Jenkins, P.J. The Expression and Function of IGFBP-3 in Normal and Malignant Breast Tissue. Anticancer Res. 2009, 29, 3785–3790. [Google Scholar]
- Marzec, K.A.; Baxter, R.C.; Martin, J.L. Targeting Insulin-Like Growth Factor Binding Protein-3 Signaling in Triple-Negative Breast Cancer. Biomed. Res. Int. 2015, 2015, 638526. [Google Scholar] [CrossRef]
- Velcheti, V.; Govindan, R. Insulin-Like Growth Factor and Lung Cancer. J. Thorac. Oncol. 2006, 1, 607–610. [Google Scholar] [CrossRef] [Green Version]
- Rajah, R.; Khare, A.; Lee, P.D.; Cohen, P. Insulin-like Growth Factor-Binding Protein-3 Is Partially Responsible for High-Serum-Induced Apoptosis in PC-3 Prostate Cancer Cells. J. Endocrinol. 1999, 163, 487–494. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Wang, Z.; Liang, Z.; Liu, J.; Shi, W.; Bai, P.; Lin, X.; Magaye, R.; Zhao, J. Expression and Clinical Significance of IGF-1, IGFBP-3, and IGFBP-7 in Serum and Lung Cancer Tissues from Patients with Non-Small Cell Lung Cancer. OncoTargets Ther. 2013, 6, 1437–1444. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Spitz, M.R.; Mistry, J.; Gu, J.; Hong, W.K.; Wu, X. Plasma Levels of Insulin-like Growth Factor-I and Lung Cancer Risk: A Case-Control Analysis. J. Natl. Cancer Inst. 1999, 91, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Grimberg, A.; Liu, B.; Bannerman, P.; El-Deiry, W.S.; Cohen, P. IGFBP-3 Mediates P53-Induced Apoptosis during Serum Starvation. Int. J. Oncol. 2002, 21, 327–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takaoka, M.; Harada, H.; Andl, C.D.; Oyama, K.; Naomoto, Y.; Dempsey, K.L.; Klein-Szanto, A.J.; El-Deiry, W.S.; Grimberg, A.; Nakagawa, H. Epidermal Growth Factor Receptor Regulates Aberrant Expression of Insulin-Like Growth Factor-Binding Protein 3. Cancer Res. 2004, 64, 7711–7723. [Google Scholar] [CrossRef] [Green Version]
- Fowlkes, J.L.; Serra, D.M. Characterization of Glycosaminoglycan-Binding Domains Present in Insulin-like Growth Factor-Binding Protein-3. J. Biol. Chem. 1996, 271, 14676–14679. [Google Scholar] [CrossRef] [Green Version]
- Fowlkes, J.L.; Thrailkill, K.M.; George-Nascimento, C.; Rosenberg, C.K.; Serra, D.M. Heparin-Binding, Highly Basic Regions within the Thyroglobulin Type-1 Repeat of Insulin-Like Growth Factor (IGF)-Binding Proteins (IGFBPs) -3, -5, and -6 Inhibit IGFBP-4 Degradation. Endocrinology 1997, 138, 2280–2285. [Google Scholar] [CrossRef]
- Booth, B.A.; Boes, M.; Andress, D.L.; Dake, B.L.; Kiefer, M.C.; Maack, C.; Linhardt, R.J.; Bar, K.; Caldwell, E.E.; Weiler, J. IGFBP-3 and IGFBP-5 Association with Endothelial Cells: Role of C-Terminal Heparin Binding Domain. Growth Regul. 1995, 5, 1–17. [Google Scholar]
- Yang, Y.W.; Yanagishita, M.; Rechler, M.M. Heparin Inhibition of Insulin-like Growth Factor-Binding Protein-3 Binding to Human Fibroblasts and Rat Glioma Cells: Role of Heparan Sulfate Proteoglycans. Endocrinology 1996, 137, 4363–4371. [Google Scholar] [CrossRef] [Green Version]
- Muterspaugh, R.; Price, D.; Esckilsen, D.; McEachern, S.; Guthrie, J.; Heyl, D.; Evans, H.G. Interaction of Insulin-Like Growth Factor-Binding Protein 3 With Hyaluronan and Its Regulation by Humanin and CD44. Biochemistry 2018, 57, 5726–5737. [Google Scholar] [CrossRef]
- Price, D.; Muterspaugh, R.; Clegg, B.; Williams, A.; Stephens, A.; Guthrie, J.; Heyl, D.; Guy Evans, H. IGFBP-3 Blocks Hyaluronan-CD44 Signaling, Leading to Increased Acetylcholinesterase Levels in A549 Cell Media and Apoptosis in a P53-Dependent Manner. Sci. Rep. 2020, 10, 5083–5099. [Google Scholar] [CrossRef] [Green Version]
- Dorandish, S.; Devos, J.; Clegg, B.; Price, D.; Muterspaugh, R.; Guthrie, J.; Heyl, D.L.; Evans, H.G. Biochemical Determinants of the IGFBP-3-Hyaluronan Interaction. FEBS Open Bio. 2020, 10, 1668–1684. [Google Scholar] [CrossRef] [PubMed]
- Price, D.; Dorandish, S.; Williams, A.; Iwaniec, B.; Stephens, A.; Marshall, K.; Guthrie, J.; Heyl, D.; Evans, H.G. Humanin Blocks the Aggregation of Amyloid-β Induced by Acetylcholinesterase, an Effect Abolished in the Presence of IGFBP-3. Biochemistry 2020, 59, 1981–2002. [Google Scholar] [CrossRef]
- Dorandish, S.; Atali, S.; Ray, R.; Al Khashali, H.; Coleman, K.-L.; Guthrie, J.; Heyl, D.; Evans, H.G. Differences in the Relative Abundance of ProBDNF and Mature BDNF in A549 and H1299 Human Lung Cancer Cell Media. Int. J. Mol. Sci. 2021, 22, 7059. [Google Scholar] [CrossRef] [PubMed]
- Atali, S.; Dorandish, S.; Devos, J.; Williams, A.; Price, D.; Taylor, J.; Guthrie, J.; Heyl, D.; Evans, H.G. Interaction of Amyloid Beta with Humanin and Acetylcholinesterase Is Modulated by ATP. FEBS Open Bio. 2020, 10, 2805–2823. [Google Scholar] [CrossRef]
- Dorandish, S.; Williams, A.; Atali, S.; Sendo, S.; Price, D.; Thompson, C.; Guthrie, J.; Heyl, D.; Evans, H.G. Regulation of Amyloid-β Levels by Matrix Metalloproteinase-2/9 (MMP2/9) in the Media of Lung Cancer Cells. Sci. Rep. 2021, 11, 9708. [Google Scholar] [CrossRef] [PubMed]
- Al Khashali, H.; Ray, R.; Coleman, K.-L.; Atali, S.; Haddad, B.; Wareham, J.; Guthrie, J.; Heyl, D.; Evans, H.G. Regulation of the Soluble Amyloid Precursor Protein α (SAPPα) Levels by Acetylcholinesterase and Brain-Derived Neurotrophic Factor in Lung Cancer Cell Media. Int. J. Mol. Sci. 2022, 23, 10746. [Google Scholar] [CrossRef]
- Ray, R.; Al Khashali, H.; Haddad, B.; Wareham, J.; Coleman, K.-L.; Alomari, D.; Ranzenberger, R.; Guthrie, J.; Heyl, D.; Evans, H.G. Regulation of Cisplatin Resistance in Lung Cancer Cells by Nicotine, BDNF, and a β-Adrenergic Receptor Blocker. Int. J. Mol. Sci. 2022, 23, 12829. [Google Scholar] [CrossRef]
- Ikonen, M.; Liu, B.; Hashimoto, Y.; Ma, L.; Lee, K.-W.; Niikura, T.; Nishimoto, I.; Cohen, P. Interaction between the Alzheimer’s Survival Peptide Humanin and Insulin-like Growth Factor-Binding Protein 3 Regulates Cell Survival and Apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 13042–13047. [Google Scholar] [CrossRef] [Green Version]
- Heyl, D.L.; Iwaniec, B.; Esckilsen, D.; Price, D.; Guttikonda, P.; Cooper, J.; Lombardi, J.; Milletti, M.; Evans, H.G. Using Small Peptide Segments of Amyloid-β and Humanin to Examine Their Physical Interactions. Protein Pept. Lett. 2019, 26, 502–511. [Google Scholar] [CrossRef]
- Evans, H.G.; Guthrie, J.W.; Jujjavarapu, M.; Hendrickson, N.; Eitel, A.; Park, Y.; Garvey, J.; Newman, R.; Esckilsen, D.; Heyl, D.L. D-Amino Acid Analogues of the Antimicrobial Peptide CDT Exhibit Anti- Cancer Properties in A549, a Human Lung Adenocarcinoma Cell Line. Protein Pept. Lett. 2017, 24, 590–598. [Google Scholar] [CrossRef]
- Ye, X.; Zhang, C.; Chen, Y.; Zhou, T. Upregulation of Acetylcholinesterase Mediated by P53 Contributes to Cisplatin-Induced Apoptosis in Human Breast Cancer Cell. J. Cancer 2015, 6, 48–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baraz, L.; Haupt, Y.; Elkin, M.; Peretz, T.; Vlodavsky, I. Tumor Suppressor P53 Regulates Heparanase Gene Expression. Oncogene 2006, 25, 3939–3947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leroy, B.; Girard, L.; Hollestelle, A.; Minna, J.D.; Gazdar, A.F.; Soussi, T. Analysis of TP53 Mutation Status in Human Cancer Cell Lines: A Reassessment. Hum. Mutat. 2014, 35, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Oren, M. The First 30 Years of P53: Growing Ever More Complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef] [Green Version]
- Hafner, A.; Bulyk, M.L.; Jambhekar, A.; Lahav, G. The Multiple Mechanisms That Regulate P53 Activity and Cell Fate. Nat. Rev. Mol. Cell Biol. 2019, 20, 199–210. [Google Scholar] [CrossRef]
- Godar, S.; Ince, T.A.; Bell, G.W.; Feldser, D.; Donaher, J.L.; Bergh, J.; Liu, A.; Miu, K.; Watnick, R.S.; Reinhardt, F.; et al. Growth-Inhibitory and Tumor- Suppressive Functions of P53 Depend on Its Repression of CD44 Expression. Cell 2008, 134, 62–73. [Google Scholar] [CrossRef] [Green Version]
- Dhar, D.; Antonucci, L.; Nakagawa, H.; Kim, J.Y.; Glitzner, E.; Caruso, S.; Shalapour, S.; Yang, L.; Valasek, M.A.; Lee, S.; et al. Liver Cancer Initiation Requires P53 Inhibition by CD44-Enhanced Growth Factor Signaling. Cancer Cell 2018, 33, 1061–1077.e6. [Google Scholar] [CrossRef] [Green Version]
- Hollowood, A.D.; Lai, T.; Perks, C.M.; Newcomb, P.V.; Alderson, D.; Holly, J.M. IGFBP-3 Prolongs the P53 Response and Enhances Apoptosis Following UV Irradiation. Int. J. Cancer 2000, 88, 336–341. [Google Scholar] [CrossRef]
- Butt, A.J.; Williams, A.C. IGFBP-3 and Apoptosis—A Licence to Kill? Apoptosis 2001, 6, 199–205. [Google Scholar] [CrossRef]
- Buckbinder, L.; Talbott, R.; Velasco-Miguel, S.; Takenaka, I.; Faha, B.; Seizinger, B.R.; Kley, N. Induction of the Growth Inhibitor IGF-Binding Protein 3 by P53. Nature 1995, 377, 646–649. [Google Scholar] [CrossRef]
- Wang, Z.; Piha-Paul, S.; Janku, F.; Subbiah, V.; Shi, N.; Gong, J.; Wathoo, C.; Shaw, K.; Hess, K.; Broaddus, R.; et al. Antiangiogenesis and Gene Aberration-Related Therapy May Improve Overall Survival in Patients with Concurrent KRAS and TP53 Hotspot Mutant Cancer. Oncotarget 2017, 8, 33796–33806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saleh, M.M.; Scheffler, M.; Merkelbach-Bruse, S.; Scheel, A.H.; Ulmer, B.; Wolf, J.; Buettner, R. Comprehensive Analysis of TP53 and KEAP1 Mutations and Their Impact on Survival in Localized- and Advanced-Stage NSCLC. J. Thorac. Oncol. 2022, 17, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant P53 as a Guardian of the Cancer Cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutant P53 as a Regulator and Target of Autophagy—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/33614491/ (accessed on 3 November 2022).
- Freed-Pastor, W.A.; Prives, C. Mutant P53: One Name, Many Proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef] [Green Version]
- Murnyák, B.; Hortobágyi, T. Immunohistochemical Correlates of TP53 Somatic Mutations in Cancer. Oncotarget 2016, 7, 64910–64920. [Google Scholar] [CrossRef] [Green Version]
- Kotler, E.; Shani, O.; Goldfeld, G.; Lotan-Pompan, M.; Tarcic, O.; Gershoni, A.; Hopf, T.A.; Marks, D.S.; Oren, M.; Segal, E. A Systematic P53 Mutation Library Links Differential Functional Impact to Cancer Mutation Pattern and Evolutionary Conservation. Mol. Cell 2018, 71, 873. [Google Scholar] [CrossRef] [Green Version]
- Pavlakis, E.; Stiewe, T. P53’s Extended Reach: The Mutant P53 Secretome. Biomolecules 2020, 10, 307. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.; Zhou, Y.; Huang, L.; Ou, W.; Wu, J.; Li, S.; Xu, J.; Feng, J.; Liu, B. TP53 Mutation Is Associated with a Poor Clinical Outcome for Non-Small Cell Lung Cancer: Evidence from a Meta-Analysis. Mol. Clin. Oncol. 2016, 5, 705–713. [Google Scholar] [CrossRef] [Green Version]
- Tsao, M.-S.; Aviel-Ronen, S.; Ding, K.; Lau, D.; Liu, N.; Sakurada, A.; Whitehead, M.; Zhu, C.-Q.; Livingston, R.; Johnson, D.H.; et al. Prognostic and Predictive Importance of P53 and RAS for Adjuvant Chemotherapy in Non Small-Cell Lung Cancer. J. Clin. Oncol. 2007, 25, 5240–5247. [Google Scholar] [CrossRef]
- Custodio, A.B.; González-Larriba, J.L.; Bobokova, J.; Calles, A.; Alvarez, R.; Cuadrado, E.; Manzano, A.; Díaz-Rubio, E. Prognostic and Predictive Markers of Benefit from Adjuvant Chemotherapy in Early-Stage Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2009, 4, 891–910. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al Khashali, H.; Wareham, J.; Ray, R.; Haddad, B.; Coleman, K.-L.; Ranzenberger, R.; McCombs, P.; Guthrie, J.; Heyl, D.; Evans, H.G. Opposing Roles of IGFBP-3 and Heparanase in Regulating A549 Lung Cancer Cell Survival. Cells 2022, 11, 3533. https://doi.org/10.3390/cells11223533
Al Khashali H, Wareham J, Ray R, Haddad B, Coleman K-L, Ranzenberger R, McCombs P, Guthrie J, Heyl D, Evans HG. Opposing Roles of IGFBP-3 and Heparanase in Regulating A549 Lung Cancer Cell Survival. Cells. 2022; 11(22):3533. https://doi.org/10.3390/cells11223533
Chicago/Turabian StyleAl Khashali, Hind, Jadziah Wareham, Ravel Ray, Ben Haddad, Kai-Ling Coleman, Robert Ranzenberger, Patrick McCombs, Jeffrey Guthrie, Deborah Heyl, and Hedeel Guy Evans. 2022. "Opposing Roles of IGFBP-3 and Heparanase in Regulating A549 Lung Cancer Cell Survival" Cells 11, no. 22: 3533. https://doi.org/10.3390/cells11223533